Introduction
The maintenance of genome integrity is fundamental
to cellular homeostasis and organismal health, yet the genome is
continually challenged by both endogenous and exogenous genotoxic
stresses. DNA damage response (DDR) pathways orchestrate detection,
signaling and repair of DNA lesions to preserve genomic stability.
Among various types of DNA damage, DNA double-strand breaks (DSBs)
represent the most lethal and deleterious lesions, with the
potential to compromise chromosome integrity and cellular viability
if unrepaired or misrepaired (1).
Inefficient or erroneous DDR leads to genome instability, a
hallmark of cancer that fuels tumor initiation, progression and
therapeutic resistance (2,3).
Germline mutations in key DDR genes, such as breast cancer gene
(BRCA) 1/2 and ataxia-telangiectasia mutated (ATM), are known to
predispose individuals to cancer by impairing efficient DSB repair,
underscoring the crucial role of DDR in tumor suppression (4-7).
Cells incur tens of DSBs per day under physiological
conditions, resulting from replication stress, oxidative damage and
metabolic byproducts (8). In
cancer cells, rapid proliferation and oncogene-induced replication
stress exacerbate DSB accumulation, while therapeutic interventions
such as ionizing radiation (IR) and chemotherapeutics further
increase the burden of DSBs (9-11).
Successfully evolving or surviving cancer cells often exhibit
enhanced or altered DNA repair competencies, which contributes to
therapeutic resistance and disease relapse (12).
Classical DSB repair in human cells primarily
involves non-homologous end joining (NHEJ) and homologous
recombination (HR). NHEJ quickly rejoins broken DNA ends with
minimal processing, while HR uses a homologous template for
error-free repair, predominantly during S/G2 phases
(13,14). However, a third repair pathway,
alternative end-joining (alt-EJ), has emerged as a distinct
mechanism with a unique biological footprint, which is also
referred to as microhomology-mediated end joining (MMEJ) or
alternative NHEJ (alt-NHEJ) or theta mediated end joining (TMEJ)
(15-19). Alt-EJ leverages microhomologous
DNA sequence at DSB ends for annealing, frequently resulting in
deletions, insertions or complex chromosomal rearrangements
(1,20,21).
Alt-EJ was initially considered a backup or
error-prone compensatory pathway used only when NHEJ or HR fails.
However, a growing body of evidence in recent years has revealed
that alt-EJ is actively engaged and tightly regulated in various
contexts within cancer cells in particular (14,22,23). The increased reliance on alt-EJ by
cancer cells is likely multi-factorial, reflecting intrinsic DDR
alterations, replication stress adaptation and selective pressures
from DNA-damaging therapies. Importantly, the inherent mutagenic
potential of alt-EJ contributes not only to genome destabilization
but also to tumor heterogeneity and evolution (15,22,24,25). Furthermore, a recent study has
discovered that alt-EJ plays an important role in maintenance of
extrachromosomal DNA in tumor cells (15).
Beyond oncogenic contexts, alt-EJ also operates in
normal physiology. During immunoglobulin class switch recombination
(CSR) in activated B cells, cytidine deaminase (AID) is induced and
creates lesions in donor and acceptor switch (S) regions that are
processed into DSBs by UNG and APE1 (26). While canonical NHEJ (c-NHEJ)
mediates most joins, restriction of c-NHEJ factors or enhanced end
resection diverts repair to alt-EJ (27,28). Alt-EJ mediated CSR exhibits a
distinctive mutational signature: Increased microhomology at
junctions with longer resection tracts, larger deletions within S
regions, templated insertions consistent with POLQ mediated end
joining and an elevated rate of inter chromosomal translocations
(29-32). These features illustrate that
alt-EJ is intrinsically mutagenic even in a programmed
developmental setting.
Despite significant advances having been made in
recent years, critical aspects of the molecular regulation of
alt-EJ, the contextual cues that determine its engagement over
canonical pathways and its precise effect on cancer genome
landscapes remain incompletely defined. This gap hampers full
exploitation of alt-EJ as a therapeutic target, although promising
evidence has shown that inhibition of alt-EJ components sensitizes
resistant tumors and enhances treatment efficacy (22,33,34).
The present review aimed to consolidate current
knowledge of alt-EJ mechanisms, including the key factors and
regulatory networks that control its activity; to dissect its dual
roles in maintaining genome integrity compared with driving
instability; and to evaluate its emerging contributions to cancer
initiation, progression and therapeutic response. Furthermore, it
highlight the therapeutic potential of targeting alt-EJ vis-à-vis
recent advances in inhibitors and biomarkers that could transform
cancer treatment paradigms.
Alt-EJ in DSB Repair
DSB repair mechanisms
DSBs are primarily repaired via three major pathways
in cancer cells: NHEJ, HR and alt-EJ, with single-strand annealing
(SSA) also used in certain circumstances (Fig. 1). These pathways differ in
mechanism, repair fidelity and cell cycle regulation, collectively
maintaining genome stability while balancing repair efficiency and
accuracy (21,35-38).
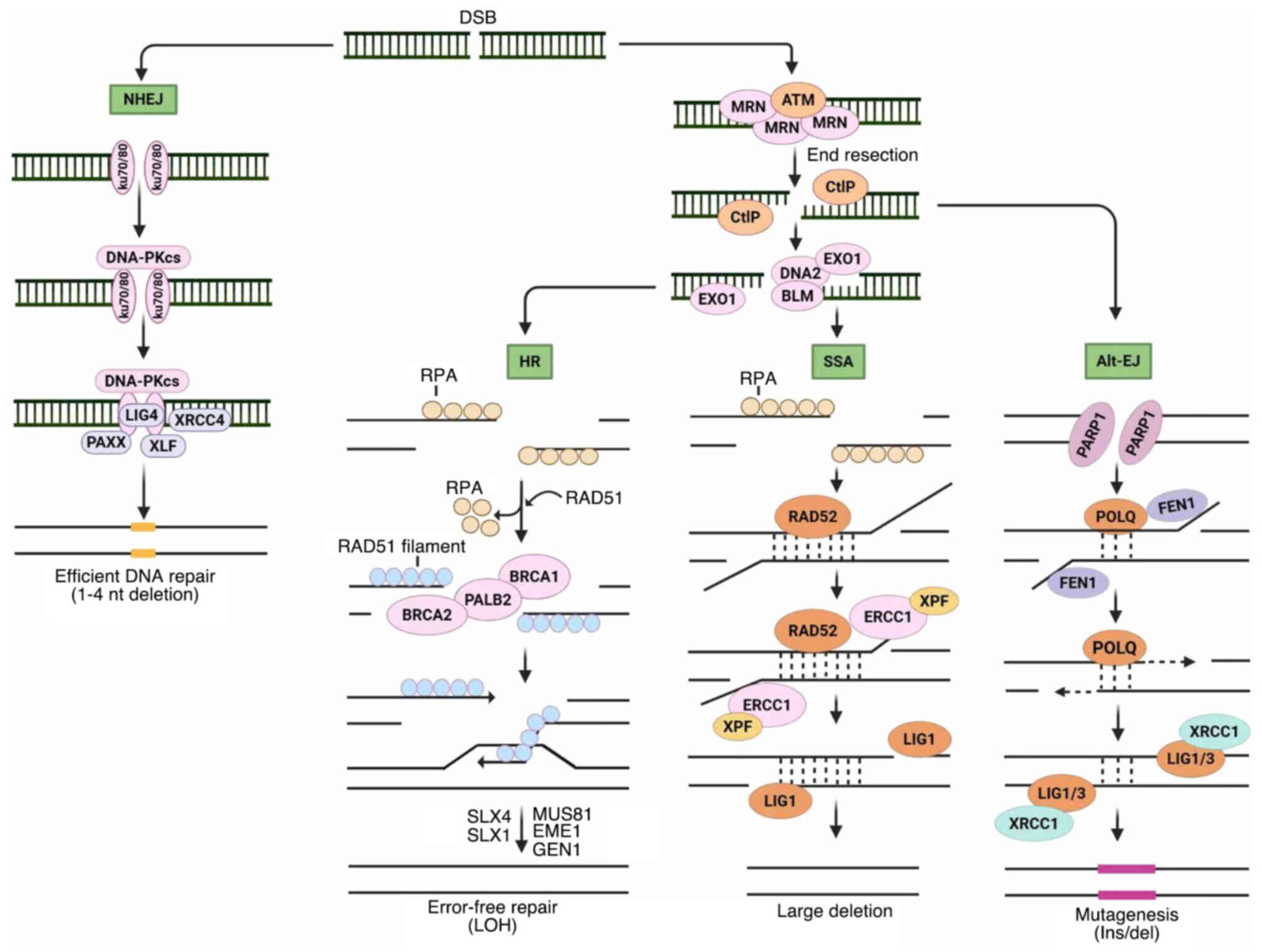 | Figure 1Major pathways for DSB repair. NHEJ
begins with Ku70-Ku80 hetero-dimer attaching to DNA ends. DNA-PKcs
recruitment and auto-phosphorylation bring the DNA ends together
and allow ligation by XRCC4-LIG4 and XLF or PAXX. Resection by MRN
complex and CtIP promotes homology-directed repair. Long-range
resection creates RPA-coated ssDNA overhangs using BLM-DNA2
helicase-nuclease or EXO1 nucleases. HR occurs when BRCA1, PALB2
and BRCA2 facilitate loading RAD51 onto ssDNA and displace RPA.
RAD51 nucleoprotein filaments invade the DNA-synthesis template
sister chromatids. Alternatively, substantial resection generates a
substrate for SSA, where RAD52 promotes homologous sequence
annealing on each DNA end. ERCC1 and XPF handle 3'single-stranded
flaps for LIG1-mediated DNA ligation. DSB resection also activates
alt-EJ via PARP1, where POLQ anneals short homologous sequences,
synthesizes DNA and re-ligates DNA ends using LIG1 or LIG3. DSB,
DNA double-strand break; NHEJ, non-homologous end joining;
DNA-PKcs, DNA-dependent protein kinase catalytic subunit; ssDNA,
single-stranded DNA; HR, homologous recombination; alt-EJ,
alternative end-joining; PARP, poly (ADP-ribose) polymerase; XRCC4,
X-ray repair cross-complementing protein 4; LIG4, DNA ligase 4;
XLF, XRCC4-like factor; PAXX, paralogue of XRCC4 and XLF; CtIP,
c-terminal-binding protein-interacting protein; RPA, replication
protein A; BLM, Bloom syndrome helicase; DNA2, DNA replication
helicase/nuclease 2; EXO1, exonuclease 1; BRCA1/2, breast cancer
gene 1/2; PALB2, partner and localizer of BRCA2; SSA, single strand
annealing; ERCC1, excision repair cross complementation group 1;
XPF, xeroderma pigmentosum group F; POLQ, DNA polymerase theta;
LIG1/3, DNA ligase1/3; Ins/del, insertion or deletion; LOH, loss of
heterogeneity; nt, nucleotide. |
NHEJ is the predominant DSB repair pathway
throughout the cell cycle and is especially active during
G1 phase. It involves recognition of broken DNA ends by
the Ku70/80 heterodimer, which recruits and activates the kinase of
DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and
Artemis to process DSB ends, followed by ligation with
XRCC4-LIG4-XLF complex (39-41). NHEJ typically re-ligates breaks
with minimal processing, which occasionally causes small insertions
or deletions.
HR offers high-fidelity repair by using a homologous
template, primarily in S/G2 phases. The MRN complex
(MRE11-RAD50-NBS1) recognizes DSBs, which activates ATM kinase,
initiates MRE11-mediated resection and produces 3'single-stranded
DNA (ssDNA) overhangs coated by RPA, facilitating replacement by
RAD51 through BRCA1/2 mediation (13). This mediates strand invasion and
template-directed repair via gene conversion or break-induced
replication. HR deficiency (HRD) tumors usually show
hypersensitivity poly (ADP-ribose) polymerase (PARP) inhibitors
(PARPi) (42-44).
SSA, although less common, uses extensive homology
between repeat sequences flanking a DSB, which contributes to
genome instability when dysregulated (38). Initiation of SSA requires
extensive 5'-3' end resection producing long 3' (ssDNA) tails bound
by RPA, followed by RAD52-mediated annealing of complementary
repeats (45,46).
Molecular mechanisms of alt-EJ
Alt-EJ was first discovered in NHEJ-deficient cells
and considered an alternate repair route for rejoining DSBs in the
presence of compromised NHEJ and HR (47,48). At present, alt-EJ has gained
prominence as a distinct, regulated pathway that contributes to
both physiological repair processes and pathological genome
instability, particularly in cancer. Alt-EJ utilizes short
microhomologous sequences, typically 2-20 base pairs, near DSB ends
to facilitate alignment and repair. Unlike NHEJ, which ligates DNA
ends with minimal processing, or HR, which uses templates for
error-free repair, alt-EJ requires DNA end resection and often
results in deletions, insertions and chromosomal rearrangements,
thereby contributing markedly to genomic instability in cancer
(49,50).
The Alt-EJ process is initiated by rapid detection
of DSBs and stabilization of broken DNA ends, predominantly
mediated by PARP1, which recruits repair factors and modulates
chromatin at break sites (51,52). The MRN complex, in concert with
CtIP, catalyzes 5'-3' resection of the DNA ends, generating 3'
ssDNA overhangs required for microhomology exposure (1,53).
When microhomology sites are spaced farther apart, exonuclease 1
(EXO1) and Bloom syndrome helicase (BLM) extend this resection,
producing longer ssDNA tracts (21,53,54). This extensive resection sharply
distinguishes alt-EJ from NHEJ, which repairs minimally processed
ends.
Alt-EJ exploits short stretches of microhomology
exposed on the complementary 3' ssDNA overhangs for annealing. This
step is inherently mutagenic, frequently generating nucleotide
deletions or insertions at repair junctions (21,49). DNA Pol θ, encoded by POLQ, is the
key mediator of this process, performing DNA synthesis that extends
and stabilizes annealed microhomologies, thereby bridging the
broken ends (55,56).
POLQ is unique in its domain architecture and
consists of an N-terminal helicase-like domain and a C-terminal
A-family polymerase domain linked by a central region. The domains
both contribute to the end-joining activity of POLQ by stabilizing
DNA synapses and catalyzing synthesis across partially paired or
mismatched templates (18).
POLQ's low-fidelity polymerase lacks 3'-5' proofreading and
tolerates base mispairing, enabling it to efficiently extend from
short microhomologies but often introducing mutations or templated
insertions that constitute the signatures of alt-EJ repair
(57,58). The final ligation step in alt-EJ
is mediated by LIG1 or LIG3, in complex with XRCC1 (59-61).
Beyond POLQ, DNA polymerase λ (Polλ), an X-family
polymerase typically involved in NHEJ, has been implicated in an
alternative alt-EJ mechanism. Polλ promotes alt-EJ independently of
canonical NHEJ factors by stabilizing DNA end synapses with minimal
base pairing and inserting nucleotides at gaps flanked by
microhomologies of 4-6 base pairs. This polymerase acts on a range
of substrates, including 5-12 nucleotide single-stranded overhangs
and small gaps, expanding the diversity of polymerases supporting
alt-EJ (62).
Following gap-filling synthesis by POLQ or Polλ,
displaced 3' ssDNA flaps must be removed to generate proper DNA
ends for ligation. The apurinic/apyrimidinic endonuclease APE2 has
recently emerged as a critical nuclease in this process. APE2
exhibits 3'-5' exonuclease and flap endonuclease activities,
enabling it to cleave 3' flaps generated during alt-EJ and
facilitate end processing necessary for subsequent repair (63-65). Intriguingly, APE2 depletion
sensitizes homologous recombination-deficient cells, highlighting
its complementary role and synthetic lethality relationship to POLQ
(63). This synergy underscores
the importance of APE2 in maintaining genome stability by enabling
POLQ-driven alt-EJ in contexts where NHEJ or HR is defective.
PARP1 recruitment is essential, particularly when
NHEJ and HR are compromised, as it promotes assembly of alt-EJ
factors at damage sites (23).
POLQ is a central determinant of alt-EJ, not only for carrying out
annealing and extension at microhomologies but also for
antagonizing HR by preventing RAD51 filament formation (66-68). The 9-1-1
(RAD9A-RAD1-HUS1)/Rad9-Hus1-Rad1-interacting nuclear orphan (RHINO)
complex also directs POLQ to DSBs during mitosis, highlighting the
cell cycle-specific regulation of alt-EJ (69).
Although microhomology usage is often considered a
defining feature of alt-EJ, numerous repair junctions formed by
this pathway lack clear microhomologous sequences. For example,
studies quantifying chromosomal aberrations suggest that up to 50%
of events attributed to alt-EJ occur without detectable
microhomology, even when they require POLQ or other essential
alt-EJ components (18,70). This may reflect variability in DNA
end resection, the influence of local sequence context, or the
capacity of alt-EJ polymerases such as POLQ to facilitate end
joining through template-independent synthesis (56,71). These findings indicate that alt-EJ
is a mechanistically flexible and heterogeneous pathway and that
reliance solely on microhomology as a marker may underestimate its
role in genome instability and cancer.
Factors that affecting Alt-EJ
The efficiency and reliance on alt-EJ in cells,
especially cancer cells, are shaped by a complex interplay between
intrinsic genomic factors, regulatory molecules, cell cycle status
and external signals (Fig. 2).
Alt-EJ activity is governed by the degree of 5'-3' resection at DSB
ends, initiated by the MRN complex with CtIP and further extended
by nucleases/helicases such as EXO1 and BLM (53,72). This resection exposes
microhomologies, with the length of ssDNA determining whether short
(2-6 bp) or longer (>10 bp) microhomologies are used (73). Factors such as 53BP1 restrain
resection, favoring classical NHEJ and loss or suppression of 53BP1
skews repair towards alt-EJ (74). Deficiencies in other factors, such
as the ssDNA-binding protein RPA, can also potentiate alt-EJ by
increasing the use of longer microhomologies and promoting
chromosomal rearrangements (73,75). Moreover, components mutated in
Fanconi Anemia (FA), a disorder characterized by defective
interstrand crosslink (ICL) repair, have been implicated in
modulating alt-EJ activity, suggesting cross-talk between
replication stress responses and alt-EJ (23,76,77).
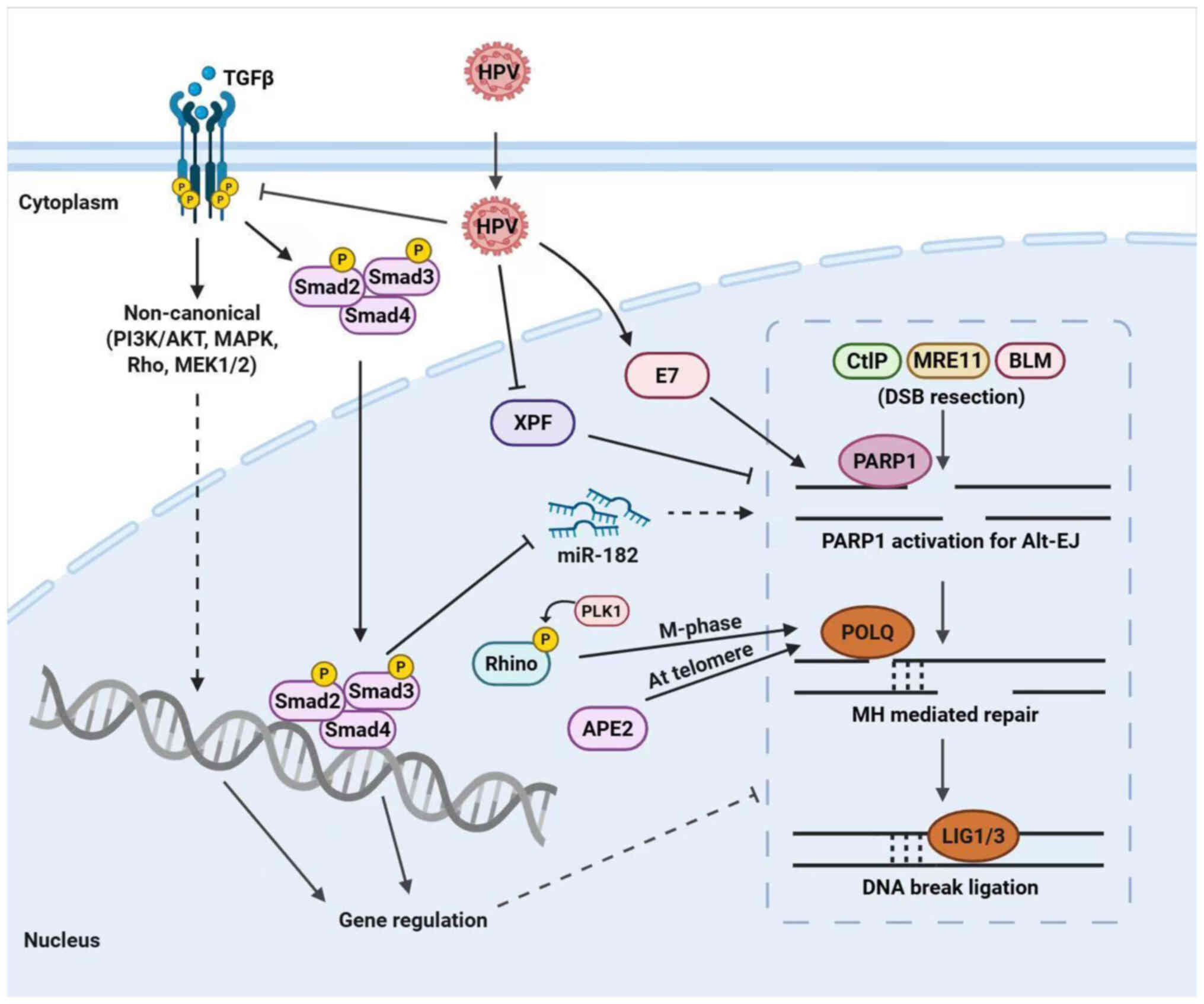 | Figure 2Schematic illustration of alt-EJ
effectors. The alt-EJ pathway is influenced by several factors.
TGFβ signaling begins in the cytoplasm and activates Smad proteins
and non-canonical pathways to regulate DDR through miR-182 and
other mechanisms (24,25,78). Loss of TGFβ signaling, such as
with the effects induced by HPV, causes enhanced alt-EJ activity
(25,79). The HPV oncoprotein E7 directs DNA
repair to alt-EJ (83). PLK1
facilitate RHINO to accumulate in the M phase recruit POLQ to the
break site (69). APE2 interacts
with POLQ in an epistatic way for alt-EJ (63). CtIP, Mre11 and BLM proteins
regulate DNA end resection, which subsequently facilitates the
process of strand annealing and the coupling of fragmented DNA ends
through the annealing of microhomologies. alt-EJ, alternative
end-joining; DDR, DNA damage response; TGFβ, transforming growth
factor-beta; Smad, suppressor of mothers against decapentaplegic;
miR, microRNA; HPV, human papillomavirus; PLK1, polo-like kinase 1;
APE2, apurinic/apyrimidinic endodeoxyribonuclease 2; RHINO,
Rad9-Hus1-Rad1-interacting nuclear orphan; POLQ, DNA polymerase θ;
CtIP, c-terminal-binding protein-interacting protein; MRE11,
Meiotic recombination 11; BLM, Bloom syndrome helicase; PARP, poly
(ADP-ribose) polymerase. |
Alt-EJ is also subject to regulation by
extracellular signals, notably TGFβ. Upon activation in the tissue
microenvironment, TGFβ ligands bind to heteromeric complexes of
serine/threonine kinase receptors, TGFβ receptor type I and type
II, initiating downstream phosphorylation cascades primarily
mediated by receptor-regulated suppressor of mother against
decapentaplegic (Smad) proteins (Smad2, Smad3 and Smad4) that
translocate to the nucleus, or by non-canonical TGFβ pathways
(Fig. 2). These TGFβ signaling
transduction pathways orchestrate a broad array of biological
functions that govern cell cycle, differentiation, apoptosis and
importantly, DDR pathways (25,78,79). Active TGFβ signaling supports
ATM-dependent DDR and suppresses alt-EJ gene expression (80). Conversely, TGFβ pathway
inhibition, frequently observed in the tumor microenvironment or
certain viral infections, upregulates alt-EJ components (POLQ,
PARP1 and LIG1) and increases reliance on this mutagenic pathway,
leading to more frequent chromosomal aberrations (25,81). Mechanistically, this effect is
partly mediated by the microRNA miR-182, which is upregulated upon
TGFβ inhibition and acts to repress key HR and NHEJ repair
effectors, thereby shifting repair pathway choice towards alt-EJ
(24).
Oncogenic viruses such as human papillomavirus (HPV)
further hijack the alt-EJ machinery. A critical step in
HPV-mediated oncogenesis is the integration of viral DNA into the
host genome, which not only ensures persistent viral replication
but also promotes genomic instability and malignant transformation.
Parfenov et al (82)
performed comprehensive genomic profiling of 279 head and neck
squamous cell carcinoma (HNSCC) specimens using next-generation
sequencing techniques and found that ~60% of HPV integration events
occurred within regions characterized by microhomology at the
junction sites. Since microhomology usage is a hallmark of alt-EJ,
the authors' study suggests that HPV exploits this error-prone
repair pathway to facilitate viral insertion and tumorigenesis.
This hypothesis is supported by several mechanistic
studies demonstrating elevated alt-EJ activity in HPV-positive
cancers (Fig. 2). For example,
Liu et al (24) reported
markedly higher expression of key alt-EJ factors in HPV-positive
HNSCC compared with HPV-negative tumors. Functional assays in that
study showed that HPV-positive tumor cells preferentially rely on
PARP1-dependent alt-EJ for repairing DSBs, highlighting a shift in
DSB repair pathway choice induced by HPV infection. Furthermore,
Liu et al (25) analyzed
transcriptomic data from The Cancer Genome Atlas (TCGA) that
comprised 243 HPV-negative and 36 HPV-positive HNSCC samples, where
the authors found unsupervised clustering based on alt-EJ gene
signatures distinctly segregated HPV-positive tumors into a cluster
characterized by high expression of alt-EJ-associated genes.
Similarly, Guix et al (79) validated this signature in
patient-derived xenografts and primary tumor tissues via NanoString
assays, finding that HPV-positive samples exhibited an upregulated
alt-EJ signature. Independent validation by Leeman et al
(83) also demonstrated that HPV
infection increases the frequency of DNA deletions flanked by
microhomology by alt-EJ. The authors' mechanistic dissection
revealed that the HPV oncoprotein E7 promotes a shift in DSB repair
pathway choice toward alt-EJ. The aforementioned evidence suggests
the existence of a strategy wherein a virus hijacks alt-EJ in its
host for its oncogenic agenda.
Crosstalk between alt-EJ and other DSB
repair pathways
The interplay between alt-EJ and the canonical DSB
repair pathways, HR and NHEJ, reflects the remarkable plasticity of
the cellular DNA damage response. Disruption or inhibition of HR or
NHEJ frequently redirects DSB repair toward alt-EJ, suggesting that
alt-EJ is a flexible and adaptable mechanism, which may be
exploited by cancer cells to maintain survival under genotoxic
stress (24,84). Studies have revealed that the
regulation of alt-EJ is a dynamic and multifaceted process
involving complex coordination between numerous signaling pathways
and repair proteins, including TGFβ, PARP1 and POLQ and factors
involved in DNA end resection (22,62,85). Such regulation is critical because
dysregulated repair pathway choice can profoundly affect genome
stability, with significant implications for tumorigenesis and
therapeutic resistance.
Although the alt-EJ mechanism was originally
described as a backup pathway to NHEJ and HR, emerging evidence now
positions alt-EJ as an independent and potentially competitive
repair mechanism that operates even in the presence of intact HR
and NHEJ pathways (1,86). Alt-EJ is preferentially engaged
during the S and G2 phases of the cell cycle when
limited DNA end resection has occurred, contributing 10-20% of DSB
repair activity in various mammalian cell contexts (48). This is particularly salient in
conditions of replication stress, where alt-EJ facilitates rapid
repair of collapsed replication forks, balancing between the
cytotoxic risk of error-prone repair and the cellular imperative to
maintain genomic integrity.
A key nexus of crosstalk exists between HR and
alt-EJ centers on POLQ, a multifunctional enzyme with both DNA
polymerase and N-terminal helicase domains endowed with ATPase and
DNA unwinding activities (50,57,87,88). POLQ's helicase domain mediates
critical molecular antagonism of HR by binding RAD51 and thereby
preventing RAD51 nucleoprotein filament assembly on RPA-coated
ssDNA, a prerequisite for HR-mediated strand invasion. This
inhibitory interaction facilitates the suppression of HR and
promotes alt-EJ, effectively channeling repair towards a more
error-prone pathway (68,88). The interaction is structurally
mediated by a disordered central domain within POLQ (residues
847-894) that is essential for RAD51 binding and displacement, thus
highlighting a direct molecular mechanism for pathway choice
regulation (68,89). However, the question of whether
POLQ's ATPase activity regulates the stability and dynamics of
RAD51 filaments remains to be elucidated.
The synthetic lethal relationship between HRD and
alt-EJ dependency further exemplifies their functional interplay.
HR-deficient tumors rely heavily on alt-EJ for repairing complex
DSBs, especially those that arise from replication fork collapse,
events characterized by single-ended DSBs that cannot be repaired
by classical NHEJ due to the absence of a second DSB end (90). Therefore, POLQ-mediated alt-EJ is
potentially vital in bridging ssDNA gaps formed during replication
stress and fork collapse, particularly important in HRD contexts or
following treatment with PARPi (85,91,92).
Single-ended DSBs typically arise when an unrepaired
single-strand break is converted into a DSB, leading to replication
fork collapse. It is also important to appreciate that single-ended
DSBs, the predominant form of DSB in unperturbed cells due to
unrepaired single-strand breaks converted into DSBs during
replication, are primarily repaired by HR (93,94). Alt-EJ repair of single-ended DSBs
tends to be detrimental, often causing chromosomal abnormalities
and increased genome instability and is thus suppressed under
normal conditions to preserve genomic integrity (94). However, alt-EJ can act as a
salvage pathway in HR-compromised scenarios, albeit at the cost of
increased mutagenesis and chromosomal rearrangements (90). This delicate balance illustrates
an intricate regulatory network where repair pathway choice is
tightly controlled to limit deleterious outcomes while facilitating
survival.
Alt-EJ effects in genome instability
Gene mutations and genomic scars
Alt-EJ often introduces insertions and deletions
(indels) at DSB repair sites. This pathway not only fosters genetic
alterations within coding regions but also leaves distinctive
'genomic scars'. These scars frequently involve microhomology
regions that facilitate end alignment prior to repair, leading to
characteristic deletions, rearrangements and templated or
non-templated insertions, which can be detected as a genomic
signature for alt-EJ activity.
In the context of cancer, the mutagenic potential of
alt-EJ is especially significant. For example, loss of TGFβ
signaling increases alt-EJ in pan-cancer, which lead to increased
genomic alterations (25).
Notably, a subset of types of cancer with upregulated alt-EJ has
shown a specific indel mutation signature, characterized by >5
base pair deletions and overlapping microhomology at deletion
boundaries (25). Furthermore, in
BRCA-mutant tumors, increased reliance on alt-EJ not only drives
tumor progression but also engenders specific mutational signatures
that can be exploited for diagnosis or therapeutic targeting. These
mutational signatures include recurrent deletions featuring
microhomology at breakpoint junctions and complex rearrangements
that impact genome stability. Such patterns serve as genomic scars
indicative of defective HR repair and are associated with increased
sensitivity to PARPi (95-98).
POLQ facilitates annealing of microhomologous
regions and contributes to the characteristic junctional mutations.
In a C. elegans study, the results analyzing ~7,000 deletion
breakpoints demonstrated that POLQ-dependent alt-EJ accounts for a
significant portion of mutations induced by alkylating agents such
as ethyl methane sulfonate and UV/TMP treatments (95). These breakpoints often include
small deletions due to microhomology and show inserted DNA
sequences, either copied from close (templated) or newly added
(non-templated) region.
The frequent insertion of short sequences at
breakpoints can be attributed to POLQ's terminal transferase
activity and its propensity for template switching processes that
generate diverse repair signatures, including partial ssDNA
intermediates (99). This
accumulation of ssDNA renders cells more susceptible to further
damage or mutations, especially under conditions of external
genotoxic stress or endogenous replicative stress. Alt-EJ leads to
exposure of ssDNA regions and increased opportunities for
nucleotide misincorporation or further damage. Notably,
hypermutagenesis tends to extend over a defined physical distance
(~7-9 kb) from the breakpoints, indicating that error-prone repair
is not confined solely to immediate junctions but can propagate
along adjacent DNA regions (100). Notably, this hypermutability
appears to be an intrinsic feature of alt-EJ and not solely
dependent on POLQ activity, suggesting that the pathway's inherent
mechanics predispose to extensive mutagenic outcomes.
Chromosomal aberrations and
translocations
Alt-EJ often results in chromosomal aberrations and
translocations. In Saccharomyces cerevisiae (budding yeast),
the presence of multiple simultaneous DSBs increases the likelihood
of promiscuous end joining via alt-EJ, generating chromosomal
translocations and complex rearrangements (101). This finding highlights the
potential for alt-EJ to misrepair disparate DNA ends. Consistently,
a high frequency of microhomology at translocation junctions has
been detected in human tumor cells, supporting a mechanistic link
between alt-EJ and chromosomal rearrangements in cancer (102,103). This microhomology enrichment at
breakpoints contrasts with the more blunt-end ligation
characteristic of NHEJ, underscoring the distinct repair signature
of alt-EJ. Furthermore, loss of TGFβ signaling increases IR-induced
chromosome aberrations in HNSCC cells, but knock-down of POLQ
suppresses the effects, suggesting a critical role for alt-EJ
(24). Cisplatin treatment also
induces chromosomal aberrations, especially in human papillomavirus
(HPV)-positive HNSCC with preference for alt-EJ (76).
To investigate the direct role of alt-EJ in
chromosomal translocation formation, studies induced targeted DSBs
on distinct chromosomes using site-specific nucleases such as
CRISPR-Cas9 or I-SceI endonuclease (104,105). Experiments performed in both
murine and human cells deficient in NHEJ factors demonstrated that
loss of key NHEJ components, notably XRCC4, substantially elevated
the frequency of reciprocal translocations up to fivefold compared
with wild-type controls (104,105). Furthermore, microhomologies were
observed at ~60% of these breakpoint junctions, strongly
implicating alt-EJ in the formation of the chromosomal aberrations.
Beyond XRCC4, other proteins critical to alt-EJ function, such as
CtIP, which initiates DNA end resection, and LIG3, which catalyzes
the final ligation, have been shown to influence chromosomal
rearrangement frequencies. For example, mouse cells depleted of
CtIP or LIG3 exhibit reduced alt-EJ activity and corresponding
decreases in chromosomal aberrations, indicating that a functional
alt-EJ machinery is required for these instability phenotypes
(70,106).
Telomere fusions
Telomeres, the specialized nucleoprotein structures
capping chromosome ends, are essential for preserving genomic
integrity by preventing chromosome ends from being recognized as
DSBs. However, critically short or dysfunctional telomeres that
result from replicative attrition or loss of protective factors
become 'uncapped', exposing chromosome ends as DNA breaks
vulnerable to erroneous repair pathways. In these contexts, alt-EJ
plays a significant role in driving telomere fusions, which
contribute to profound genome instability. Indeed, telomere fusion
events mediated by alt-EJ have been observed in aggressive cancers
including glioblastomas, where they correlate with poor prognosis
and increased genomic chaos (107-109).
Recent investigations have provided critical
mechanistic insights into alt-EJ mediators in telomere fusion. For
example, one study that used mouse embryonic fibroblasts lacking
TRF2 and Ku demonstrated robust NHEJ-independent telomere fusions
that are highly dependent on POLQ and this POLQ-dependent fusion
process is sensitive to inhibition by PARPi, highlighting a
potential therapeutic vulnerability in tumors reliant on alt-EJ for
telomere maintenance (69).
Importantly, the same study revealed that APE2 acts as an epistatic
partner of POLQ in executing these fusion events, as simultaneous
depletion of POLQ and APE2 did not further reduce telomere fusions
compared with single depletions. These findings not only strengthen
evidence that POLQ is a central player in alt-EJ-mediated telomere
fusion but also identify APE2 as a novel cooperating factor in this
pathway.
Consistent with the pronounced effects of alt-EJ in
genome instability, recent discoveries have also increasingly
linked aberrant activation of alt-EJ to cancer development,
progression and therapeutic resistance. In HRD tumors, such as
those harboring BRCA1/2 mutations, reliance on alt-EJ fosters the
accumulation of microhomology-associated deletions and templated
insertions, generating genomic scars that drive tumor evolution and
heterogeneity (25,55,95,98). These genomic features are
associated with poor clinical prognosis and have been proposed as
predictive biomarkers for response to genotoxic therapies and
PARPi. Large-scale pan-cancer transcriptomic analyses have further
revealed that tumors with elevated alt-EJ activity frequently
exhibit high expression of POLQ and PARP1, correlating with
increased chromosomal instability and resistance to standard
treatments (22,25,68,98). Together, these findings support
the emerging view that alt-EJ is an active contributor to malignant
transformation. Understanding these oncogenic consequences of
alt-EJ and factors that affect alt-EJ activity are critical for
harnessing its components as potential diagnostic markers and novel
therapeutic targets in cancer.
Potential cancer targets in Alt-EJ
DDR contributes to cancer cell resistance to
genotoxic therapies. Numerous cancers exhibit an abnormal
dependence on alt-EJ, creating unique therapeutic vulnerabilities
that can be exploited by targeting DDR components involved in
alt-EJ (Table I) (110,111). Among alt-EJ factors, PARP1
stands out as a well-validated and promising target in oncology.
Multiple PARPi have been FDA-approved or are currently in clinical
trials for diverse types of cancer, particularly tumors harboring
HRD (112-114). PARPi exemplify synthetic
lethality, where cancer cells defective in HR, commonly due to
BRCA1/2 mutations in breast and ovarian cancers, become selectively
vulnerable to PARP1 inhibition. Beyond BRCA mutations, emerging
evidence reveals that defects in multiple DDR components, including
ATM and SMAD4, also confer hypersensitivity to PARPi (115,116).
 | Table IInhibition effects of alt-EJ targets
in cancer. |
Table I
Inhibition effects of alt-EJ targets
in cancer.
| First author/s,
year | Target | Inhibitor | Cancer type | Therapeutic
effects | (Refs.) |
|---|
| Jagsi et al,
2018 | PARP1 | Veliparib | Breast Cancer | Increases
radiosensitivity in breast cancer patients | (121) |
| de Bono et
al, 2020 | PARP1 | Olaparib | Prostate
cancer | Improve
progression-free survival in patients with metastatic
castration-resistant prostate cancer | (147) |
| Liu et al,
2018 | PARP1 | Olaparib | Bladder cancer | Increases
radiosensitivity in cancer cell line | (116) |
| De Haan et
al, 2019 | PARP1 | Olaparib | Breast cancer,
NSCLC, HNSCC | Enhances
radiosensitivity in phase1clinical trial of the patients | (120) |
| Weaver et
al, 2015 | PARP1 | Veliparib | HNSCC | Reduces cell
viability and mouse xenograft tumor growth in vitro | (117) |
| Wang et al,
2020 | PARP1 | Niraparib | HNSCC | Increases
radiosensitivity in cancer cell lines and increased proton vs.
photon RBE | (148) |
| Zhou et al,
2021 | POLQ | Novobiocin | Breast cancer,
ovarian cancer | Enhances
chemosensitivity to PARPi in cancer cell lines and mouse xenograft
and patient derived xenograft model | (131) |
| Zatreanu et
al, 2021 | POLQ | ART558 | Breast cancer | Enhances synthetic
lethality and the effect of PARPi in BRCA1-mutant cell in
vivo and in vitro | (132) |
|
Rodriguez-Berriguete et al,
2023 | POLQ | ART558
ART899 | Colorectal cancer,
NSCLC, bladder cancer | Increases the
radiosensitivity of cancer cell lines and mouse xenograft
model | (130) |
| Fried et al,
2024 | POLQ | RTx-161 | Breast cancer | Induces synthetic
lethality and enhances the effect of PARPi in HRD cells | (133) |
| Hossain et
al, 2021 | APE2 | Celastrol | Pancreatic
cancer | Enhances the effect
of chemotherapy drugs | (145) |
| Chen et al,
2008 | LIG1/3 | L67, L82, L189 | Breast cancer,
colon cancer | Enhances
radiosensitivity and cytotoxicity to DNA damaging agent | (140) |
| Tobin et al,
2013 | LIG1/3
PARP1 | L67
NU1025 | CML | Enhances combine
sensitivity of DNA repair inhibitors | (143) |
| Liu et al,
2018 | TGFBRI
PARP1 | LY2157299
Olaparib | HNSCC | Increases
radiosensitivity of a mouse tumor model | (24) |
| Hintelmann et
al, 2021 | PARP1
Weel | Olaparib
Adavosertib | HNSCC | Increases
radiosensitivity to cancer cell | (149) |
| Zuo et al,
2023 | XPF PARP1 | F06 Olaparib | HNSCC | Induces
chemosensitivity in a mouse tumor model | (76) |
| Molkentine et
al, 2021 | PARP1
Chk1 Weel | Niraparib MK-8776,
MK-1775 | HNSCC | Increases
radiosensitivity to cancer cell and xenograft tumor model | (127) |
In HNSCC, functional deficiencies in key DSB repair
genes such as DNA-PKcs and BRCA2, particularly in HPV-positive
tumors, sensitize cells to PARPi. Weaver et al (117) demonstrated that PARP inhibition
with veliparib impaired cell survival and delayed tumor growth by
exploiting these DDR deficiencies. Furthermore, metastatic
castration-resistant prostate cancer with frequent HR pathway
alterations has been increasingly targeted with PARPi in clinical
studies (118).
Due to the importance of PARP1 in DSB repair,
especially for alt-EJ, PARPi are expected to have synergistic
effects with IR. In addition to DSBs, IR also generates other types
of DNA damage, such as single strand break (SSB) or base lesions.
PARPi are able to convert these DNA lesions to DSBs, thus
increasing the DSB load and producing synergistic effects with
radiotherapy (119). Indeed,
PARPi-enhanced therapeutic efficacy has been detected across
multiple solid tumors (116,118,120,121). These synergistic effects are
detected not only in tumors treated with low linear energy transfer
(LET) IR such as X-rays, but also in those treated with high-LET
particle radiation. It is well established that high-LET IR induces
complex clustered DNA damage comprising both SSBs and DSBs and
alt-EJ may play an important role in the repair of these complex
DNA damages (122). PARPi
enhances the radiosensitivity of cancer cells exposed to particle
irradiation in various studies (123-126). Combinatorial PARP1-targeting
strategies are also quickly advancing. For example, co-targeting
PARP1 and cell cycle checkpoint kinases such as CHK1 or Wee1
displays differential radiosensitization related to HPV status:
PARP1 plus CHK1 inhibition enhances radiosensitivity in
HPV-positive cells, whereas PARP1 plus Wee1 inhibition is more
effective in HPV-negative tumors (127). Zuo et al (76) found that, due to the increased
reliance of XPF-deficient HNSCC on alt-EJ to repair ICLs,
inhibiting both PARP1 and the endonuclease XPF enhanced the effects
of cisplatin on HPV-negative HNSCC in vitro and in
vivo. Another recent study has revealed that polymerase α
(POLA1) inhibition, in combination with PARP inhibition, synergizes
to increase replication stress and DSB accumulation in
BRCA1-deficient backgrounds, sensitizing cancer cells to PARPi
treatment (128). This suggests
that disruption of replication processes can further compromise
genome stability, amplifying alt-EJ dependency. Such findings
underscore the importance of personalized strategies that account
for tumor genotype and DDR pathway context, for which alt-EJ
addiction is an important factor.
The clinical success of PARPi has firmly established
the potential of targeting alt-EJ in cancer therapy. Beyond PARP1,
POLQ has emerged as a critical, targetable player central to the
alt-EJ mechanism. Results from preclinical studies have
demonstrated that POLQ inhibitors have significant anti-tumor
effects in DDR-deficient contexts or under DNA damage overload
during genotoxic treatments (129,130). One breakthrough in POLQ-targeted
therapy came with the identification of novobiocin, an antibiotic
repurposed as a selective POLQ inhibitor through high-throughput
small-molecule library screening. Novobiocin directly binds the
ATPase domain of POLQ, blocking its recruitment to DNA damage sites
and inhibiting alt-EJ repair (131). Functionally, novobiocin
preferentially induces the death of BRCA-deficient cells and
synergizes with PARPi to exacerbate DNA repair defects. Notably,
novobiocin suppresses tumorigenesis in BRCA1-deleted
triple-negative breast cancer mouse models and in patient-derived
xenografts that have developed resistance to PARPi therapy as a
consequence of 53BP1 loss, highlighting that POLQ inhibition may
overcome the resistance mechanisms.
Similarly to novobiocin, the investigational
compound ART558 demonstrates nanomolar affinity for POLQ and
selectively inhibits proliferation in BRCA2-deficient cancer cells
in vitro (132). ART558
also effectively targets PARPi-resistant BRCA1-null cells and
organoids, a trait shared with the next-generation POLQ inhibitor
ART812, which has offered improved bioavailability and
pharmacokinetics in animal models. ART812 markedly suppresses
growth of PARP inhibitor-resistant BRCA1-knock-out xenografts,
reinforcing the clinical translation potential of POLQ inhibitors
for overcoming therapeutic resistance.
More recent advances include a new class of POLQ
polymerase-specific inhibitors, represented by RTx-161 and RTx-152,
which exhibit highly potent inhibitory activity (IC50
values of 4-6 nM) and demonstrate broad efficacy in eliminating HRD
tumors, as well as overcoming PARPi resistance in genetically
diverse backgrounds, including some HR-proficient tumors (133). These inhibitors differ
mechanistically by targeting the polymerase function rather than
the ATPase activity, effectively trapping POLQ on DNA and
preventing repair completion.
Beyond monotherapy, POLQ inhibition shows
considerable promise as a radiosensitizing strategy. Preclinical
evidence indicates that POLQ inhibitors such as ART558 and its
derivative ART899 potentiate the cytotoxic effects of IR across
diverse tumor models, especially under hypoxic or S-phase-enriched
conditions common in solid tumors, where conventional therapies
often falter or resistance emerges (134). Encouragingly, a Phase I clinical
trial (NCT04991480) is currently underway to evaluate safety and
efficacy of combining POLQ inhibition with radiotherapy, which may
open new avenues for more precise and less toxic cancer treatments
(130).
Synthetic-lethal interactions between POLQ and key
DDR genes (for example, BRCA1, BRCA2 and ATM) have been detected in
in vitro and in vivo models, emphasizing the
therapeutic potential of targeting POLQ in genetically defined
cancer populations (56,68,135). Despite these advances, further
investigations are imperative to deepen our mechanistic
understanding of POLQ-mediated alt-EJ, optimize inhibitor potency
and refine biomarkers to predict treatment response. Such efforts
will ultimately bridge molecular insights with clinical oncology,
thereby enhancing personalized medicine approaches that exploit
alt-EJ dependence for cancer therapy.
Although PARP1 and POLQ remain the most extensively
studied therapeutic targets within the alt-EJ pathway, several
other factors integral to alt-EJ-mediated DSB repair have emerged
as promising candidates for cancer therapy. Flap endonuclease 1
(FEN1) is an essential nuclease that processes 5'-flap structures
generated during DNA end resection and strand displacement
synthesis within alt-EJ (136,137). FEN1's activity is crucial for
the removal of displaced DNA flaps that otherwise hinder efficient
alt-EJ. Loss of FEN1 function critically impairs alt-EJ efficiency,
paralleling the loss of POLQ in HRD cells and resulting in
synthetic lethality (56,68,136). Importantly, novel FEN1
inhibitors, structurally related to hydroxyurea derivatives, have
demonstrated selective cytotoxicity against HRD cancer cells
without substantial toxicity to normal cells, underscoring their
therapeutic potential (56,68,136).
FANCD2, a central player in the FA pathway, have
been identified in synthetic lethal interactions with POLQ
function. FANCD2 also promotes POLQ recruitment to DNA breaks,
facilitating alt-EJ activation (138). Preclinical studies have
demonstrated that dual depletion of POLQ and FANCD2 markedly
sensitizes cancer cells to cisplatin and PARP inhibitors,
particularly in lung and ovarian cancer models. For example, short
interfering RNA-mediated co-suppression of POLQ and FANCD2 resulted
in markedly increased cell death and tumor volume reduction in
xenograft models, highlighting the synthetic lethal potential of
targeting this axis (68,139).
DNA ligases, particularly LIG1 and LIG3, are
critical components of the alt-EJ pathway. Some years ago, Chen
et al (140) reported
that structure-based drug design led to the development of small
molecules that inhibit human DNA ligases by targeting their
DNA-binding domains. For instance, compounds such as L82
selectively inhibit LIG1; L67 inhibits both LIG1 and LIG3; and L189
targets LIG1, LIG3 and LIG4. These inhibitors have demonstrated
efficacy in vitro by impairing DNA repair processes. In
cell-based studies, L67 and L189 exhibited cytotoxicity and
synergized with DNA-damaging agents, particularly in cancer cell
lines, highlighting their potential to sensitize tumors to
chemotherapy.
LIG1 has been identified as a synthetic lethal
target in cancers harboring BRCA1 mutations. Using CRISPR/Cas9
screening and validation assays, researchers have shown that
BRCA1-mutant cells depend heavily on LIG1 activity for survival,
whereas BRCA1/2 wild-type cells are less affected. Notably, LIG1's
catalytic function is essential for this dependency, suggesting
that its inhibition disrupts sealing of single-strand DNA nicks
during alt-EJ. Depletion of LIG1 leads to accumulation of
unrepaired DNA nicks and results in tumor stasis in xenograft
models. These findings demonstrate that targeting LIG1 can exploit
the synthetic lethality associated with BRCA1 deficiency, making it
a promising therapeutic avenue in such malignancies (141).
In addition, resistance to tyrosine kinase
inhibitors such as imatinib in chronic myeloid leukemia, as well as
resistance to endocrine therapies in certain breast cancers, have
been linked to reliance on the alt-EJ pathway. Resistant cells
often exhibit elevated levels of PARP1 along with DNA ligases,
especially LIG3. Combined inhibition of PARP1 and LIG3 impairs
alt-EJ-mediated repair, markedly reducing the survival of these
therapy-resistant cancer cells. Importantly, the degree of
sensitivity to this combination correlates with the expression
levels of PARP1 and ligases in both cell lines and patient-derived
samples, supporting their potential as biomarkers and therapeutic
targets (142,143).
Another player recently implicated in alt-EJ is
APE2. APE2 facilitates alt-EJ by processing damaged DNA ends and
modulating telomere fusions (63), which positions APE2 as a promising
therapeutic target (63,136,144). The first APE2 inhibitor
identified, Celastrol, originally characterized for its
anti-inflammatory properties, has been shown to inhibit APE2's
ssDNA binding and 3'-5' exonuclease activity, thereby attenuating
ATR checkpoint activation in pancreatic cancer models (145). Given APE2's role in processing
the 3'-DNA-protein adducts generated by PARPi, APE2 inhibition may
augment PARPi efficacy to overcome resistance in both
BRCA-deficient and proficient tumors.
Alessandra Brambati et al (69) recently described RHINO as an
M-phase accumulated protein that facilitates POLQ recruitment to
DSBs, thereby promoting alt-EJ-mediated repair. This finding
uncovers a temporal regulation of alt-EJ repair activity during the
cell cycle and suggests that RHINO could be a novel candidate
target for cancer therapy. Modulation of RHINO function may provide
new avenues for targeted intervention, particularly in cancers
addicted to alt-EJ due to HRD or other reasons.
Conclusion and future prospective
Alt-EJ plays a critical and complex role in genome
instability and cancer biology. Mounting evidence highlights alt-EJ
not merely as a backup repair pathway but as a pivotal contributor
to both tumor development and therapeutic response. Alt-EJ operates
with inherently lower fidelity, leading to mutational signatures
characterized by deletions, insertions and chromosomal
rearrangements, which drive genome instability and can contribute
to therapeutic resistance in cancer.
The present review emphasized that alt-EJ is
regulated by intricate molecular mechanisms influenced by both
intrinsic cellular components and extrinsic factors, underscoring
its role as a highly coordinated and context-dependent repair
pathway rather than a simple fail-safe mechanism. Recognizing these
complexities presents unique challenges and opportunities: Future
research should therefore focus on deciphering the precise
molecular basis of alt-EJ-mediated mutagenesis, which will be
crucial for optimizing therapeutic strategies aimed at mitigating
its deleterious effects while exploiting its vulnerabilities to
enhance cancer cell destruction.
Mutations in DDR genes prevalent in numerous types
of cancer often increases alt-EJ reliance (14,146). This dependency offers a
significant therapeutic window. Specifically targeting alt-EJ
components such as POLQ, PARP1 and emerging factors such as FEN1
and APE2 can selectively control HRD tumors. Recent advances in
understanding alt-EJ mechanisms, coupled with novel therapeutic
approaches including combination regimens with radiotherapy or
immunotherapy, hold great promise for improving outcomes in these
clinically challenging cancers.
Despite recent advances, substantial gaps remain
about the molecular determinants of tumor reliance on alt-EJ and
the heterogeneity of responses to DDR-targeting therapies.
Coordinated, systematic studies are needed to define how alt-EJ
interacts with other repair pathways, translate this biology into
predictive and pharmacodynamic biomarkers and uncover the routes by
which tumors evade therapy. This foundation will enable context
specific treatment strategies that exploit alt-EJ inhibition to
maximize antitumor efficacy while minimizing toxicity.
Translating these insights into clinical benefit
requires clinically deployable assays that quantify alt-EJ
dependence and confirm on target inhibition, alongside a rigorous
understanding of resistance to alt-EJ-directed agents, including
POLQ and PARP1 inhibitors. Anticipated resistance mechanisms
include on target alterations, pathway rewiring toward NHEJ or HR,
adaptation to replication stress and pharmacologic tolerance;
sensitive early indicators should guide timely countermeasures and
adaptive trial design. Clarifying how alt-EJ-driven lesions
interface with immune checkpoint signaling and with chromatin and
epigenetic regulation of end resection will inform combinations
with immunotherapy and epigenetic agents. Equally important is
optimization of dose, fractionation and sequencing for pairings
with radiotherapy and other DDR regulators, supported by
standardized analytic pipelines to measure alt-EJ activity across
platforms and over time. Addressing these priorities will enable
biomarker guided patient selection, anticipate therapeutic
resistance and deliver precise, durable exploitation of alt-EJ
vulnerabilities in the clinic.
In conclusion, alt-EJ represents a promising and
expanding frontier for cancer therapy. By deepening our mechanistic
understanding and integrating emerging molecular insights with
innovative drug development, the field is poised to translate
alt-EJ targeting into effective, personalized interventions that
improve both prognosis and quality of life for patients with
alt-EJ-dependent malignancies.
Availability of data and materials
Not applicable.
Authors' contributions
Conceptualization was by QL, investigation was by
QL and NA. Writing and original draft preparation was by NA, LM and
QL. Writing, reviewing and editing was by QL, NA, LM and XL.
Visualization was by NA and QL. Supervision was by QL, LM and XL.
Funding acquisition was by QL, XL and LM.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work the authors
used AI tools in order to improve language and readability and
subsequently the authors reviewed and edited the content as needed
and take full responsibility for the content of the present
manuscript.
Acknowledgements
The authors thank Dr Lei Wang from the
International Cancer Center, Shenzhen University (China), for his
support and advices.
Funding
The present study was supported by National Natural Science
Foundation of China (grant nos. 82373212 and 82073007 to QL; grant
no. 82203967 to LM); Guangdong Basic and Applied Basic Research
Foundation (grant no. 2023A1515011945 to LM); Shenzhen Science and
Technology Program (grant no. JCYJ20240813142112017 to LM). HaiYa
Young Scientist Foundation of Shenzhen University General Hospital
(grant no. 2025-HY006 to XL).
References
|
1
|
Liu Q, Lopez K, Murnane J, Humphrey T and
Barcellos-Hoff MH: Misrepair in context: TGFβ regulation of DNA
repair. Front Oncol. 9:7992019. View Article : Google Scholar
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Negrini S, Gorgoulis VG and Halazonetis
TD: Genomic instability-an evolving hallmark of cancer. Nat Rev Mol
Cell Biol. 11:220–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Driscoll M: Diseases associated with
defective responses to DNA damage. Cold Spring Harb Perspect Biol.
4:a0127732012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taylor AMR, Rothblum-Oviatt C, Ellis NA,
Hickson ID, Meyer S, Crawford TO, Smogorzewska A, Pietrucha B,
Weemaes C and Stewart GS: Chromosome instability syndromes. Nat Rev
Dis Primers. 5:642019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chae YK, Anker JF, Carneiro BA, Chandra S,
Kaplan J, Kalyan A, Santa-Maria CA, Platanias LC and Giles FJ:
Genomic landscape of DNA repair genes in cancer. Oncotarget.
7:23312–23321. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma J, Setton J, Lee NY, Riaz N and Powell
SN: The therapeutic significance of mutational signatures from DNA
repair deficiency in cancer. Nat Commun. 9:32922018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vilenchik MM and Knudson AG: Endogenous
DNA Double-strand breaks: Production, fidelity of repair, and
induction of cancer. Proc Natl Acad Sci USA. 100:12871–12876. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Trenner A and Sartori AA: Harnessing DNA
double-strand break repair for cancer treatment. Front Oncol.
9:13882019. View Article : Google Scholar
|
|
10
|
Linders AN, Dias IB, López Fernández T,
Tocchetti CG, Bomer N and Van der Meer P: A review of the
pathophysiological mechanisms of doxorubicin-induced cardiotoxicity
and aging. NPJ Aging. 10:92024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qian J, Liao G, Chen M, Peng RW, Yan X, Du
J, Huang R, Pan M, Lin Y, Gong X, et al: Advancing cancer therapy:
New frontiers in targeting DNA damage response. Front Pharmacol.
15:14743372024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jasin M and Rothstein R: Repair of strand
breaks by homologous recombination. Cold Spring Harb Perspect Biol.
5:a0127402013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Q, Zuo N, Li X, Deng Y, Wei L and Ma
L: Novel insights into DNA damage repair defects in HPV-positive
head and neck squamous cell carcinoma: From the molecular basis to
therapeutic opportunities. Genome Instability Dis. 4:255–265. 2023.
View Article : Google Scholar
|
|
15
|
Kang X, Li X, Zhou J, Zhang Y, Qiu L, Tian
C, Deng Z, Liang X, Zhang Z, Du S, et al: Extrachromosomal DNA
replication and maintenance couple with DNA damage pathway in
tumors. Cell. 188:3405–3421.e27. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ramsden DA, Carvajal-Garcia J and Gupta
GP: Mechanism, cellular functions and cancer roles of
polymerase-theta-mediated DNA end joining. Nat Rev Mol Cell Biol.
23:125–140. 2022. View Article : Google Scholar
|
|
17
|
Newman JA, Cooper CD, Aitkenhead H and
Gileadi O: Structure of the helicase domain of DNA polymerase theta
reveals a possible role in the microhomology-mediated end-joining
pathway. Structure. 23:2319–2330. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kent T, Chandramouly G, McDevitt SM,
Ozdemir AY and Pomerantz RT: Mechanism of microhomology-mediated
end-joining promoted by human DNA polymerase θ. Nat Struct Mol
Biol. 22:230–237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dueva R and Iliakis G: Alternative
pathways of non-homologous end joining (NHEJ) in genomic
instability and cancer. Transl Cancer Res. 2:163–177. 2013.
|
|
20
|
Daley JM and Wilson TE: Rejoining of DNA
double-strand breaks as a function of overhang length. Mol Cell
Biol. 25:896–906. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang HH, Pannunzio NR, Adachi N and
Lieber MR: Non-homologous DNA end joining and alternative pathways
to double-strand break repair. Nat Rev Mol Cell Biol. 18:495–506.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Patterson-Fortin J and D'Andrea AD:
Exploiting the microhomology-mediated end-joining pathway in cancer
therapy. Cancer Res. 80:4593–4600. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Howard SM, Yanez DA and Stark JM: DNA
damage response factors from diverse pathways, including DNA
crosslink repair, mediate alternative end joining. PLoS Genetics.
11:e10049432015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Q, Ma L, Jones T, Palomero L, Pujana
MA, Martinez-Ruiz H, Ha PK, Murnane J, Cuartas I, Seoane J, et al:
Subjugation of TGFβ signaling by human papilloma virus in head and
neck squamous cell carcinoma shifts DNA repair from homologous
recombination to alternative end joining. Clin Cancer Res.
24:6001–6014. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Q, Palomero L, Moore J, Guix I, Espín
R, Aytés A, Mao JH, Paulovich AG, Whiteaker JR, Ivey RG, et al:
Loss of TGFβ signaling increases alternative end-joining DNA repair
that sensitizes to genotoxic therapies across cancer types. Sci
Transl Med. 13:eabc44652021. View Article : Google Scholar
|
|
26
|
Xu Z, Zan H, Pone EJ, Mai T and Casali P:
Immunoglobulin class-switch DNA recombination: Induction, targeting
and beyond. Nat Rev Immunol. 12:517–531. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zan H, Tat C, Qiu Z, Taylor JR, Guerrero
JA, Shen T and Casali P: Rad52 competes with Ku70/Ku86 for binding
to S-region DSB ends to modulate antibody class-switch DNA
recombination. Nat Commun. 8:142442017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boboila C, Yan C, Wesemann DR, Jankovic M,
Wang JH, Manis J, Nussenzweig A, Nussenzweig M and Alt FW:
Alternative end-joining catalyzes class switch recombination in the
absence of both Ku70 and DNA ligase 4. J Exp Med. 207:417–427.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robert I, Dantzer F and Reina-San-Martin
B: Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses
IgH/c-myc translocations during immunoglobulin class switch
recombination. J Exp Med. 206:1047–1056. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ortega R, Bitler BG and Arnoult N:
Multiple functions of PARP1 in the repair of DNA double strand
breaks. DNA Repair (Amst). 152:1038732025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boboila C, Oksenych V, Gostissa M, Wang
JH, Zha S, Zhang Y, Chai H, Lee CS, Jankovic M, Saez LM, et al:
Robust chromosomal DNA repair via alternative end-joining in the
absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc
Natl Acad Sci USA. 109:2473–2478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saha T, Sundaravinayagam D and Di Virgilio
M: Charting a DNA repair roadmap for immunoglobulin class switch
recombination. Trends Biochem Sci. 46:184–199. 2021. View Article : Google Scholar
|
|
33
|
Espín R, Medina-Jover F, Sigüenza-Andrade
J, Farran-Matas S, Mateo F, Figueras A, Sanz RT, Vicent GP, Shabbir
A, Ruiz-Auladell L, et al: Harnessing transcriptional regulation of
alternative end-joining to predict cancer treatment. NAR Cancer.
7:zcaf0072025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng B, Ding Z, Hong Y, Wang Y, Zhou Y,
Chen J, Peng X and Zeng C: Research progress in DNA damage response
(DDR)-Targeting modulators: From hits to clinical candidates. Eur J
Med Chem. 287:1173472025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wright WD, Shah SS and Heyer WD:
Homologous recombination and the repair of DNA double-strand
breaks. J Biol Chem. 293:10524–10535. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ranjha L, Howard SM and Cejka P: Main
steps in DNA Double-strand break repair: An introduction to
homologous recombination and related processes. Chromosoma.
127:187–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lieber MR: The mechanism of double-strand
DNA break repair by the nonhomologous DNA end-joining pathway. Annu
Rev Biochem. 79:181–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bhargava R, Onyango DO and Stark JM:
Regulation of single-strand annealing and its role in genome
maintenance. Trends Genet. 32:566–575. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zha S, Guo C, Boboila C, Oksenych V, Cheng
HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, et al: ATM
damage response and XLF repair factor are functionally redundant in
joining DNA breaks. Nature. 469:250–254. 2011. View Article : Google Scholar :
|
|
40
|
Ochi T, Blackford AN, Coates J, Jhujh S,
Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, et
al: PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote
DNA double-strand break repair. Science. 347:185–188. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu X, Shao Z, Jiang W, Lee BJ and Zha S:
PAXX promotes KU accumulation at DNA breaks and is essential for
end-joining in XLF-deficient mice. Nat Commun. 8:138162017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Voutsadakis IA and Stravodimou A:
Homologous recombination defects and mutations in DNA damage
response (DDR) genes besides BRCA1 and BRCA2 as breast cancer
biomarkers for PARP inhibitors and other DDR targeting therapies.
Anticancer Res. 43:967–981. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wen H, Feng Z, Ma Y, Liu R, Ou Q, Guo Q,
Shen Y and Wu X, Shao Y, Bao H and Wu X: Homologous recombination
deficiency in diverse cancer types and its correlation with
platinum chemotherapy efficiency in ovarian cancer. BMC Cancer.
22:5502022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Takamatsu S, Murakami K and Matsumura N:
Homologous recombination deficiency unrelated to platinum and PARP
inhibitor response in cell line libraries. Sci Data. 11:1712024.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vu TV, Das S, Nguyen CC, Kim J and Kim JY:
Single-strand annealing: Molecular mechanisms and potential
applications in CRISPR-Cas-based precision genome editing.
Biotechnol J. 17:21004132022. View Article : Google Scholar
|
|
46
|
Liang CC, Greenhough LA, Masino L, Maslen
S, Bajrami I, Tuppi M, Skehel M, Taylor IA and West SC: Mechanism
of single-stranded DNA annealing by RAD52-RPA complex. Nature.
629:697–703. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wyatt DW, Feng W, Conlin MP, Yousefzadeh
MJ, Roberts SA, Mieczkowski P, Wood RD, Gupta GP and Ramsden DA:
Essential roles for polymerase θ-mediated end joining in the repair
of chromosome breaks. Mol Cell. 63:662–673. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Truong LN, Li Y, Shi LZ, Hwang PY, He J,
Wang H, Razavian N, Berns MW and Wu X: Microhomology-mediated End
Joining and Homologous Recombination share the initial end
resection step to repair DNA double-strand breaks in mammalian
cells. Proc Natl Acad Sci USA. 110:7720–7725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Saito S, Maeda R and Adachi N: Dual loss
of human POLQ and LIG4 abolishes random integration. Nat Commun.
8:161122017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wood RD and Doublié S: DNA polymerase θ
(POLQ), double-strand break repair, and cancer. DNA Repair (Amst).
44:22–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chatterjee N and Walker GC: Mechanisms of
DNA damage, repair, and mutagenesis. Environ Mol Mutagen.
58:235–263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Suskiewicz MJ, Zobel F, Ogden TEH, Fontana
P, Ariza A, Yang JC, Zhu K, Bracken L, Hawthorne WJ, Ahel D, et al:
HPF1 completes the PARP active site for DNA damage-induced
ADP-ribosylation. Nature. 579:598–602. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhao F, Kim W, Kloeber JA and Lou Z: DNA
end resection and its role in DNA replication and DSB repair choice
in mammalian cells. Exp Mol Med. 52:1705–1714. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Daley JM, Jimenez-Sainz J, Wang W, Miller
AS, Xue X, Nguyen KA, Jensen RB and Sung P: Enhancement of
BLM-DNA2-mediated long-range DNA end resection by CtIP. Cell Rep.
21:324–332. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carvajal-Garcia J, Cho JE, Carvajal-Garcia
P, Feng W, Wood RD, Sekelsky J, Gupta GP, Roberts SA and Ramsden
DA: Mechanistic basis for microhomology identification and genome
scarring by polymerase theta. Proc Natl Acad Sci USA.
117:8476–8485. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mateos-Gomez PA, Gong F, Nair N, Miller
KM, Lazzerini-Denchi E and Sfeir A: Mammalian polymerase θ promotes
alternative NHEJ and suppresses recombination. Nature. 518:254–257.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Black SJ, Kashkina E, Kent T and Pomerantz
RT: DNA polymerase θ: A unique multifunctional end-joining machine.
Genes. 7:672016. View Article : Google Scholar
|
|
58
|
Li C, Maksoud LM and Gao Y: Structural
basis of error-prone DNA synthesis by DNA polymerase θ. Nat Commun.
16:20632025. View Article : Google Scholar
|
|
59
|
Masani S, Han L, Meek K and Yu K:
Redundant function of DNA ligase 1 and 3 in alternative end-joining
during immunoglobulin class switch recombination. Proc Natl Acad
Sci USA. 113:1261–1266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Audebert M, Salles B and Calsou P:
Involvement of poly (ADP-ribose) polymerase-1 and XRCC1/DNA ligase
III in an alternative route for DNA double-strand breaks rejoining.
J Biol Chem. 279:55117–55126. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Soni A, Siemann M, Grabos M, Murmann T,
Pantelias GE and Iliakis G: Requirement for Parp-1 and DNA ligases
1 or 3 but not of Xrcc1 in chromosomal translocation formation by
backup end joining. Nucl Acids Res. 42:6380–6392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chandramouly G, Jamsen J, Borisonnik N,
Tyagi M, Calbert ML, Tredinnick T, Ozdemir AY, Kent T, Demidova EV,
Arora S, et al: Polλ promotes microhomology-mediated end-joining.
Nat Struct Mol Biol. 30:107–114. 2023. View Article : Google Scholar
|
|
63
|
Fleury H, MacEachern MK, Stiefel CM, Anand
R, Sempeck C, Nebenfuehr B, Maurer-Alcalá K, Ball K, Proctor B III,
Belan O, et al: The APE2 nuclease is essential for DNA
double-strand break repair by microhomology-mediated end joining.
Mol Cell. 83:1429–1445.e8. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lin Y, McMahon A, Driscoll G, Bullock S,
Zhao J and Yan S: Function and molecular mechanisms of APE2 in
genome and epigenome integrity. Mutat Res Rev Mutat Res.
787:1083472021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kumar S, Talluri S, Pal J, Yuan X, Lu R,
Nanjappa P, Samur MK, Munshi NC and Shammas MA: Role of
apurinic/apyrimidinic nucleases in the regulation of homologous
recombination in myeloma: Mechanisms and translational
significance. Blood Cancer J. 8:922018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chan SH, Yu AM and McVey M: Dual roles for
DNA polymerase theta in alternative end-joining repair of
double-strand breaks in Drosophila. PLoS Genet. 6:e10010052010.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yousefzadeh MJ, Wyatt DW, Takata K, Mu Y,
Hensley SC, Tomida J, Bylund GO, Doublié S, Johansson E, Ramsden
DA, et al: Mechanism of suppression of chromosomal instability by
DNA polymerase POLQ. PLoS Genet. 10:e10046542014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ceccaldi R, Liu JC, Amunugama R, Hajdu I,
Primack B, Petalcorin MI, O'Connor KW, Konstantinopoulos PA,
Elledge SJ, Boulton SJ, et al: Homologous-recombination-deficient
tumours are dependent on Polθ-mediated repair. Nature. 518:258–262.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Brambati A, Sacco O, Porcella S, Heyza J,
Kareh M, Schmidt JC and Sfeir A: RHINO directs MMEJ to repair DNA
breaks in mitosis. Science. 381:653–660. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang Y and Jasin M: An essential role for
CtIP in chromosomal translocation formation through an alternative
end-joining pathway. Nat Struct Mol Biol. 18:80–84. 2011.
View Article : Google Scholar
|
|
71
|
Seki M, Masutani C, Yang LW, Schuffert A,
Iwai S, Bahar I and Wood RD: High-efficiency bypass of DNA damage
by human DNA polymerase Q. EMBO J. 23:4484–4494. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ceccaldi R and Cejka P: Mechanisms and
regulation of DNA end resection in the maintenance of genome
stability. Nat Rev Mol Cell Biol. 26:586–599. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Deng SK, Gibb B, De Almeida MJ, Greene EC
and Symington LS: RPA antagonizes microhomology-mediated repair of
DNA double-strand breaks. Nat Struct Mol Biol. 21:405–412. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zimmermann M, Lottersberger F, Buonomo SB,
Sfeir A and de Lange T: 53BP1 regulates DSB repair using Rif1 to
control 5' end resection. Science. 339:700–704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chen C, Umezu K and Kolodner RD:
Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator
mutants due to mutagenic lesions processed by double-strand-break
repair. Mol Cell. 2:9–22. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zuo N, Ma L, Liu T, Hu W, Luo Y, Meng H,
Ren Q, Deng Y, Wei L and Liu Q: Human papillomavirus associated XPF
deficiency increases alternative end joining and cisplatin
sensitivity in head and neck squamous cell carcinoma. Oral Oncol.
140:1063672023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shahid M, Azfaralariff A, Zubair M,
Abdulkareem Najm A, Khalili N, Law D, Firasat S and Fazry S: In
silico study of missense variants of FANCA, FANCC and FANCG genes
reveals high risk deleterious alleles predisposing to Fanconi
anemia pathogenesis. Gene. 812:1461042022. View Article : Google Scholar
|
|
78
|
Barcellos-Hoff MH and Yom SS: Revisiting
the TGFβ paradox: Insights from HPV-driven cancer and the DNA
damage response. Nat Rev Cancer. 25:534–544. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Guix I, Liu Q, Pujana MA, Ha P, Piulats J,
Linares I, Guedea F, Mao JH, Lazar A, Chapman J, et al: Validation
of anticorrelated TGFβ signaling and alternative end-joining DNA
repair signatures that predict response to genotoxic cancer
therapy. Clin Cancer Res. 28:1372–1382. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kirshner J, Jobling MF, Pajares MJ, Ravani
SA, Glick AB, Lavin MJ, Koslov S, Shiloh Y and Barcellos-Hoff MH:
Inhibition of TGFbeta1 signaling attenutates ATM activity in
response to genotoxic stress. Cancer Res. 66:10861–10869. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chowdhury S, Kennedy JJ, Ivey RG, Murillo
OD, Hosseini N, Song X, Petralia F, Calinawan A, Savage SR, Berry
AB, et al: Proteogenomic analysis of chemo-refractory high-grade
serous ovarian cancer. Cell. 187:10162024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Parfenov M, Pedamallu CS, Gehlenborg N,
Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova
EV, Wilkerson MD, et al: Characterization of HPV and host genome
interactions in primary head and neck cancers. Proc Natl Acad Sci
USA. 111:15544–15549. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Leeman JE, Li Y, Bell A, Hussain SS,
Majumdar R, Rong-Mullins X, Blecua P, Damerla R, Narang H,
Ravindran PT, et al: Human papillomavirus 16 promotes
microhomology-mediated end-joining. Proc Natl Acad Sci USA.
116:21573–21579. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Eccleston J, Schrader CE, Yuan K,
Stavnezer J and Selsing E: Class switch recombination efficiency
and junction microhomology patterns in Msh2-, Mlh1-, and
Exo1-deficient mice depend on the presence of mu switch region
tandem repeats. J Immunol. 183:1222–1228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Mann A, Ramirez-Otero MA, De Antoni A,
Hanthi YW, Sannino V, Baldi G, Falbo L, Schrempf A, Bernardo S,
Loizou J and Costanzo V: POLθ prevents MRE11-NBS1-CtIP-dependent
fork breakage in the absence of BRCA2/RAD51 by filling
lagging-strand gaps. Mol Cell. 82:4218–4231.e8. 2022. View Article : Google Scholar
|
|
86
|
Ceccaldi R, Rondinelli B and D'Andrea AD:
Repair pathway choices and consequences at the double-strand break.
Trends Cell Biol. 26:52–64. 2016. View Article : Google Scholar :
|
|
87
|
Sfeir A and Symington LS:
Microhomology-mediated end joining: A back-up survival mechanism or
dedicated pathway? Trends Biochem Sci. 40:701–714. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Brambati A, Barry RM and Sfeir A: DNA
polymerase theta (Polθ)-an error-prone polymerase necessary for
genome stability. Curr Opin Genet Dev. 60:119–126. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
McVey M and Lee SE: MMEJ repair of
double-strand breaks (director's cut): Deleted sequences and
alternative endings. Trends Genet. 24:529–538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wang Z, Song Y, Li S, Kurian S, Xiang R,
Chiba T and Wu X: DNA polymerase θ (POLQ) is important for repair
of DNA double-strand breaks caused by fork collapse. J Biol Chem.
294:3909–3919. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Belan O, Sebald M, Adamowicz M, Anand R,
Vancevska A, Neves J, Grinkevich V, Hewitt G, Segura-Bayona S,
Bellelli R, et al: POLQ seals post-replicative ssDNA gaps to
maintain genome stability in BRCA-deficient cancer cells. Mol Cell.
82:4664–4680.e9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Schrempf A, Bernardo S, Verge E, Ramirez
Otero MA, Wilson J, Kirchhofer D, Timelthaler G, Ambros AM, Kaya A,
Wieder M, et al: POLθ processes ssDNA gaps and promotes replication
fork progression in BRCA1-deficient cells. Cell Rep. 41:1117162022.
View Article : Google Scholar
|
|
93
|
Chanut P, Britton S, Coates J, Jackson SP
and Calsou P: Coordinated nuclease activities counteract Ku at
single-ended DNA double-strand breaks. Nat Commun. 7:128892016.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Britton S, Chanut P, Delteil C, Barboule
N, Frit P and Calsou P: ATM antagonizes NHEJ proteins assembly and
DNA-ends synapsis at single-ended DNA double strand breaks. Nucleic
Acids Res. 48:9710–9723. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
van Schendel R, van Heteren J, Welten R
and Tijsterman M: Genomic scars generated by polymerase theta
reveal the versatile mechanism of alternative end-joining. PLoS
Genet. 12:e10063682016. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gou R, Dong H and Lin B: Application and
reflection of genomic scar assays in evaluating the efficacy of
platinum salts and PARP inhibitors in cancer therapy. Life Sci.
261:1184342020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hanscom T, Woodward N, Batorsky R, Brown
AJ, Roberts SA and McVey M: Characterization of sequence contexts
that favor alternative end joining at Cas9-induced double-strand
breaks. Nucleic Acids Res. 50:7465–7478. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Rempel E, Kluck K, Beck S, Ourailidis I,
Kazdal D, Neumann O, Volckmar AL, Kirchner M, Goldschmid H, Pfarr
N, et al: Pan-cancer analysis of genomic scar patterns caused by
homologous repair deficiency (HRD). NPJ Precis Oncol. 6:362022.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yu AM and McVey M: Synthesis-dependent
microhomology-mediated end joining accounts for multiple types of
repair junctions. Nucleic Acids Res. 38:5706–5717. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sinha S, Li F, Villarreal D, Shim JH, Yoon
S, Myung K, Shim EY and Lee SE: Microhomology-mediated end joining
induces hypermutagenesis at breakpoint junctions. PLoS Genet.
13:e10067142017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Villarreal DD, Lee K, Deem A, Shim EY,
Malkova A and Lee SE: Microhomology directs diverse DNA break
repair pathways and chromosomal translocations. PLoS Genet.
8:e10030262012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chiarle R, Zhang Y, Frock RL, Lewis SM,
Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al:
Genome-wide translocation sequencing reveals mechanisms of
chromosome breaks and rearrangements in B cells. Cell. 147:107–119.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Brunet E, Simsek D, Tomishima M, DeKelver
R, Choi VM, Gregory P, Urnov F, Weinstock DM and Jasin M:
Chromosomal translocations induced at specified loci in human stem
cells. Proc Natl Acad Sci USA. 106:10620–10625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ghezraoui H, Piganeau M, Renouf B, Renaud
JB, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA,
Giovannangeli C, et al: Chromosomal translocations in human cells
are generated by canonical nonhomologous end-joining. Mol Cell.
55:829–842. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Simsek D and Jasin M: Alternative
end-joining is suppressed by the canonical NHEJ component
Xrcc4-ligase IV during chromosomal translocation formation. Nat
Struct Mol Biol. 17:410–416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Simsek D, Brunet E, Wong SY, Katyal S, Gao
Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, et al: DNA ligase
III promotes alternative nonhomologous end-joining during
chromosomal translocation formation. PLoS Genet. 7:e10020802011.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cesare AJ, Hayashi MT, Crabbe L and
Karlseder J: The telomere deprotection response is functionally
distinct from the genomic DNA damage response. Mol Cell.
51:141–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Gu L, Liu M, Zhang Y, Zhou H, Wang Y and
Xu ZX: Telomere-related DNA damage response pathways in cancer
therapy: Prospective targets. Front Pharmacol. 15:13791662024.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Maciejowski J and de Lange T: Telomeres in
cancer: Tumour suppression and genome instability. Nat Rev Mol Cell
Biol. 18:175–186. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bai Y, Wang W and Wang J: Targeting DNA
repair pathways: Mechanisms and potential applications in cancer
therapy. Genome Instability Dis. 1:318–338. 2020. View Article : Google Scholar
|
|
111
|
Xu X, Nowsheen S and Deng M: Exploring the
DNA damage response pathway for synthetic lethality. Genome
Instability Dis. 4:98–120. 2023. View Article : Google Scholar
|
|
112
|
Choi W and Lee ES: Therapeutic targeting
of DNA damage response in cancer. Int J Mol Sci. 23:17012022.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gourley C, Balmaña J, Ledermann JA, Serra
V, Dent R, Loibl S, Pujade-Lauraine E and Boulton SJ: Moving from
poly (ADP-ribose) polymerase inhibition to targeting DNA repair and
DNA damage response in cancer therapy. J Clin Oncol. 37:2257–2269.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Guo Y, Fan B and Li M: PARP molecular
functions and applications of PARP inhibitors in cancer treatment.
Genome Instability Dis. 4:137–153. 2023. View Article : Google Scholar
|
|
115
|
Neeb A, Herranz N, Arce-Gallego S, Miranda
S, Buroni L, Yuan W, Athie A, Casals T, Carmichael J, Rodrigues DN,
et al: Advanced prostate cancer with ATM loss: PARP and ATR
inhibitors. Eur Urol. 79:200–211. 2021. View Article : Google Scholar
|
|
116
|
Liu Q, Gheorghiu L, Drumm M, Clayman R,
Eidelman A, Wszolek MF, Olumi A, Feldman A, Wang M, Marcar L, et
al: PARP-1 inhibition with or without ionizing radiation confers
reactive oxygen species-mediated cytotoxicity preferentially to
cancer cells with mutant TP53. Oncogene. 37:2793–2805. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Weaver AN, Cooper TS, Rodriguez M,
Trummell HQ, Bonner JA, Rosenthal EL and Yang ES: DNA double strand
break repair defect and sensitivity to poly ADP-ribose polymerase
(PARP) inhibition in human papillomavirus 16-positive head and neck
squamous cell carcinoma. Oncotarget. 6:26995–27007. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Staniszewska M, Iking J, Lueckerath K,
Hadaschik B, Herrmann K, Ferdinandus J and Fendler WP: Drug and
molecular radiotherapy combinations for metastatic castration
resistant prostate cancer. Nucl Med Biol. 96:101–111. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nonnekens J, van Kranenburg M, Beerens CE,
Suker M, Doukas M, van Eijck CH, de Jong M and van Gent DC:
Potentiation of peptide receptor radionuclide therapy by the PARP
inhibitor olaparib. Theranostics. 6:1821–1832. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
De Haan R, Van Werkhoven E, Van Den Heuvel
MM, Peulen HMU, Sonke GS, Elkhuizen P, van den Brekel MWM,
Tesselaar MET, Vens C, Schellens JHM, et al: Study protocols of
three parallel phase 1 trials combining radical radiotherapy with
the PARP inhibitor olaparib. BMC Cancer. 19:9012019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Jagsi R, Griffith KA, Bellon JR, Woodward
WA, Horton JK, Ho A, Feng FY, Speers C, Overmoyer B, Sabel M, et
al: Concurrent veliparib with chest wall and nodal radiotherapy in
patients with inflammatory or locoregionally recurrent breast
cancer: The TBCRC 024 phase I multicenter study. J Clin Oncol.
36:1317–1322. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Hou Z, Yu T, Yi Q, Du Y, Zhou L, Zhao Y,
Wu Y, Wu L, Wang T and Bian P: High-complexity of DNA double-strand
breaks is key for alternative end-joining choice. Commun Biol.
7:9362024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Carter R, Nickson C, Thompson J, Kacperek
A, Hill M and Parsons J: Complex DNA damage induced by high linear
energy transfer Alpha-particles and protons triggers a specific
cellular DNA damage response. Int J Radiat Oncol Biol Phys.
100:776–784. 2017. View Article : Google Scholar
|
|
124
|
Hirai T, Shirai H, Fujimori H, Okayasu R,
Sasai K and Masutani M: Radiosensitization effect of poly
(ADP-ribose) polymerase inhibition in cells exposed to low and high
liner energy transfer radiation. Cancer Sci. 103:1045–1050. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Césaire M, Ghosh U, Austry JB, Muller E,
Cammarata FP, Guillamin M, Caruso M, Castéra L, Petringa G, Cirrone
GAP and Chevalier F: Sensitization of chondrosarcoma cells with
PARP inhibitor and high-LET radiation. J Bone Oncol. 17:1002462019.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Dong M, Luo H, Liu R, Zhang J, Yang Z,
Wang D, Wang Y, Chen J, Ou Y, Zhang Q and Wang X:
Radiosensitization of osteosarcoma cells using the PARP inhibitor
olaparib combined with X-rays or carbon ions. J Cancer. 15:6992024.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Molkentine JM, Molkentine DP, Bridges KA,
Xie T, Yang L, Sheth A, Heffernan TP, Clump DA, Faust AZ, Ferris
RL, et al: Targeting DNA damage response in head and neck cancers
through abrogation of cell cycle checkpoints. Int J Radiat Biol.
97:1121–1128. 2021. View Article : Google Scholar
|
|
128
|
Machacova Z, Chroma K, Lukac D,
Protivankova I and Moudry P: DNA polymerase α-primase facilitates
PARP inhibitor-induced fork acceleration and protects
BRCA1-deficient cells against ssDNA gaps. Nat Commun. 15:73752024.
View Article : Google Scholar
|
|
129
|
Russo TDB, Mujacic C, Di Giovanni E,
Vitale MC, Ferrante Bannera C, Randazzo U, Contino S, Bono M,
Gristina V, Galvano A, et al: Polθ: Emerging synthetic lethal
partner in homologous recombination-deficient tumors. Cancer Gene
Ther. 31:1619–1631. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Rodriguez-Berriguete G, Ranzani M, Prevo
R, Puliyadi R, Machado N, Bolland HR, Millar V, Ebner D, Boursier
M, Cerutti A, et al: Small-molecule Polθ inhibitors provide safe
and effective tumor radiosensitization in preclinical models. Clin
Cancer Res. 29:1631–1642. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhou J, Gelot C, Pantelidou C, Li A, Yücel
H, Davis RE, Färkkilä A, Kochupurakkal B, Syed A, Shapiro GI, et
al: A first-in-class polymerase theta inhibitor selectively targets
homologous-recombination-deficient tumors. Nat Cancer. 2:598–610.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zatreanu D, Robinson HM, Alkhatib O,
Boursier M, Finch H, Geo L, Grande D, Grinkevich V, Heald RA and
Langdon S: Polθ inhibitors elicit BRCA-gene synthetic lethality and
target PARP inhibitor resistance. Nat Commun. 12:36362021.
View Article : Google Scholar
|
|
133
|
Fried W, Tyagi M, Minakhin L, Chandramouly
G, Tredinnick T, Ramanjulu M, Auerbacher W, Calbert M, Rusanov T
and Hoang T: Discovery of a small-molecule inhibitor that traps
Polθ on DNA and synergizes with PARP inhibitors. Nat Commun.
15:28622024. View Article : Google Scholar
|
|
134
|
Higgins GS, Prevo R, Lee YF, Helleday T,
Muschel RJ, Taylor S, Yoshimura M, Hickson ID, Bernhard EJ and
McKenna WG: A small interfering RNA screen of genes involved in DNA
repair identifies tumor-specific radiosensitization by POLQ
knockdown. Cancer Res. 70:2984–2993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Shima N, Munroe RJ and Schimenti JC: The
mouse genomic instability mutation chaos1 is an allele of Polq that
exhibits genetic interaction with Atm. Mol Cell Biol.
24:10381–10389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Mengwasser KE, Adeyemi RO, Leng Y, Choi
MY, Clairmont C, D'Andrea AD and Elledge SJ: Genetic screens reveal
FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol Cell.
73:885–899.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sharma S, Javadekar S, Pandey M,
Srivastava M, Kumari R and Raghavan S: Homology and enzymatic
requirements of microhomology-dependent alternative end joining.
Cell Death Dis. 6:e1697. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Li J, Ko JMY, Dai W, Yu VZ, Ng HY,
Hoffmann JS and Lung ML: Depletion of DNA polymerase theta inhibits
tumor growth and promotes genome instability through the
cGAS-STING-ISG pathway in esophageal squamous cell carcinoma.
Cancers (Basel). 13:32042021. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Dai C-H, Chen P, Li J, Lan T, Chen YC,
Qian H, Chen K and Li MY: Co-inhibition of pol θ and HR genes
efficiently synergize with cisplatin to suppress
cisplatin-resistant lung cancer cells survival. Oncotarget.
7:65157–65170. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Chen X, Zhong S, Zhu X, Dziegielewska B,
Ellenberger T, Wilson GM, MacKerell AD Jr and Tomkinson AE:
Rational design of human DNA ligase inhibitors that target cellular
DNA replication and repair. Cancer Res. 68:3169–3177. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Martires LCM, Ahronian LG, Pratt CB, Das
NM, Zhang X, Whittington DA, Zhang H, Shen B, Come J, McCarren P,
et al: LIG1 is a synthetic lethal target in BRCA1 mutant cancers.
Mol Cancer Ther. 24:618–627. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Tobin LA, Robert C, Nagaria P, Chumsri S,
Twaddell W, Ioffe OB, Greco GE, Brodie AH, Tomkinson AE and Rassool
FV: Targeting abnormal DNA repair in therapy-resistant breast
cancers. Mol Cancer Res. 10:96–107. 2012. View Article : Google Scholar :
|
|
143
|
Tobin LA, Robert C, Rapoport AP, Gojo I,
Baer MR, Tomkinson AE and Rassool FV: Targeting abnormal DNA
double-strand break repair in tyrosine kinase inhibitor-resistant
chronic myeloid leukemias. Oncogene. 32:1784–1793. 2013. View Article : Google Scholar
|
|
144
|
Álvarez-Quilón A, Wojtaszek JL, Mathieu
MC, Patel T, Appel CD, Hustedt N, Rossi SE, Wallace BD, Setiaputra
D, Adam S, et al: Endogenous DNA 3' blocks are vulnerabilities for
BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease.
Mol Cell. 78:1152–1165.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Hossain MA, Lin Y, Driscoll G, Li J,
McMahon A, Matos J, Zhao H, Tsuchimoto D, Nakabeppu Y, Zhao J and
Yan S: APE2 is a general regulator of the ATR-Chk1 DNA damage
response pathway to maintain genome integrity in pancreatic cancer
cells. Front Cell Dev Biol. 9:7385022021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Barcellos-Hoff MH and Gulley JL: Molecular
pathways and mechanisms of TGFβ in cancer therapy. Clin Cancer Res.
29:2025–2033. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
de Bono J, Mateo J, Fizazi K, Saad F,
Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D, et al:
Olaparib for metastatic castration-resistant prostate cancer. N
Engl J Med. 382:2091–2102. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Wang L, Cao J, Wang X, Lin E, Wang Z, Li
Y, Li Y, Chen M, Wang X, Jiang B, et al: Proton and photon
radiosensitization effects of niraparib, a PARP-1/-2 inhibitor, on
human head and neck cancer cells. Head Neck. 42:2244–2256. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Hintelmann K, Berenz T, Kriegs M,
Christiansen S, Gatzemeier F, Struve N, Petersen C, Betz C,
Rothkamm K, Oetting A and Rieckmann T: Dual inhibition of PARP and
the intra-S/G2 cell cycle checkpoints results in highly effective
radiosensitization of HPV-positive HNSCC cells. Front Oncol.
11:6836882021. View Article : Google Scholar : PubMed/NCBI
|














