Introduction
Breast cancer (BC), a common malignancy, is the
leading cause of cancer-related death in women worldwide (1). Although some progress has been made
in understanding the pathogenesis and treatment of BC (2-6),
its pathogenesis is complex and involves abnormal regulation of a
variety of genes and signaling pathways; thus, further studies are
still needed. The family of protein tyrosine phosphatases (PTPs)
rich in proline, glutamic acid, serine and threonine includes PTP
non-receptor type 18 (PTPN18), PTPN22 and PTPN12 (7). Members of this family have a key
role in various physiological and pathological processes of cells
(7). For instance, previous
studies by the authors confirmed the notable role of PTPN22 in
immune regulation (8,9) as well as signaling events triggered
by the synergistic inactivation of Src kinase by PTPN18 and C-Src
kinase (10). As a classical PTP,
PTPN18 can also regulate multiple signal transduction pathways by
regulating the phosphorylation of cellular Abelson tyrosine kinase,
a key signaling protein tyrosine kinase (11). The interaction between PTPN18 and
proline-serine-threonine phosphatase interacting protein 1/2 or
P190 Rho GTPase-activating protein confirms its function in
regulating the cytoskeleton (11-14). Additionally, it has been shown
that PTPN18 negatively regulates human epidermal growth factor
receptor 2 (HER2) to inhibit BC progression (15,16). Furthermore, a recent study by the
authors found that nuclear PTPN18 exerts antitumor effects in BC by
targeting ETS proto-oncogene 1 (ETS1) to inhibit transforming
growth factor-β signaling and epithelial-mesenchymal transition
(EMT) (17). Notably, patients
with BC harboring high PTPN18 expression have an improved prognosis
and longer overall survival (OS) (17), which suggests that PTPN18 may play
an important role in BC progression.
Cyclin E, a key molecule of the cell cycle
regulatory network, functions in the G1 and S phases and mainly
binds to and activates cyclin-dependent kinase 2 (CDK2) (18). Cyclin E/CDK2 complexes
phosphorylate numerous substrates to control important cellular
processes (18,19). There are two E-type cyclins, E1
and E2, which are considered to primarily exert overlapping
functions (18). However, it has
been reported that cyclin E1 and cyclin E2 have different emphasis
on regulatory patterns and functions (20). High levels of cyclin E protein
lead to poor prognosis, reduced survival and treatment resistance
in patients with cancer (21,22). Additionally, cyclin E
amplification/upregulation is one of the mechanisms of trastuzumab
resistance in patients with HER2+ BC (23). HER2 is an important target of
PTPN18 in inhibiting BC (15,16), suggesting that there may be a link
between PTPN18 and cyclin E.
Based on the potential importance of PTPN18 in the
field of oncology as well as its broad role in the regulation of
cellular function, it is necessary to further explore its role in
BC and its underlying mechanisms. Therefore, through phenotypic
functional experiments, the present study first confirmed that
PTPN18 can exert antitumor effects by promoting apoptosis,
inhibiting metastasis and proliferation and causing S phase cell
cycle arrest in BC cells. Subsequently, it was found that S-phase
arrest caused by PTPN18 may be via the downregulation of cyclin E
expression. Further mechanistic dissection revealed that cyclin E
is an important substrate for PTPN18 in exerting antitumor effects.
Taken together, the mechanism of BC development and progression
identified in the present study provides an additional
comprehensive and extensive theoretical basis for the prevention
and treatment of this disease.
Materials and methods
Cell culture and transfection
The Michigan Cancer Foundation-7 (MCF7) and
Metastatic Derivative of Anaplastic BC-231 (MDA-MB-231) cell lines
were obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences and maintained in Dulbecco's modified
Eagle's medium (HyClone; Cytiva) supplemented with 10% fetal bovine
serum (FBS; HyClone; Cytiva) and 1% penicillin/streptomycin at 37°C
in a humidified incubator with 5% CO2. Interference with
PTPN18 expression levels was achieved by transfecting (37°C, 6 h)
cells with PTPN18-3HA (1 µg/µl) and PTPN18 small
interfering RNA (20 µM) (siRNA; Suzhou GenePharma Co., Ltd.)
using Lipo6000 (Beyotime Institute of Biotechnology), according to
the manufacturer's instructions. To achieve an improved knockdown
efficiency, the knockdown siRNA was transfected again after 24 h of
initial transfection. The siRNA sequences used are listed in
Table SI.
Western blotting
First, total protein was extracted from cells using
precooled radioimmunoprecipitation assay (RIPA) buffer (Beyotime
Biotechnology) containing 1% protease inhibitor and phosphatase
inhibitor (Selleck Chemicals) and then quantified by bicinchoninic
acid assay. Equal amounts of protein (10 µg) were separated
by 10% SDS-PAGE at a constant voltage (80 V) until the blue band of
the loading buffer approached the lower edge of the gel. Proteins
were then transferred to PVDF membranes using the sandwich method
at a constant voltage of 100 V for 100 min at 4°C. Membranes were
blocked with 5% bovine serum albumin (Beyotime Institute of
Biotechnology) in Tris-buffered saline with 0.1% Tween 20 (TBST)
for 1 h at room temperature, then incubated overnight at 4°C with
blocking buffer plus primary antibodies. Subsequent washing with
TBST was followed by incubation with secondary antibodies for 1 h
at room temperature. Goat anti-rabbit IgG (heavy and light chain)
antibody or horse anti-mouse IgG antibody conjugated with
horseradish peroxidase secondary antibody was selected for
chemiluminescence detection according to the primary antibody
species. Finally, the protein bands were visualized and
semi-quantified using the ChemiDoc XRS + Gel Imaging Analysis
System (Bio-Rad Laboratories, Inc.) and Image Lab 5.2.1 software
(6). The antibody information is
listed in Table SII.
Co-immunoprecipitation (Co-IP) assay
Treated cells were lysed in pre-chilled RIPA lysis
buffer containing protease inhibitor and phosphatase inhibitor
cocktails for 10-30 min. Experiments involving tyrosine
phosphorylation or PTPN18 C229S were stimulated with EGF 100 ng/ml
for 30 min before cells were collected. The samples were
centrifuged at 12,000 × g for 15 min at 4°C to remove cell debris,
then the supernatant was incubated with the appropriate primary
antibody for 2-4 h or overnight at 4°C. The conjugated product was
incubated with 30-50 µl Protein A/G agarose beads (Beyotime
Biotechnology) for 2-4 h at 4°C the following day. The agarose
beads were washed five times with cold lysis buffer, the
supernatant was discarded and the beads were boiled with 1X SDS
loading buffer for 10 min prior to western blotting.
Flow cytometry apoptosis assay
The apoptosis of BC cells was observed using flow
cytometry. Cells were washed twice with pre-chilled
phosphate-buffered saline (PBS) and centrifuged at 500 × g for 5
min at 4°C to collect 1-2.5×106 cells. The cells were
resuspended in 100 µl of 1X Binding Buffer (BD Biosciences)
and divided into two aliquots, one without Annexin V-FITC/PI and
the other with 5 µl Annexin V-FITC and 5 µl PI
Staining Solution (BD Biosciences), then mixed gently. The
individual groups were stained with PI or FITC only for subsequent
adjustment of compensation. The samples were protected from the
light and the reaction was allowed to proceed at room temperature
for 15 min. Next, 400 µl of 1X Binding Buffer was added and
mixed well with the samples, which were then filtered with gauze
and detected by BD Accuri™ C6 Plus flow cytometry (BD
Biosciences) within 1 h. For each sample, 10,000 events were
analyzed and the experiment was independently repeated three times
(24). Data were analyzed and
processed using the FlowJo 10.8.1 (FlowJo LLC; BD Biosciences)
software.
Nuclear staining with
4′,6-diamidino-2-phenylindole (DAPI)
In vitro apoptosis was examined by DAPI
staining. MCF-7 cells that had previously been transfected for 48 h
were seeded into 24-well plates for overnight incubation and
attachment. The cells were then washed with PBS and fixed with 4%
paraformaldehyde for 10 min at room temperature. The fixative was
removed, then the cells were washed with PBS and stained with 2
µg/ml DAPI (Beyotime Institute of Biotechnology) for 15 min
at room temperature in the dark. Changes in nuclear morphology were
examined by fluorescence microscopy and images were collected.
Cell cycle assay
Cell cycle distribution was determined using an
Accuri C6 flow cytometer (BD Biosciences). The collected cells were
fixed with pre-chilled 70-80% ethanol overnight at 4°C, centrifuged
to remove the fixative and washed once with 1 ml PBS. The samples
were then divided into two aliquots after the supernatant had been
removed: One aliquot was incubated at room temperature without
PI/RNase for 15 min; the other was incubated at room temperature
with 500 µl PI/RNase (BD Biosciences) in the dark for 15
min. The cells were then filtered with a 200-mesh filter and
detected by flow cytometry. FlowJo 10.8.1 (FlowJo LLC; BD
Biosciences) or ModFit LT 4.0 software (Verity Software House) was
used to process the data (25).
Cell viability assay
Cell viability was monitored using the MTS assay, a
modified version of the classical MTT assay that has the same core
principle (26). The treated
cells (2,000 cells/100 µl medium) were digested and
transferred to 96-well plates, with four duplicate wells set up for
each treatment group, and the absorbance was detected after 0, 12,
24, 36 and 48 h. In each well, an average of 20 µl of
CellTiter 96® AQueous One Solution Reagent (Promega
Corporation) was added to 100 µl of medium. After gentle
mixing and 2 h of reaction in a 37°C and 5% CO2
incubator, the absorbance at 490 nm was measured.
Colony formation assay
Treated cells were seeded into 6-well plates at a
density of 2,000 cells/well. After the cells adhered overnight, the
medium was replaced every 3 days for 14 days. The cells were then
fixed with 4% paraformaldehyde (room temperature, 30 min) and
stained with 0.1% crystal violet (Beyotime Institute of
Biotechnology) for 20 min at room temperature. Finally, the plates
were washed with water and air dried at room temperature. The
colonies were recorded using ImageJ 1.53e software (National
Institutes of Health).
Transwell invasion assay
Conditioned medium (500 µl) containing 20%
FBS was added to the lower chamber of a 24-well plate. Next, 100
µl of diluted BD Matrigel was added to each upper chamber.
After the Matrigel had solidified at 37°C, a digested and diluted
serum-free cell suspension was added to the upper chamber, and the
plates were incubated at 37°C for 24-48 h. The inserts (8
µm) were then washed with PBS and fixed with a 5%
paraformaldehyde solution for 30 min. The cells were stained with
crystal violet (0.1%) at room temperature for 30 min, then observed
and counted under an inverted fluorescence microscope (Leica
Microsystems GmbH) operating in white light mode.
Wound healing assay
After creating a scratch with a 200-µl
sterilized tip, the cells were washed three times with sterilized
PBS, the impurities and cell debris were washed off, and fresh low
serum (1%) culture medium was added to continue the cell culture.
Images were collected at 0, 24 and 48 h and the change in scratch
size was recorded. The experiment was repeated three times. ImageJ
software was used to assess the scratch area and for subsequent
experimental data processing.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted using TRIzol and
cDNA synthesis was performed using the All-in-One cDNA Synthesis
SuperMix kit (Selleck Chemicals) following the manufacturer's
instructions. In a total reaction volume of 20 µl per well,
the 2X SYBR Green qPCR Master Mix kit (Selleck Chemicals) was used
for qPCR. The PCR amplification protocol consisted of
pre-denaturation at 95°C for 5 min; followed by 40 cycles of
denaturation at 95°C for 15 sec, annealing at 55°C for 30 sec, and
extension at 72°C for 30 sec. A total of 3-5 replicate wells were
set up per group. The mean quantification cycle (Cq) values were
collected for each reaction and relative expression was quantified
using the 2−ΔΔCq method (27). The sequences of the qPCR primers
used in the present study are listed in Table SIII.
Associated database analysis
Changes in the expression levels of total PTPN18
protein were analyzed through the 'Proteomics' catalog of the
UALCAN database (https://ualcan.path.uab.edu/cgi-bin/CPTAC-Result.pl?genenam=PTPN18&ctype=Breast)
(28). 'Expression DIY' in the
Gene Expression Profiling Interactive Analysis (GEPIA) database
(http://gepia2.cancer-pku.cn/#index)
was used to obtain gene expression related information in the box
plot and stage plot formats. Correlation analysis (GEPIA) was used
to test the association of PTPN18 or CDKN1A/B with CCNE1/2 gene.
Survival analysis (GEPIA) was used to obtain gene and OS or
disease-free survival data (29).
Additionally, gene expression levels and OS or relapse-free
survival data were obtained from the BC category of the
Kaplan-Meier Plotter database (https://kmplot.com/analysis/index.php?p=service&cancer=breast)
(30). PTPN18 correlation
analysis was performed using breast invasive carcinoma and HiSeq
RNA data from the LinkedOmics platform (https://www.linkedomics.org/admin.php) (31). Protein interaction networks for
cyclin E1 and cyclin E2 were obtained from the STRING database
(https://cn.string-db.org/) (32).
Statistical analysis
Data are presented as the mean ± SD of at least
three independent experiments. Data analysis was performed using
GraphPad Prism software 8.0.2 (Dotmatics). Data were analyzed using
unpaired Student's t-test or one-way analysis of variance with
Tukey's multiple comparison test, as appropriate. P<0.05 was
considered to indicate a statistically significant difference.
Results
PTPN18 expression is associated with
BC
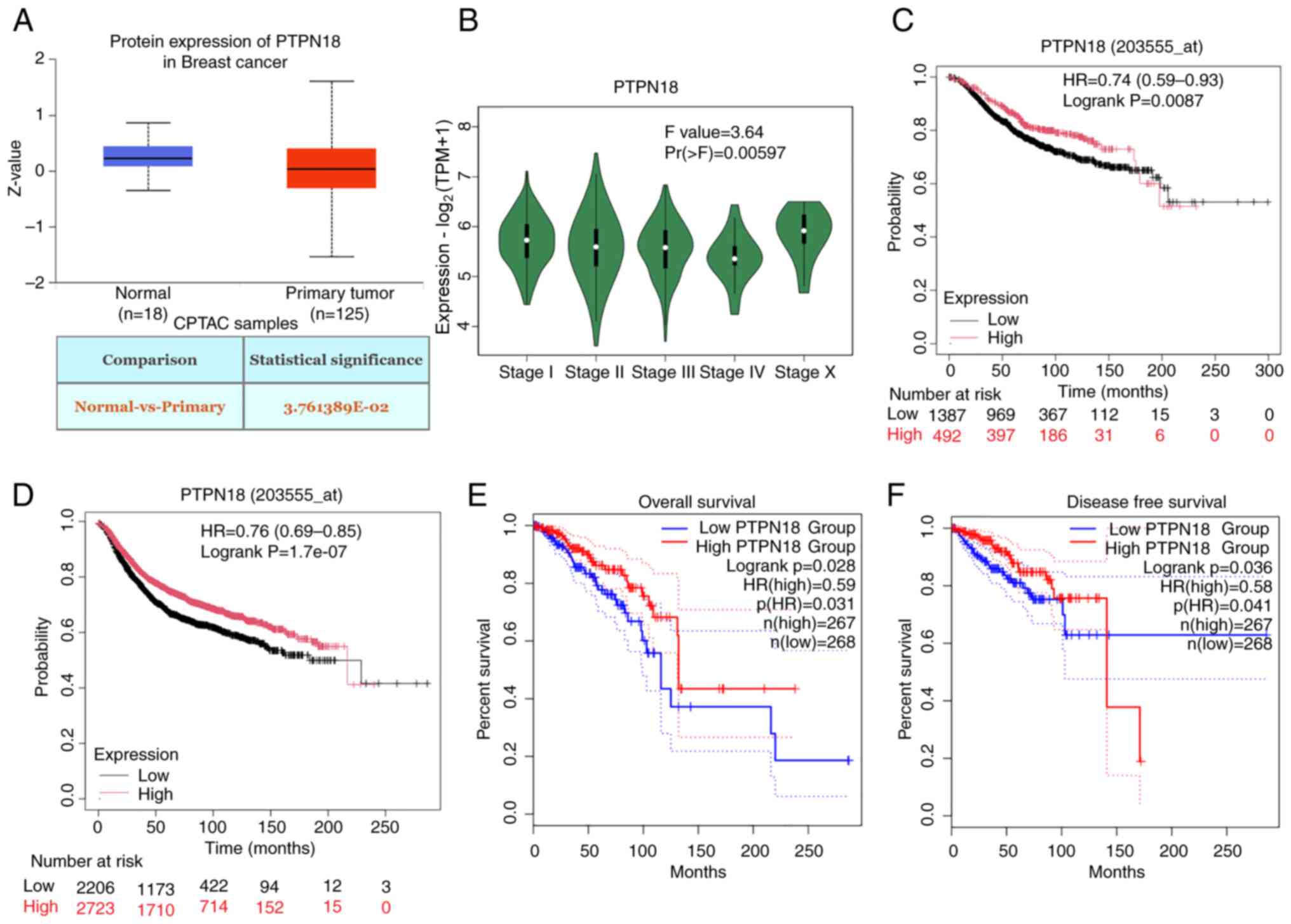
Proteomic expression profiling using the UALCAN
database showed that PTPN18 protein expression levels are decreased
in BC (Fig. 1A). In addition, a
close link between PTPN18 gene expression and the stage of BC was
identified (Fig. 1B). Statistical
analyses using the Kaplan-Meier Plotter and GEPIA databases showed
that high levels of PTPN18 improved the survival and prognosis of
patients with BC (Fig. 1C-F),
indicating that PTPN18 may play a tumor suppressor role in this
disease. Therefore, the function and mechanism of PTPN18 in BC was
further investigated.
PTPN18 suppresses the metastasis of BC
cells
PTPN18 is widely expressed in BC cell lines
(15,16). In the present study, the western
blot results showed that PTPN18 was expressed at higher levels in
MCF7 and MDA-MB-231 cells (Fig.
S1A), which represent estrogen receptor and progesterone
receptor double-positive and triple-negative BC cell lines,
respectively. Overexpression and knockdown of PTPN18 in MCF7 cells
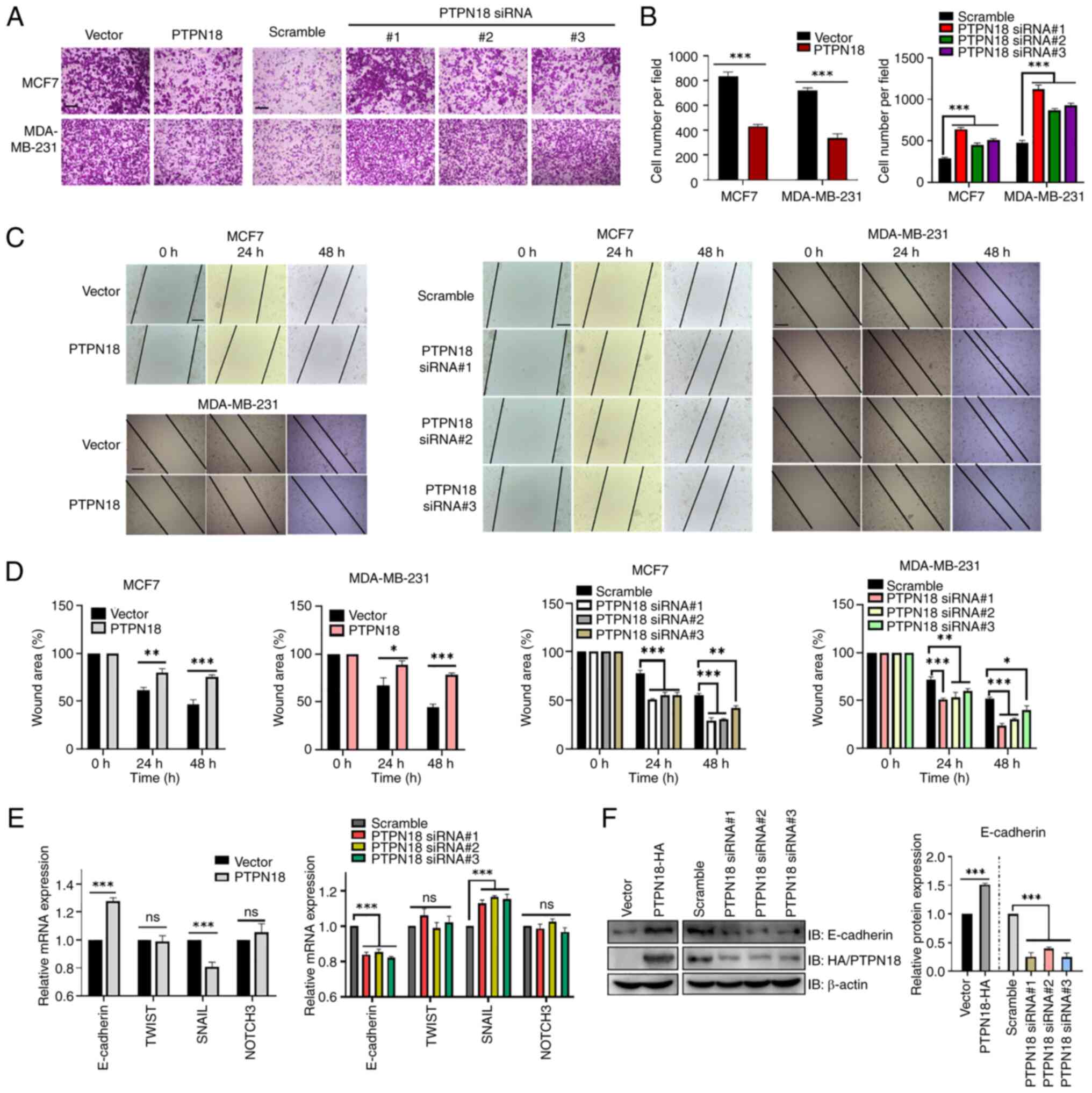
at different time points after transfection is shown in Fig. S1B and C. Next, the effect of
PTPN18 on cancer cell invasion was investigated using a Transwell
chamber assay. The results showed that overexpression of PTPN18
effectively inhibited the invasion of cancer cells, while knockdown
of endogenous PTPN18 expression enhanced invasion (Fig. 2A and B). Consistent with these
results, in vitro cell wound healing assays demonstrated
that PTPN18 expression inhibited tumor cell migration (Fig. 2C and D). Analysis of the mRNA
levels of genes involved in cell metastasis revealed that
E-cadherin gene expression increased and Snail family
transcriptional repressor 1 expression decreased in the PTPN18
overexpression group compared with the vector group, while the
TWIST basic helix-loop-helix transcription factor 1 and Notch
receptor 3 levels remained essentially unchanged (Fig. 2E). The trend was roughly the
opposite between the knockdown and overexpression groups (Fig. 2E). Subsequent verification of the
translation levels of the E-cadherin EMT marker protein revealed
changes consistent with the transcription levels (Fig. 2F).
PTPN18 inhibits BC cell proliferation and
induces apoptosis
The key to cancer treatment is inhibiting the
metastasis and proliferation of cancer cells and inducing
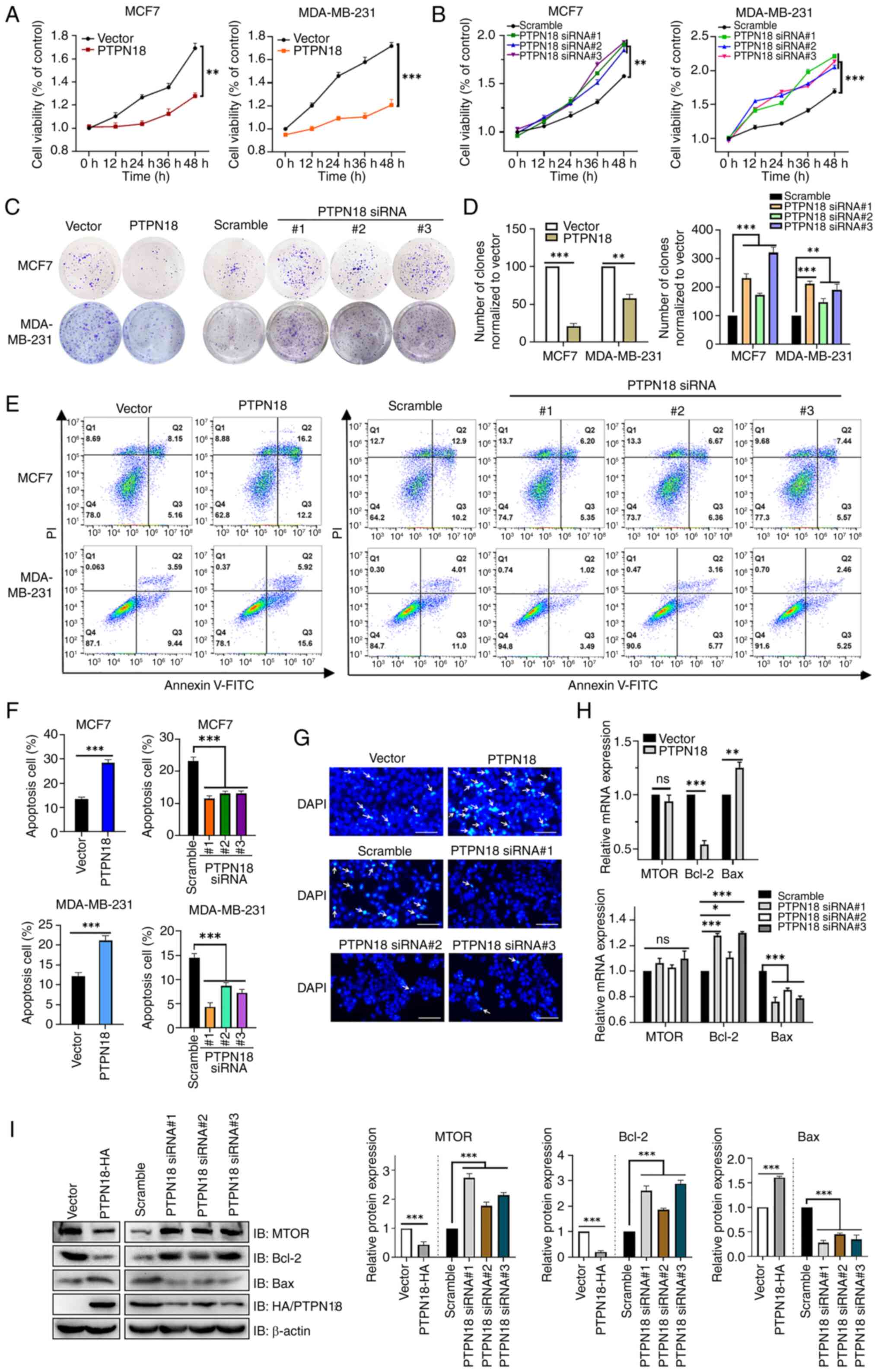
apoptosis. The MTS assay results demonstrated that PTPN18
overexpression inhibited cell viability, an inhibitory effect that
exhibited an increasing cumulative trend over time (Fig. 3A). Although the MCF7 and
MDA-MB-231 cells in the PTPN18 knockdown groups showed a high cell
viability at different time points, the overall trend was the
opposite to that of the overexpression group (Fig. 3B). This conclusion was further
verified by a colony formation assay (Fig. 3C and D).
The flow cytometric results identified that PTPN18
overexpression significantly increased the proportion of BC cells
undergoing apoptosis, while the three knockdowns of PTPN18 revealed
different degrees of apoptosis' inhibition (Fig. 3E and F). In addition, the MCF7 BC
cell line was stained with DAPI and subsequent fluorescence
microscopy showed significantly increased signs of apoptotic cell
death, such as chromatin condensation and fragmentation, in the
PTPN18 overexpression group compared with the control cells
(Fig. 3G). Furthermore, RT-qPCR
(Fig. 3H) and western blot
(Fig. 3I) results showed that
PTPN18 inhibited the expression of mechanistic target of rapamycin
kinase (mTOR) mainly at the protein level. Additionally, PTPN18
promoted Bax expression and suppressed Bcl-2 expression (Fig. 3H and I). Collectively, these
results confirmed that PTPN18 inhibited proliferation and promoted
the apoptosis of BC cells.
PTPN18 can induce S phase cell cycle
arrest in BC by downregulating the expression of cyclin E
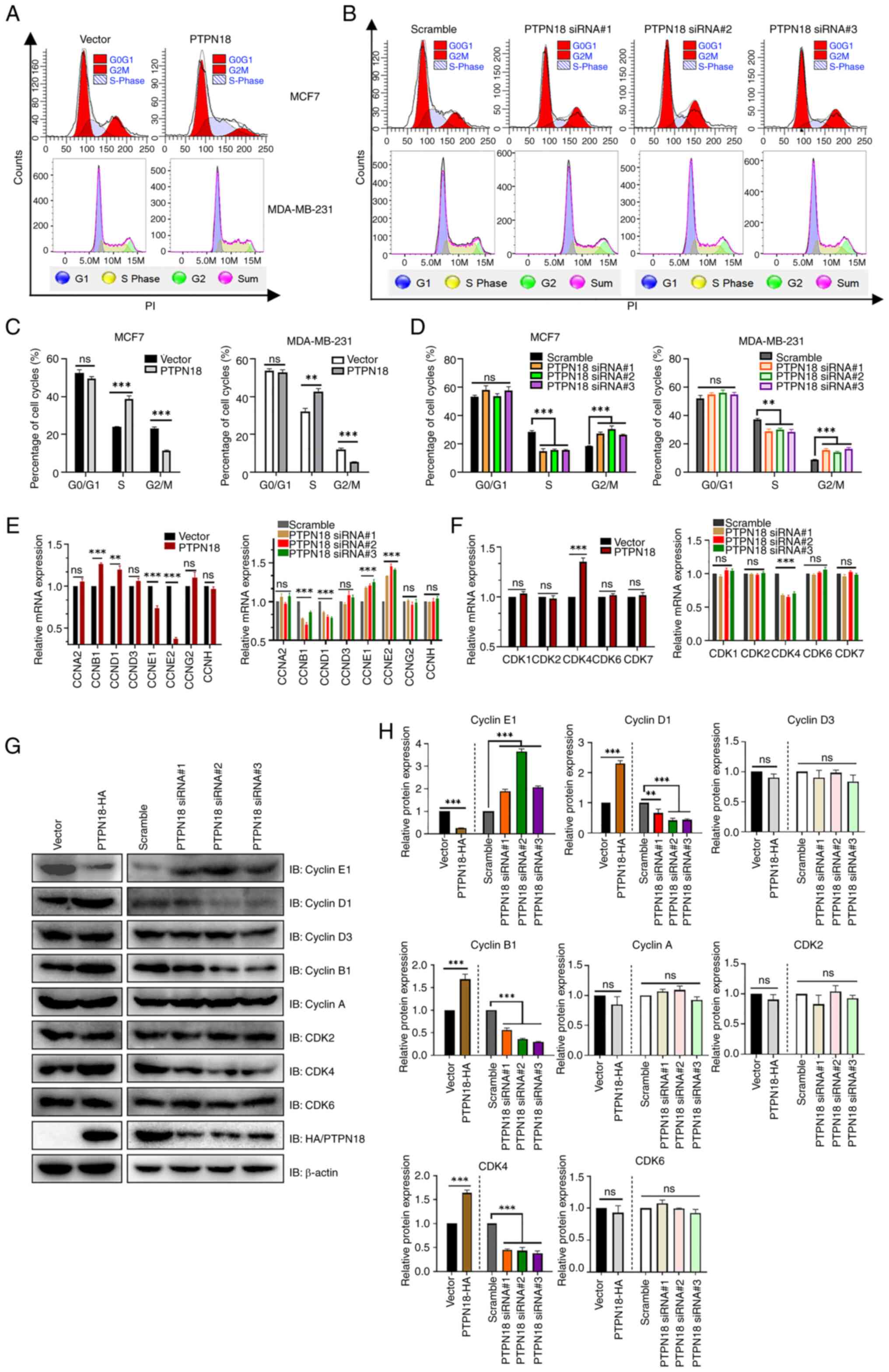
Next, the effect of PTPN18 on the cell cycle of BC
cells was further investigated. The results of the cell cycle flow
cytometry experiments revealed that PTPN18 overexpression
significantly increased the number of cells in the S phase and
decreased the number of cells in the G2/M phase but did not
significantly change the number of cells in the G0/G1 phase
(Fig. 4A and C). However, the
PTPN18 knockdown groups showed the opposite trend (Fig. 4B and D), indicating that PTPN18
could induce cell cycle arrest in the S phase and affect the cell
cycle progression of BC cells. In addition, PTPN18 overexpression
significantly decreased cyclin E1 expression and increased cyclin
D1, cyclin B1 and CDK4 expression at both the transcriptional and
translational levels (Fig. 4E-H).
These results suggested that significant downregulation of cyclin E
may be an important factor in PTPN18 overexpression-induced S phase
cell cycle arrest in BC. The aforementioned phenotypic functional
experiments demonstrated that PTPN18 could play a tumor suppressive
role in BC cells by promoting apoptosis and inhibiting
proliferation, cell cycle, migration and invasion.
PTPN18 and the cell cycle are
significantly and negatively correlated in BC
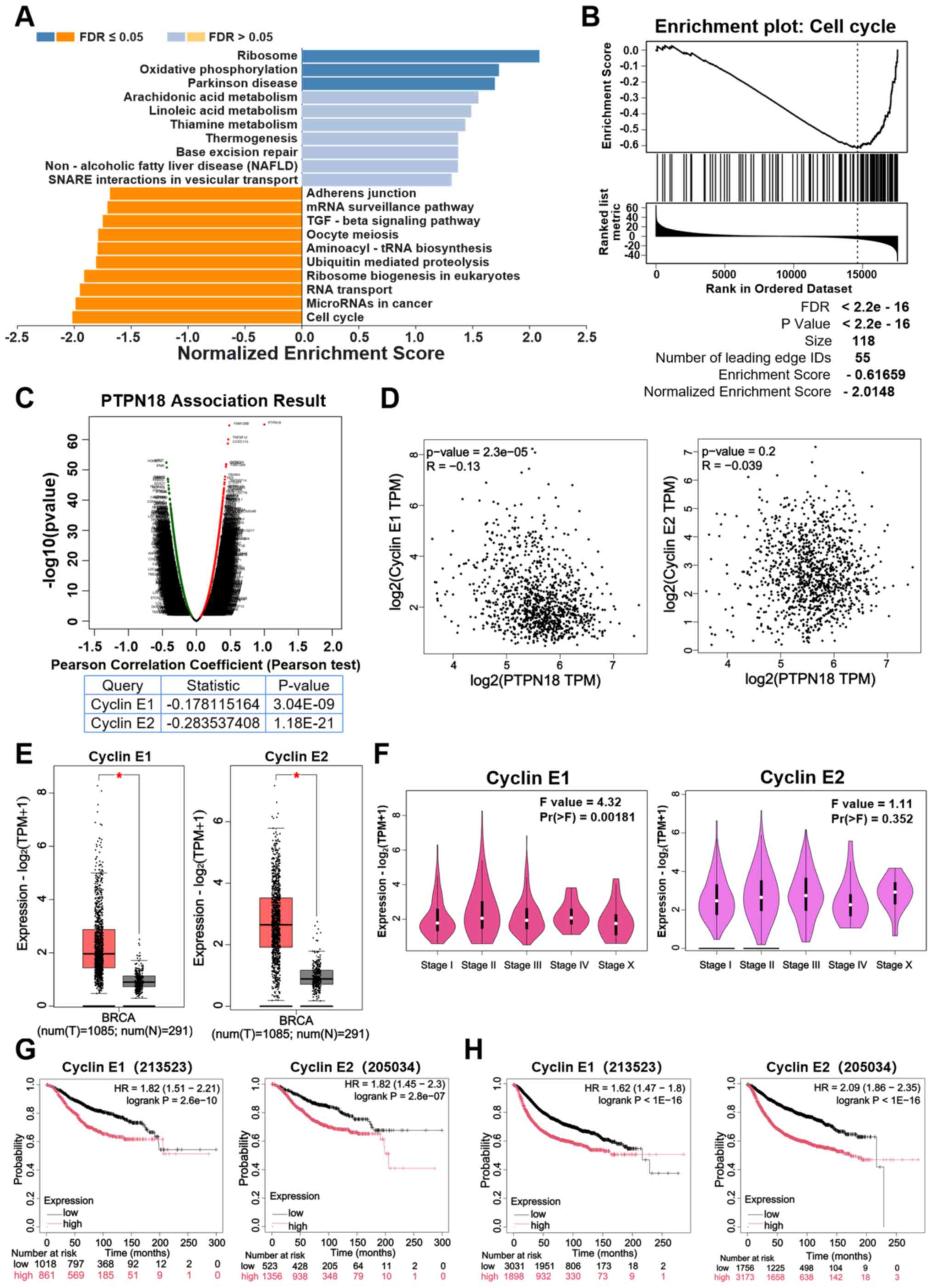
To illustrate the biological function of PTPN18 in
BC development, RNA sequencing gene enrichment analysis of PTPN18
in BC was performed using the LinkedOmics database. The results
revealed that PTPN18 showed a close and significantly negative
correlation with the cell cycle in BC (Fig. 5A). Additionally, the results of
the cell cycle gene enrichment analysis showed that PTPN18 may
affect cell cycle progression by inhibiting gene expression of cell
cycle pathways (Fig. 5B). Volcano
plots revealed the genes that were positively or negatively
correlated with PTPN18 (Fig. 5C).
Pearson correlation coefficient analysis suggested that PTPN18 was
significantly negatively correlated with cyclins E1 and E2
(Fig. 5C). Furthermore, the GEPIA
database gene correlation analysis results also demonstrated that
PTPN18 was negatively correlated with cyclins E1 and E2 (Fig. 5D). The cyclin E1 and E2 expression
levels were found to be significantly increased in BC tissues
compared with normal tissues (Fig.
5E). Cyclin E1 expression was significantly different in
different clinical stages of BC and may play a role in disease
stage progression; however, cyclin E2 expression was weakly
associated with stage, suggesting that the functions of these two
proteins in disease progression may be different (Fig. 5F). The results of the survival
analyses using the Kaplan-Meier Plotter and GEPIA databases
identified that high expression of cyclin E was associated with a
reduced OS and the poor prognosis of patients with BC (Fig. 5G and H; Fig. S2A and B). These analyses support
the hypothesis that PTPN18 may affect the BC cell cycle through
regulation of the cyclin E pathway. These results echo the
aforementioned experimental results and reinforce the rationality
and necessity of studying the mechanism of cyclin E downregulation
due to PTPN18 overexpression.
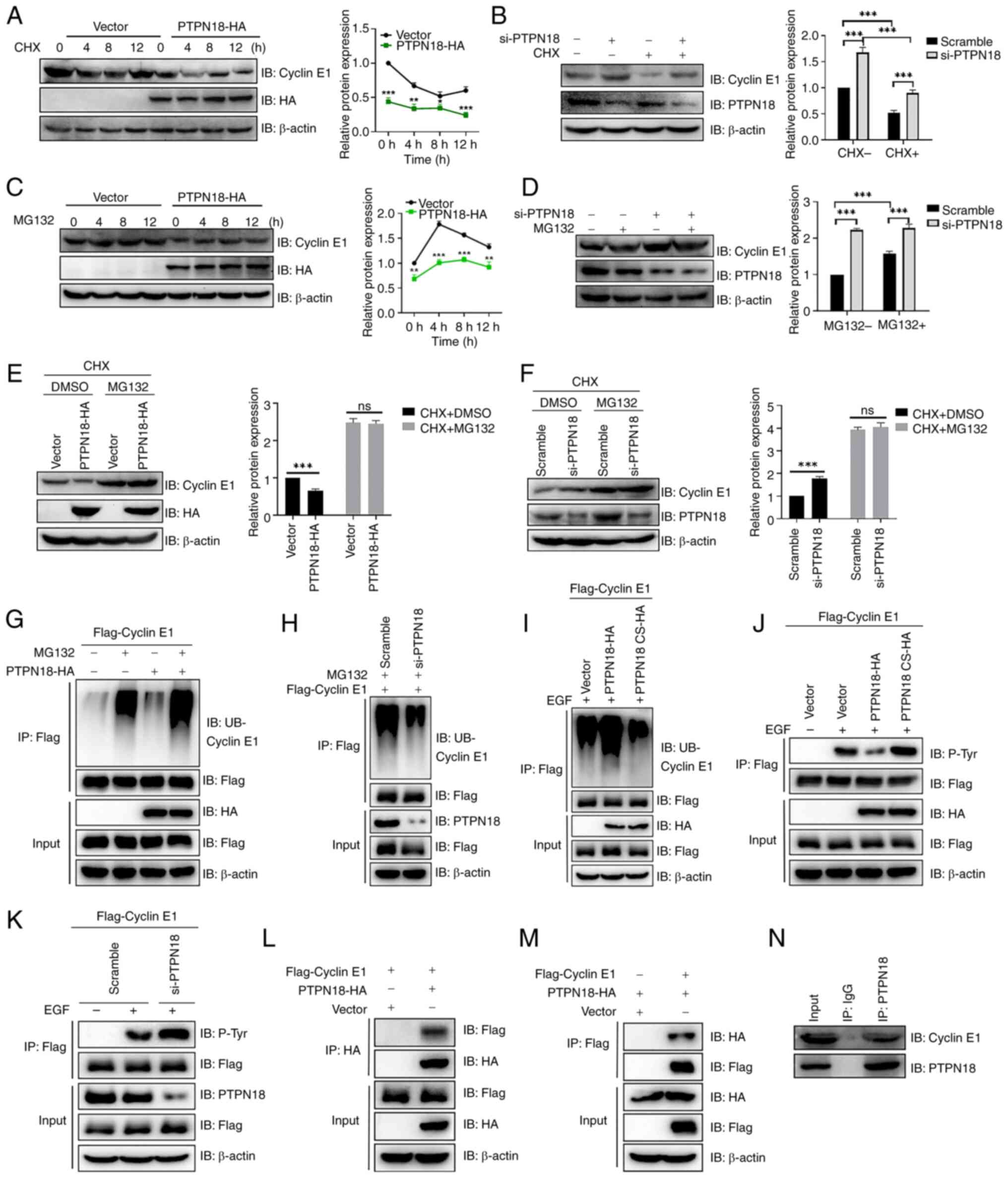
PTPN18 can interact with cyclin E1 and
promote its proteasomal degradation
In view of the notable role of PTPN18 in the BC cell
cycle, the present study further explored the molecular mechanism
by which PTPN18 causes cell cycle arrest in the S phase. First,
treatment with cycloheximide (CHX), an inhibitor of protein
synthesis, revealed that overexpression of PTPN18 promoted cyclin
E1 degradation and knockdown of PTPN18 inhibited cyclin E1
degradation (Fig. 6A and B).
Furthermore, MG132 proteasome inhibitor inhibited cyclin E1
degradation induced by PTPN18 overexpression and enhanced the
inhibitory effect on cyclin E1 degradation in the knockdown group
(Fig. 6C and D). When CHX and
MG132 were combined, the effect of PTPN18 on the cyclin E1 protein
levels was not significantly different from that of the control
(Fig. 6E and F).
Next, the effect of PTPN18 on cyclin E1
ubiquitination was examined. The results demonstrated that PTPN18
overexpression markedly increased the ubiquitination level of
cyclin E1 and that PTPN18 knockdown reduced the cyclin E1
ubiquitination level after MG132 treatment (Fig. 6G and H). Since PTPN18 is a PTP, it
was found that the ubiquitination of cyclin E1 by PTPN18 requires
enzymatic activity (Fig. 6I).
Using western blotting to detect the pan-tyrosine phosphorylation
level of cyclin E1, it was found that PTPN18 was able to
dephosphorylate the tyrosine of cyclin E1, indicating that cyclin
E1 is a substrate of PTPN18 (Fig. 6J
and K). Next, the interaction between PTPN18 and cyclin E1 was
verified by forward and reverse Co-IP experiments as well as by
examination of the endogenous interaction in MCF7 cells (Fig. 6L-N). In summary, the results
revealed that PTPN18 can bind to cyclin E1 and promotes its
degradation through the ubiquitin-proteasome pathway.
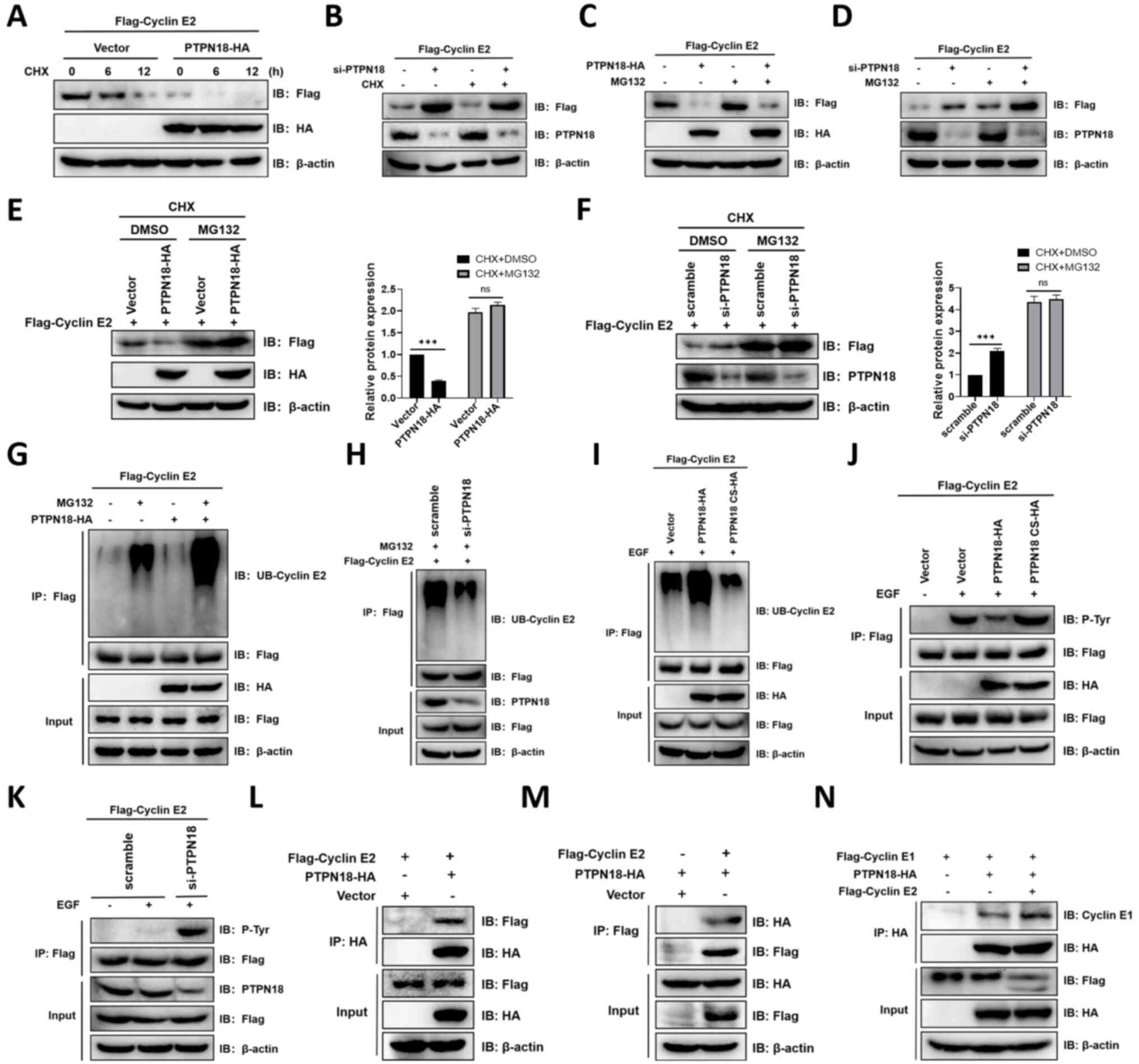
PTPN18 downregulates the expression of
cyclin E2 protein via the ubiquitin-proteasome pathway
Next, the present study explored the effect of
PTPN18 on cyclin E2, another isoform of cyclin E. It was found that
overexpression of PTPN18 decreased the cyclin E2 protein levels
after treatment with CHX (Fig.
7A). However, the cyclin E2 protein levels were restored when
MG132, a proteasome inhibitor, was added (Fig. 6C). When PTPN18 expression was
knocked down with siRNA, the opposite effects on the cyclin E2
protein levels were observed (Fig. 7B
and D). The combined use of CHX and MG132 nearly abolished the
effect of PTPN18 on the cyclin E2 protein levels compared with the
control (Fig. 7E and F).
Examination of the cyclin E2 ubiquitination levels revealed that
MG132 enhanced the PTPN18-mediated protein ubiquitination of cyclin
E2, whereas knockdown of PTPN18 reduced the cyclin E2
ubiquitination levels (Fig. 7G and
H). Similar to the cyclin E1 results, inactivation treatment
with PTPN18 C229S reduced the ubiquitination and increased the
phosphorylation of cyclin E2, indicating that cyclin E2 is also a
substrate for PTPN18 (Fig. 7I-K).
Additionally, forward and reverse Co-IP experiments verified the
interaction of PTPN18 with cyclin E2 (Fig. 7L and M). These results indicated
that PTPN18 can also promote the degradation of cyclin E2 through
the ubiquitin-proteasome pathway. Given that both cyclins E1 and E2
can bind PTPN18, it was further explored whether there is a
competitive relationship between their interactions. The Co-IP
results showed that the addition of cyclin E2 did not reduce the
binding of PTPN18 to cyclin E1, suggesting cyclin E2 did not
interfere with the binding of cyclin E1 to PTPN18 (Fig. 7N).
PTPN18 can regulate the protein
expression levels of CDK inhibitor 1A (CDKN1A, also known as
p21Cip1) and CDK inhibitor 1B (CDKN1B, also known as p27Kip1)
through the phosphatidylinositol 3-kinase (PI3K)/protein kinase B
(AKT) signaling pathway leading to cell cycle arrest
To further explore the signaling pathways through
which PTPN18 regulates the cell cycle, key genes associated with
the cell cycle according to the KEGG database were selected to
examine the effects of PTPN18 on their transcript levels. The
results demonstrated that PTPN18 overexpression produced
significant changes in the phosphoinositide-dependent protein
kinase 1 (PDPK1), phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit γ (PI3CG), forkhead box O3 (FOXO3),
mitogen-activated protein kinase 8 (MAPK8), CDKN1A, growth arrest
and DNA damage-inducible protein α (GADD45A), adenomatous polyposis
coli protein (APC) and E1A-binding protein p300 (EP300) transcript
levels, indicating that PTPN18 may have a wide range of
multi-target and multi-pathway roles in gene transcription
regulation (Fig. S3A-H).
Analysis using STRING, a functional protein association network
database, showed that CDKN1A and CDKN1B closely interacted with
cyclins E1 (CCNE1) and E2 (CCNE2) (Fig. 8A and B). The Kendall statistical
analysis results from the GEPIA database also showed that CDKN1A
and CDKN1B had significant negative correlations with cyclin E1 and
CDKN1A had a negative correlation with cyclin E2 (Fig. 8C and D). Further protein level
experiments showed that PTPN18 overexpression significantly
decreased the tyrosine phosphorylation levels of PI3K p85 and AKT
and the protein expression levels of PI3K P110β compared with the
controls, whereas the protein levels of FOXO1, CDKN1A and CDKN1B
were significantly increased (Fig.
8E-H). Decreased levels of AKT tyrosine phosphorylation
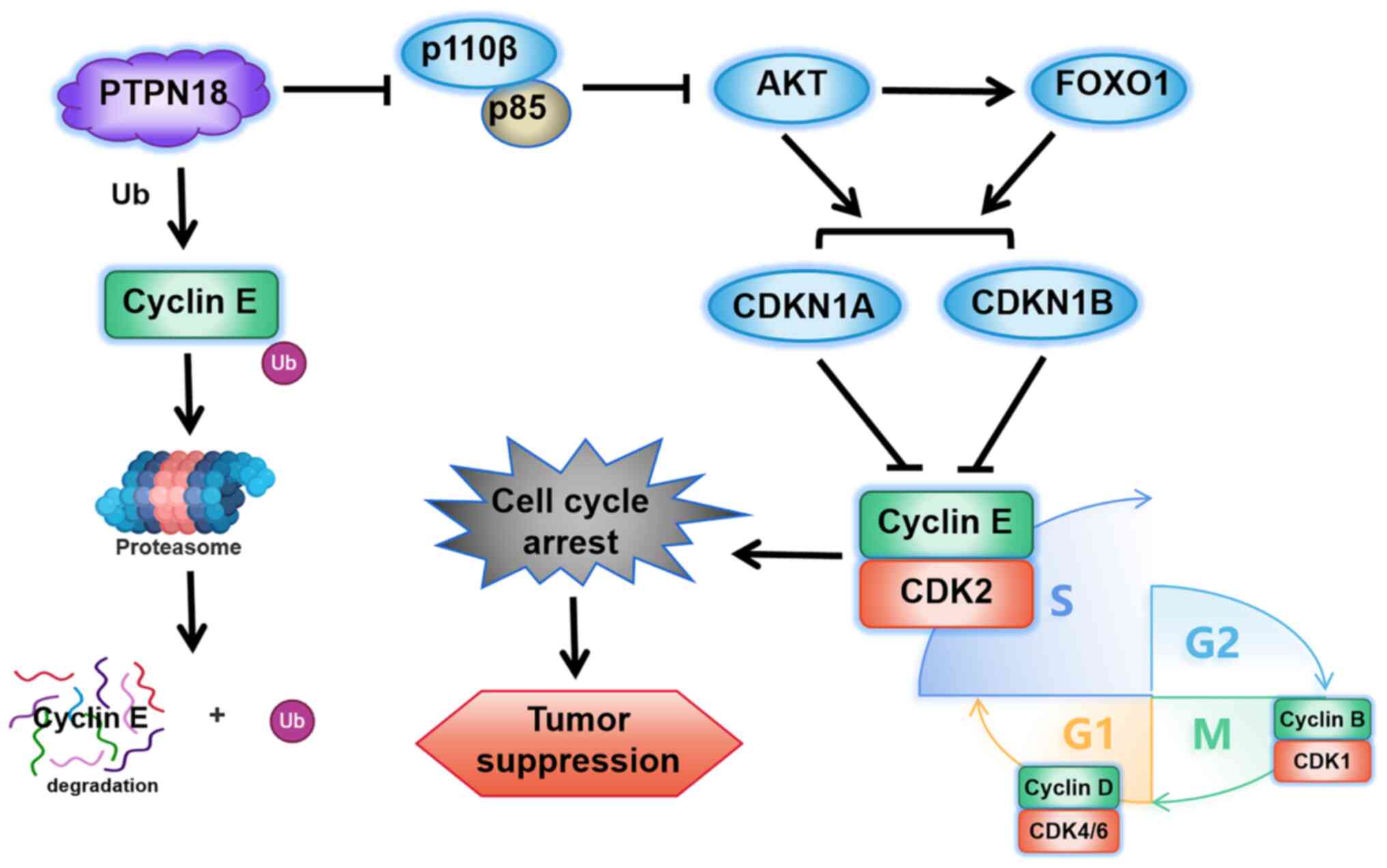
resulted in reduced inhibition of FOXO and increased FOXO
expression, followed by increased CDKN1A and CDKN1B protein levels,
resulting in cyclin inhibition and cell cycle arrest (Figs. 8E-H and 9). The results of the endogenous PTPN18
knockdown experiments were generally consistent with those of the
overexpression experiments (Fig.
8E-H). Taken together, the protein level results confirmed that
the antitumor effect of PTPN18 may be closely related to the
PI3K/AKT signaling pathway.
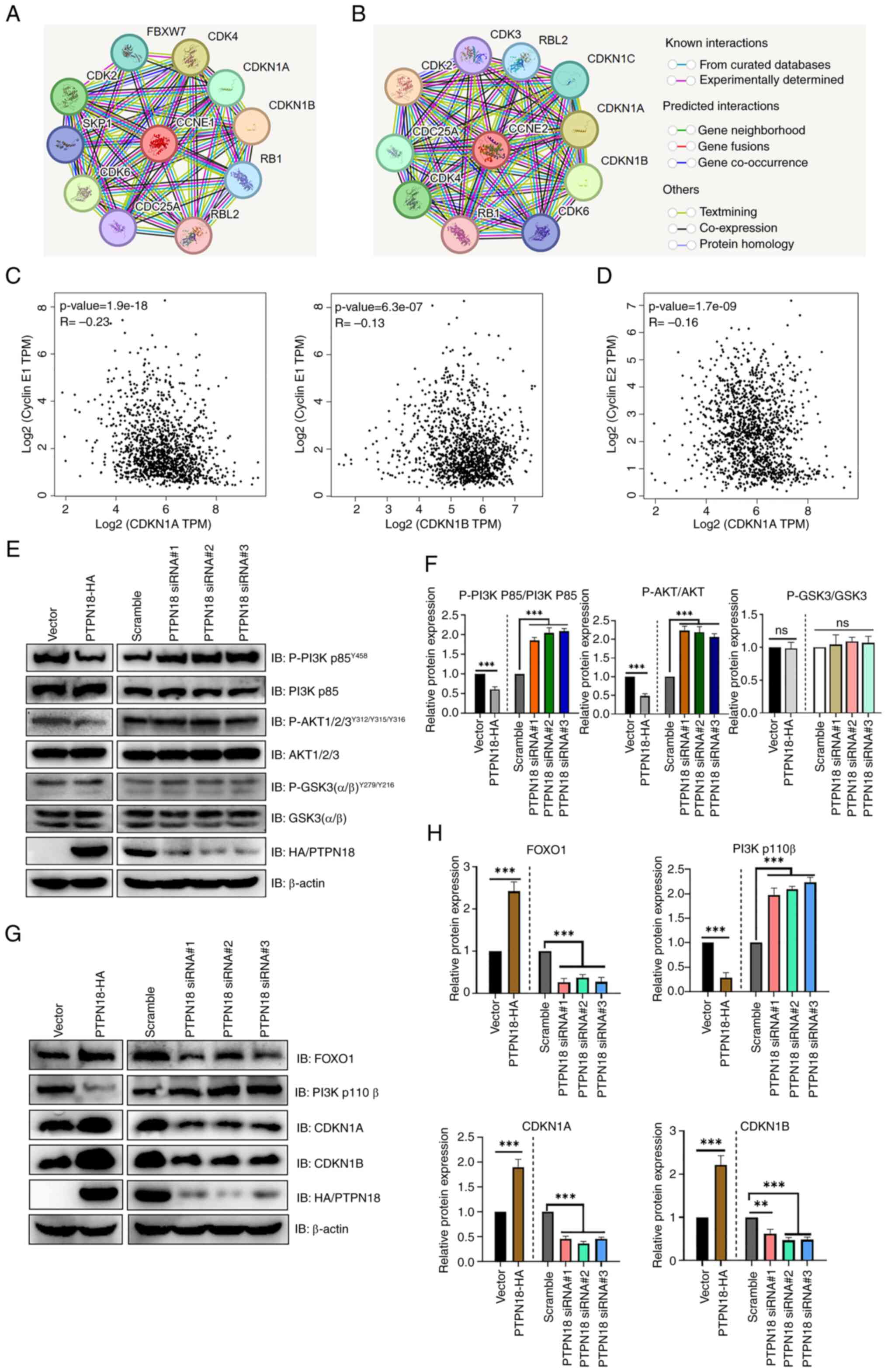 | Figure 8PTPN18 can regulate the expression
levels of CDKN1A and CDKN1B proteins through the PI3K/AKT signaling
pathway leading to cell cycle arrest. (A and B) STRING database
analysis showed proteins that interact with (A) cyclin E1 (CCNE1)
and (B) E2 (CCNE2). (C) GEPIA database analysis of the CDKN1A and
CDKN1B association with cyclin E1. (D) Correlation analysis between
CDKN1A and cyclin E2 using the GEPIA database. (E) Western blot
analysis of the PI3K/AKT signaling pathway protein expression
levels in MCF-7 cells. Levels of P-PI3K p85, P-AKT and P-GSK3 were
monitored after a 30-min stimulation with EGF (100 ng/ml). (F)
Relative quantitative analysis of the PI3K/AKT signaling pathway
protein expression levels. (G) Western blotting demonstrated the
expression levels of FOXO1, PI3K p110β, CDKN1A and CDKN1B. (H) The
bar graph was obtained by normalizing the levels to β-actin. Data
are shown as the mean ± SD from three technical replicates
following 48 h of overexpression or 72 h of knockdown.
**P<0.01 and ***P<0.001. PTPN18,
protein tyrosine phosphatase non-receptor 18; GEPIA, Gene
Expression Profiling Interactive Analysis; PI3K,
phosphatidylinositol 3-kinase; AKT, protein kinase B; CDKN1A
(p21Cip1), cyclin-dependent kinase inhibitor 1A; CDKN1B (p27Kip1),
cyclin-dependent kinase inhibitor 1B; P-, phosphorylation; FOXO1,
forkhead box O1; ns, not significant; P-, phosphorylated; siRNA,
small interfering RNA. |
Discussion
PTPN18 is expressed in a variety of normal tissues
and is ubiquitous in the nucleus and cytoplasm of cells, with low
tissue specificity (29,33,34). The core function of PTPN18 is to
act as a negative regulator of receptor tyrosine kinase signaling
and regulate a variety of cellular processes including cell
proliferation, differentiation, the mitotic cycle and oncogenic
transformation by dephosphorylating key receptors such as HER2
(7,10-17). It has also been shown that PTPN18
is a protective factor against obesity, hyperlipidemia and type 2
diabetes (35). More notably,
PTPN18 is closely related to the development of BC and its high
expression improves the survival and prognosis of patients with
this disease (15-17,29,30); therefore, it is necessary to
dissect the specific mechanism of action of PTPN18 in BC. In the
present study, through tumor phenotypic functional assays, it was
found that overexpression of PTPN18 inhibited metastasis and
proliferation, promoted apoptosis and led to cell cycle arrest in
BC cells. Although MCF-7 cells lack caspase-3 activity, PTPN18 may
still achieve pro-apoptotic effects by negatively regulating the
PI3K/AKT pathway, regulating Bcl-2 family protein balance,
increasing ROS levels, or activating lysosomal enzymes, triggering
compensatory activation of other effector caspases or
mitochondria-mediated caspase-independent apoptosis (36-38). Annexin V/PI double staining
results directly verified the existence of this apoptotic
phenotype.
Since the cell cycle is the key regulatory hub of
cell proliferation, apoptosis and metastasis (39), as well as owing to the notable
role of PTPN18 in the BC cell cycle, subsequent experiments in the
present study focused on exploring the molecular mechanisms by
which PTPN18 affects cell cycle changes to dissect its tumor
suppressor mechanism. It was found that PTPN18 not only interacted
with cyclin E1 and downregulated its protein expression and
tyrosine phosphorylation levels, but it also acted on cyclin E2
with the same effect, indicating that PTPN18 may be a specific and
broad-spectrum regulator of the cyclin E family. Moreover, PTPN18
binding to cyclin E1 was not perturbed by cyclin E2. This property
may allow PTPN18 to comprehensively inhibit cyclin E
family-mediated abnormal proliferation, avoid isoform compensation
and improve the scope and stability of its antitumor effects.
Cyclin E can bind to the CDK1 and CDK2 protein kinases and
participate in the regulation of the G1 phase, G1/S phase
transition and S phase progression (18-23,40,41). As an important regulatory protein
in the S phase, the decrease of cyclin E expression and activity
may directly lead to the inability of retinoblastoma protein (RB)
to maintain a highly phosphorylated state (39-41), resulting in S phase-related
proteins not being successfully synthesized, the accumulation of
cells in the S phase and the failure of cells to transition to the
G2/M phase. The effect of PTPN18 on RB phosphorylation status and
the key downstream targets of cyclin E/CDK2 activity decrease will
be the next research focus to dissect the mechanism of S phase
arrest caused by PTPN18 affecting DNA replication.
In the present study, it was found that PTPN18
overexpression enhanced the expression of cyclin B1, cyclin D1 and
CDK4. Elevated cyclin B1 expression promotes the formation and
activation of maturation-promoting factor, thereby accelerating
G2/M phase transition and cell division, resulting in a decrease in
the number of G2/M phase cells (42-44). Cyclin B can competitively bind to
CDK2 and CDK1 with cyclin E, affecting the stability of the cyclin
E/CDK complex and arresting the cell cycle in the S phase (19,42,43). The cyclin D-CDK4/6 complex is the
main regulatory complex of the G1 phase of the cell cycle and
phosphorylates RB (45). The
increase of cyclin D1 and CDK4 may compensate for the effect of
cyclin E in G1 phase and alleviate the stress trend caused by the
accelerated cycle progression in G2/M phase, allowing G1 phase to
transform normally and maintain dynamic balance.
In the present study, PTPN18 significantly affected
the transcript levels of PDPK1, PI3CG, FOXO3, MAPK8, CDKN1A,
GADD45A, APC and EP300. FOXO and CDKN1A are downstream targets of
the PI3K/AKT pathway (46) and
PI3K/AKT can regulate GADD45A through the FOXO pathway (47). AKT may play a role in the
regulation of a variety of cellular activities through the
transcriptional coactivator EP300 (48) and MAPK8 can regulate the AKT
pathway (49). Additionally, dual
PI3K/mTOR inhibition is a potential therapeutic strategy for APC
and PIK3CA mutant colorectal cancer (50). Given that these significantly
altered genes are all associated with the PI3K/AKT pathway, the
present study examined the regulation of the PI3K/AKT pathway by
PTPN18 at the protein level. The results of the present study
indicated that PTPN18 significantly decreased the tyrosine
phosphorylation levels of PI3K p85 and AKT and the protein levels
of P110β as well as markedly increased the protein levels of FOXO1,
CDKN1A and CDKN1B. Decreased tyrosine phosphorylation levels of
PI3K p85 and AKT leads to inhibition of their function. P110β
stimulates cell proliferation and upregulation in the wild-type
state is oncogenic (51). A
reduction in P110β expression directly leads to the weakening of
its cancer-promoting effect and a relative increase in
intracellular PI3K p85 content; excess free PI3K p85 inhibits the
PI3K/AKT pathway (52). Combined
with the characteristics of PTPN18 protein tyrosine phosphatase and
experimental data, it is speculated that PTPN18 may regulate
PI3K/AKT pathway by directly dephosphorylating the tyrosine sites
of PI3K P85 and/or AKT, and then inhibit BC progression through
FOXO1-CDKN1A/CDKN1B-Cyclin E axis (Fig. 9); this hypothesis is consistent
with the consensus of field studies (15,46,51-55), but experiments are still needed to
verify the specific substrate and whether there are other potential
targets.
Unlike previous studies in which PTPN18 has been
shown to exert antitumor effects in BC through targets such as HER2
or ETS1 (15-17), a novel anticancer target of
PTPN18, cyclin E, was discovered in the present study. The current
literature suggests that cyclin E depletion improves the
chemosensitivity of BC cells to DNA-damaging agents (56). Patients with tumors harboring high
cyclin E expression have a reduced benefit from tamoxifen treatment
compared with patients with low tumor expression (57). However, the present study found
that PTPN18 could downregulate cyclin E expression and activity,
which is likely to be one of the key mechanisms by which PTPN18
synergistic drug treatment results in an improved prognosis and
longer survival of patients with BC. Needless to say, the role and
molecular mechanism of PTPN18 in the efficacy of conventional
anticancer drugs is an important guide for future research. In the
future, it may be possible to focus on exploring how to develop new
therapeutic strategies by enhancing the activity of PTPN18 or
mimicking its mechanism of action.
In summary, the antitumor effect of PTPN18 should be
the result of a combination of numerous mechanistic pathways and
substrates (15-17). Based on the current findings
alone, it is not sufficient to clarify whether PTPN18 can be used
as a reliable marker for BC diagnosis. Subsequently, a large number
of experiments are required to analyze the relationship between
PTPN18 expression level or activity and different
clinicopathological features of BC and explore its potential as a
diagnostic marker. Because the original intention of this paper was
to dissect the protective mechanism of high PTPN18 expression on OS
in patients with BC, and its function in normal breast cells may be
at rest or physiological homeostasis, it was not investigated.
However, the function and mechanism of PTPN18 in normal breast cell
lines should not be ignored, which is an important supplement to
comprehensively elucidate its role in breast physiological
homeostasis and the development of BC.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
NZ performed the majority of the experiments,
analyzed the data and wrote and edited the manuscript. TW, BB and
XZ constructed the plasmids. WX and WC performed the reverse
transcription-quantitative PCR and data analysis. YY and BW
directed the study, analyzed and approved all of the data, and
wrote and edited the manuscript. NZ and BW confirm the authenticity
of all the raw data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
Co-IP
|
co-immunoprecipitation
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
AKT
|
protein kinase B
|
|
HER2
|
human epidermal growth factor receptor
2
|
|
MCF7
|
Michigan Cancer Foundation-7
|
|
MDA-MB-231
|
Metastatic Derivative of Anaplastic
Breast Cancer-231
|
|
WB
|
western blot
|
|
TBST
|
Tris-buffered saline with Tween 20
|
|
RIPA
|
radioimmunoprecipitation assay
|
|
PBS
|
phosphate-buffered saline
|
|
FBS
|
fetal bovine serum
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
EMT
|
epithelial-mesenchymal transition
|
|
mTOR
|
mechanistic target of rapamycin
kinase
|
|
CDK
|
cyclin-dependent kinase
|
|
RB
|
retinoblastoma protein
|
|
GEPIA
|
Gene Expression Profiling Interactive
Analysis
|
|
CHX
|
cycloheximide
|
|
PDPK1
|
phosphoinositide-dependent protein
kinase 1
|
|
PI3CG
|
phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit γ
|
|
FOXO
|
forkhead box O
|
|
MAPK8
|
mitogen-activated protein kinase
8
|
|
GADD45A
|
growth arrest and DNA
damage-inducible protein α
|
|
APC
|
adenomatous polyposis coli
protein
|
|
EP300
|
E1A-binding protein p300
|
|
CDKN1A (p21Cip1)
|
CDK inhibitor 1A
|
|
CDKN1B (p27Kip1)
|
CDK inhibitor 1B
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 32100992) and Key Laboratory of
Bioresource Research and Development of Liaoning (grant no.
2022JH13/10200026).
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Xiong X, Zheng LW, Ding Y, Chen YF, Cai
YW, Wang LP, Huang L, Liu CC, Shao ZM and Yu KD: Breast cancer:
Pathogenesis and treatments. Signal Transduct Target Ther.
10:492025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Radenkovic S, Konjevic G, Jurisic V,
Karadzic K, Nikitovic M and Gopcevic K: Values of MMP-2 and MMP-9
in tumor tissue of basal-like breast cancer patients. Cell Biochem
Biophys. 68:143–152. 2014. View Article : Google Scholar
|
|
4
|
Radenkovic S, Milosevic Z, Konjevic G,
Karadzic K, Rovcanin B, Buta M, Gopcevic K and Jurisic V: Lactate
dehydrogenase, catalase, and superoxide dismutase in tumor tissue
of breast cancer patients in respect to mammographic findings. Cell
Biochem Biophys. 66:287–295. 2013. View Article : Google Scholar
|
|
5
|
Konjević G, Jurisić V and Spuzić I:
Association of NK cell dysfunction with changes in LDH
characteristics of peripheral blood lymphocytes (PBL) in breast
cancer patients. Breast Cancer Res Treat. 66:255–263. 2001.
View Article : Google Scholar
|
|
6
|
Radenkovic S, Konjevic G, Gavrilovic D,
Stojanovic-Rundic S, Plesinac-Karapandzic V, Stevanovic P and
Jurisic V: pSTAT3 expression associated with survival and
mammographic density of breast cancer patients. Pathol Res Pract.
215:366–372. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Veillette A, Rhee I, Souza CM and Davidson
D: PEST family phosphatases in immunity, autoimmunity, and
autoinflammatory disorders. Immunol Rev. 228:312–324. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bai B, Wang T, Zhang X, Ba X, Zhang N,
Zhao Y, Wang X, Yu Y and Wang B: PTPN22 activates the PI3K pathway
via 14-3-3τ in T cells. FEBS J. 290:4562–4576. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang X, Yu Y, Bai B, Wang T, Zhao J,
Zhang N, Zhao Y, Wang X and Wang B: PTPN22 interacts with EB1 to
regulate T-cell receptor signaling. FASEB J. 34:8959–8974. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang B, Lemay S, Tsai S and Veillette A:
SH2 domain-mediated interaction of inhibitory protein tyrosine
kinase Csk with protein tyrosine phosphatase-HSCF. Mol Cell Biol.
21:1077–1088. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cong F, Spencer S, Côté JF, Wu Y, Tremblay
ML, Lasky LA and Goff SP: Cytoskeletal protein PSTPIP1 directs the
PEST-type protein tyrosine phosphatase to the c-Abl kinase to
mediate Abl dephosphorylation. Mol Cell. 6:1413–1423. 2000.
View Article : Google Scholar
|
|
12
|
Dowbenko D, Spencer S, Quan C and Lasky
LA: Identification of a novel polyproline recognition site in the
cytoskeletal associated protein, proline serine threonine
phosphatase interacting protein. J Biol Chem. 273:989–996. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu Y, Dowbenko D and Lasky LA: PSTPIP 2, a
second tyrosine phosphorylated, cytoskeletal-associated protein
that binds a PEST-type protein-tyrosine phosphatase. J Biol Chem.
273:30487–30496. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shiota M, Tanihiro T, Nakagawa Y, Aoki N,
Ishida N, Miyazaki K, Ullrich A and Miyazaki H: Protein tyrosine
phosphatase PTP20 induces actin cytoskeleton reorganization by
dephosphorylating p190 RhoGAP in rat ovarian granulosa cells
stimulated with follicle-stimulating hormone. Mol Endocrinol.
17:534–549. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang HM, Xu YF, Ning SL, Yang DX, Li Y, Du
YJ, Yang F, Zhang Y, Liang N, Yao W, et al: The catalytic region
and PEST domain of PTPN18 distinctly regulate the HER2
phosphorylation and ubiquitination barcodes. Cell Res.
24:1067–1090. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gensler M, Buschbeck M and Ullrich A:
Negative regulation of HER2 signaling by the PEST-type
protein-tyrosine phosphatase BDP1. J Biol Chem. 279:12110–12116.
2004. View Article : Google Scholar
|
|
17
|
Wang T, Ba X, Zhang X, Zhang N, Wang G,
Bai B, Li T, Zhao J, Zhao Y, Yu Y and Wang B: Nuclear import of
PTPN18 inhibits breast cancer metastasis mediated by MVP and
importin β2. Cell Death Dis. 13:7202022. View Article : Google Scholar
|
|
18
|
Chu C, Geng Y, Zhou Y and Sicinski P:
Cyclin E in normal physiology and disease states. Trends Cell Biol.
31:732–746. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Siu KT, Rosner MR and Minella AC: An
integrated view of cyclin E function and regulation. Cell Cycle.
11:57–64. 2012. View Article : Google Scholar :
|
|
20
|
Caldon CE and Musgrove EA: Distinct and
redundant functions of cyclin E1 and cyclin E2 in development and
cancer. Cell Div. 5:22010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hwang HC and Clurman BE: Cyclin E in
normal and neoplastic cell cycles. Oncogene. 24:2776–2786. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fagundes R and Teixeira LK: Cyclin E/CDK2:
DNA replication, replication stress and genomic instability. Front
Cell Dev Biol. 9:7748452021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scaltriti M, Eichhorn PJ, Cortés J,
Prudkin L, Aura C, Jiménez J, Chandarlapaty S, Serra V, Prat A,
Ibrahim YH, et al: Cyclin E amplification/overexpression is a
mechanism of trastuzumab resistance in HER2+ breast cancer
patients. Proc Natl Acad Sci USA. 108:3761–3766. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Radenković N, Milutinović M, Nikodijević
D, Jovankić J and Jurišić V: Sample preparation of adherent cell
lines for flow cytometry: protocol optimization-our experience with
SW-480 colorectal cancer cell line. Indian J Clin Biochem.
40:74–79. 2025. View Article : Google Scholar
|
|
25
|
Vuletic A, Konjevic G, Milanovic D,
Ruzdijic S and Jurisic V: Antiproliferative effect of
13-cis-retinoic acid is associated with granulocyte differentiation
and decrease in cyclin B1 and Bcl-2 protein levels in G0/G1
arrested HL-60 cells. Pathol Oncol Res. 16:393–401. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scherbakov AM, Vorontsova SK, Khamidullina
AI, Mrdjanovic J, Andreeva OE, Bogdanov FB, Salnikova DI, Jurisic
V, Zavarzin IV and Shirinian VZ: Novel pentacyclic derivatives and
benzylidenes of the progesterone series cause anti-estrogenic and
antiproliferative effects and induce apoptosis in breast cancer
cells. Invest New Drugs. 41:142–152. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Chandrashekar DS, Karthikeyan SK, Korla
PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne
U, et al: UALCAN: An update to the integrated cancer data analysis
platform. Neoplasia. 25:18–27. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang Z, Kang B, Li C, Chen T and Zhang Z:
GEPIA2: An enhanced web server for large-scale expression profiling
and interactive analysis. Nucleic Acids Res. 47(W1): W556–W560.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Posta M and Győrffy B: Pathway-level
mutational signatures predict breast cancer outcomes and reveal
therapeutic targets. Br J Pharmacol. 182:5734–5747. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vasaikar S, Straub P, Wang J and Zhang B:
LinkedOmics: analyzing multi-omics data within and across 32 cancer
types. Nucleic Acids Res. 46(D1): D956–D963. 2018. View Article : Google Scholar :
|
|
32
|
Szklarczyk D, Kirsch R, Koutrouli M,
Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT,
Pyysalo S, et al: The STRING database in 2023: Protein-protein
association networks and functional enrichment analyses for any
sequenced genome of interest. Nucleic Acids Res. 51(D1): D638–D646.
2023. View Article : Google Scholar
|
|
33
|
Kim YW, Wang H, Sures I, Lammers R,
Martell KJ and Ullrich A: Characterization of the PEST family
protein tyrosine phosphatase BDP1. Oncogene. 13:2275–2279.
1996.PubMed/NCBI
|
|
34
|
Cheng J, Daimaru L, Fennie C and Lasky LA:
A novel protein tyrosine phosphatase expressed in
lin(lo)CD34(hi)Sca(hi) hematopoietic progenitor cells. Blood.
88:1156–1167. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li W, Zhong Q, Deng N, Zhou X, Wang H,
Ouyang J, Guan Z, Cheng B, Xiang L, Huang Y, et al: Sphingolipid
metabolism-related genes for the diagnosis of metabolic syndrome by
integrated bioinformatics analysis and Mendelian randomization
identification. Diabetol Metab Syndr. 17:2342025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cuvillier O, Nava VE, Murthy SK, Edsall
LC, Levade T, Milstien S and Spiegel S: Sphingosine generation,
cytochrome c release, and activation of caspase-7 in
doxorubicin-induced apoptosis of MCF7 breast adenocarcinoma cells.
Cell Death Differ. 8:162–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang S, Huang J, Liu P, Li J and Zhao S:
Apoptosis-inducing factor (AIF) nuclear translocation mediated
caspase-independent mechanism involves in X-ray-induced MCF-7 cell
death. Int J Radiat Biol. 93:270–278. 2017. View Article : Google Scholar
|
|
38
|
Pozo-Guisado E, Merino JM, Mulero-Navarro
S, Lorenzo-Benayas MJ, Centeno F, Alvarez-Barrientos A and
Fernandez-Salguero PM: Resveratrol-induced apoptosis in MCF-7 human
breast cancer cells involves a caspase-independent mechanism with
downregulation of Bcl-2 and NF-kappaB. Int J Cancer. 115:74–84.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu J, Peng Y and Wei W: Cell cycle on the
crossroad of tumorigenesis and cancer therapy. Trends Cell Biol.
32:30–44. 2022. View Article : Google Scholar
|
|
40
|
Ekholm-Reed S, Mendez J, Tedesco D,
Zetterberg A, Stillman B and Reed SI: Deregulation of cyclin E in
human cells interferes with prereplication complex assembly. J Cell
Biol. 165:789–800. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Caldon CE, Sergio CM, Sutherland RL and
Musgrove EA: Differences in degradation lead to asynchronous
expression of cyclin E1 and cyclin E2 in cancer cells. Cell Cycle.
12:596–605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Crncec A, Lau HW, Ng LY, Ma HT, Mak JPY,
Choi HF, Yeung TK and Poon RYC: Plasticity of mitotic cyclins in
promoting the G2-M transition. J Cell Biol. 224:e2024092192025.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li J, Qian WP and Sun QY: Cyclins
regulating oocyte meiotic cell cycle progression†. Biol Reprod.
101:878–881. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao SC, Dong MZ, Zhao BW, Liu SL, Guo JN,
Sun SM, Li YY, Xu YH and Wang ZB: Fangchinoline inhibits mouse
oocyte meiosis by disturbing MPF activity. Toxicol In Vitro.
99:1058762024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
VanArsdale T, Boshoff C, ArndtK T and
Abraham RT: Molecular pathways: Targeting the cyclin D-CDK4/6 axis
for cancer treatment. Clin Cancer Res. 21:2905–2910. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kloet DEA, Polderman PE, Eijkelenboom A,
Smits LM, van Triest MH, van den Berg MCW, Groot Koerkamp MJ, van
Leenen D, Lijnzaad P, Holstege FC and Burgering BMT: FOXO target
gene CTDSP2 regulates cell cycle progression through Ras and
p21(Cip1/Waf1). Biochem J. 469:289–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Amente S, Zhang J, Lavadera ML, Lania L,
Avvedimento EV and Majello B: Myc and PI3K/AKT signaling
cooperatively repress FOXO3a-dependent PUMA and GADD45a gene
expression. Nucleic Acids Res. 39:9498–9507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen J, Halappanavar SS, St-Germain JR,
Tsang BK and Li Q: Role of Akt/protein kinase B in the activity of
transcriptional coactivator p300. Cell Mol Life Sci. 61:1675–1683.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sunayama J, Tsuruta F, Masuyama N and
Gotoh Y: JNK antagonizes Akt-mediated survival signals by
phosphorylating 14-3-3. J Cell Biol. 170:295–304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Foley TM, Payne SN, Pasch CA, Yueh AE, Van
De Hey DR, Korkos DP, Clipson L, Maher ME, Matkowskyj KA, Newton MA
and Deming DA: Dual PI3K/mTOR inhibition in colorectal cancers with
APC and PIK3CA mutations. Mol Cancer Res. 15:317–327. February
9–2017.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dbouk HA and Backer JM: A beta version of
life: p110β takes center stage. Oncotarget. 1:729–733. 2010.
View Article : Google Scholar
|
|
52
|
Geering B, Cutillas PR, Nock G, Gharbi SI
and Vanhaesebroeck B: Class IA phosphoinositide 3-kinases are
obligate p85-p110 heterodimers. Proc Natl Acad Sci USA.
104:7809–7814. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Luo J and Cantley LC: The negative
regulation of phosphoinositide 3-kinase signaling by p85 and its
implication in cancer. Cell Cycle. 4:1309–1312. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zheng Y, Peng M, Wang Z, Asara JM and
Tyner AL: Protein tyrosine kinase 6 directly phosphorylates AKT and
promotes AKT activation in response to epidermal growth factor. Mol
Cell Biol. 30:4280–4292. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen R, Kim O, Yang J, Sato K, Eisenmann
KM, McCarthy J, Chen H and Qiu Y: Regulation of Akt/PKB activation
by tyrosine phosphorylation. J Biol Chem. 276:31858–31862. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen J and Wang G: Cyclin E expression and
chemotherapeutic sensitivity in breast cancer cells. J Huazhong
Univ Sci Technolog Med Sci. 26:565–566. 2006. View Article : Google Scholar
|
|
57
|
Waltersson MA, Askmalm MS, Nordenskjöld B,
Fornander T, Skoog L and Stål O: Altered expression of cyclin E and
the retinoblastoma protein influences the effect of adjuvant
therapy in breast cancer. Int J Oncol. 34:441–448. 2009.PubMed/NCBI
|