Multiple myeloma (MM) is an incurable hematologic
malignancy characterized by clonal expansion and accumulation of
plasma cells in the bone marrow (1,2).
It is associated with the production of abnormal antibodies,
referred to as monoclonal proteins or M proteins (3). Also known as plasma cell myeloma or
simply myeloma, MM accounts for ~1% of all types of cancer and 15%
of hematologic malignancies, with an incidence that increases with
age and is higher in men than in women (4-7).
Despite remarkable progress in its treatment, MM remains largely
incurable, posing significant challenges because of its complex
pathogenesis, frequent diagnostic delays caused by nonspecific
symptoms and the almost inevitable development of treatment
resistance (8,9).
The treatment paradigm for MM has undergone a
profound transformation, moving from conventional chemotherapy to a
multi-agent, mechanism-based approach. Foundational regimens
combining proteasome inhibitors (such as bortezomib and
carfilzomib), immunomodulatory drugs (IMiDs; such as lenalidomide
and pomalidomide) and corticosteroids formed the first major wave
of therapeutic progress (10-17). More recently, immunotherapies,
including monoclonal antibodies (such as daratumumab targeting
CD38) and T-cell redirecting agents such as bispecific antibodies
and chimeric antigen receptor (CAR) T-cell therapy have further
improved patient outcomes (18-21). Despite these advances, relapse and
drug resistance remains a nearly universal challenge, highlighting
the remarkable adaptability of MM cells and the urgent need for
strategies that target the molecular basis of resistance (22-24).
These challenges are further compounded by the
disease's complexity. Diagnosis is often delayed because of
nonspecific initial symptoms (such as fatigue, bone pain and
anemia) (4,25-27) and its pathogenesis involves a
multifaceted interplay of genetic, epigenetic and bone marrow
microenvironment factors that promote myeloma cell survival and
drug resistance (1,4,28-30). The incomplete understanding of
these resistance mechanisms limits the development of curative
therapies for relapsed/refractory MM (18,20,31-33). Therefore, gaining deeper insights
into MM biology from new perspectives is critical for improving
patient outcomes.
Epitranscriptomics, the study of
post-transcriptional RNA modifications, has emerged as a crucial
layer of gene regulation (34-36). More than 170 chemical
modifications have been identified and these alterations profoundly
influence RNA metabolism, including stability, splicing and
translation (37,38). Among them, N6-methyladenosine
(m6A), the most abundant internal mRNA modification, has
garnered significant attention for its roles in normal biology and
cancer pathogenesis (39,40). Growing evidence implicates
dysregulated m6A modification in solid tumors and
hematological malignancies, including MM, where it influences
disease pathogenesis, therapeutic response and drug resistance
(41-43).
Eligible records included original research
articles, preclinical studies and clinical or translational reports
that examined i) m6A regulators or
m6A-dependent mechanisms in MM models or patient
samples, or ii) pharmacological or genetic modulation of
m6A pathways with potential therapeutic relevance.
Studies from other malignancies were included selectively when they
provided key mechanistic insight or information on inhibitor
development/chemical tractability and such evidence is explicitly
identified as non-MM where discussed. Studies were excluded if they
were i) published in non-English languages, ii) conference
abstracts without accompanying full-text primary data, iii)
retracted publications, or iv) not directly relevant to the core
focus on RNA modification biology in MM.
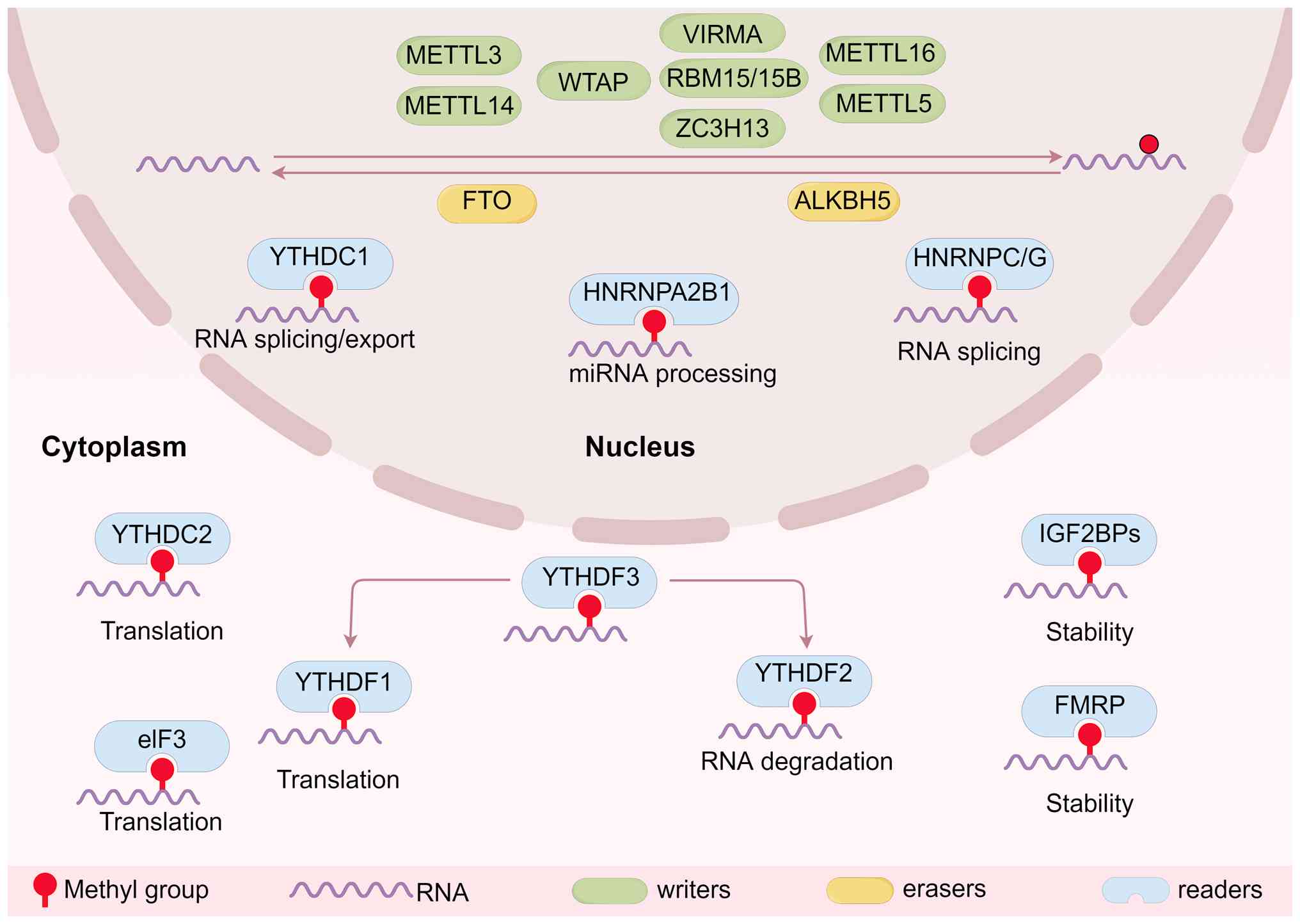
Traditionally, epigenetic regulation has focused on
DNA methylation and histone modifications as key determinants of
gene expression (44-46). The emergence of epitranscriptomics
has now revealed reversible chemical modifications on RNA as a
critical additional layer of gene regulation (47,48). Among known RNA modifications,
m6A is the most prevalent internal modification on
eukaryotic mRNA (49). The
presence of m6A on a transcript serves as a direct
binding platform for specific reader proteins, which markedly
influence the RNA's fate by regulating its splicing, stability,
export and translation (40,50,51). Another key mechanism of action
involves the ability of m6A to function as an
'm6A switch', in which the modification alters the RNA's
secondary structure to reveal binding sites for proteins that would
otherwise be inaccessible (52,53). This allows for rapid,
post-transcriptional fine-tuning of gene expression in response to
cellular signals, a process frequently dysregulated in cancer.
Additionally, heterogeneous nuclear
ribonucleoproteins (hnRNPs), such as heterogeneous nuclear
ribonucleoprotein C/G (hnRNPC/G) and heterogeneous nuclear
ribonucleoprotein A2/B1 (hnRNPA2B1), recognize m6A
modifications and influence miRNA processing and splicing events by
facilitating the recruitment of splicing factors and modulating
alternative splicing patterns (63,64). Eukaryotic initiation factor 3 can
bind to m6A sites, particularly near the 5' untranslated
region (UTR) of mRNAs, to directly promote translation initiation
in a cap-independent manner, enhancing the translation of specific
transcripts under certain conditions (65). Fragile X mental retardation
protein is another recognized m6A reader that plays a
role in neuronal mRNA localization and stability, thereby
influencing synaptic function and plasticity (66). This regulatory capacity of
m6A modification, which spans mRNA splicing,
localization, stability and translation efficiency, allows cells to
finely tune gene expression in response to developmental cues and
environmental stresses.
Other writers also contribute to MM pathogenesis in
pre-clinical contexts. For example, METTL5 has been shown to drive
MM progression by enhancing the translation of selenoproteins such
as selenophosphate synthetase 2 (SEPHS2), which is critical
for mitigating oxidative stress (121). The accessory writer protein
VIRMA (KIAA1429) is overexpressed in MM and experimental evidence
suggests it promotes tumorigenesis by enhancing m6A
modification and expression of the oncogene forkhead box M1
(FOXM1) via the reader YTHDF1 (122). Beyond regulating oncogene
expression, VIRMA has been linked to suppression of ferroptosis to
promote MM cell survival (123).
Mechanistically, VIRMA is stabilized by the lncRNA FEZF1-AS1
and, in turn, enhances m6A-dependent translation of OTU
deubiquitinase, ubiquitin aldehyde binding 1 (OTUB1). OTUB1
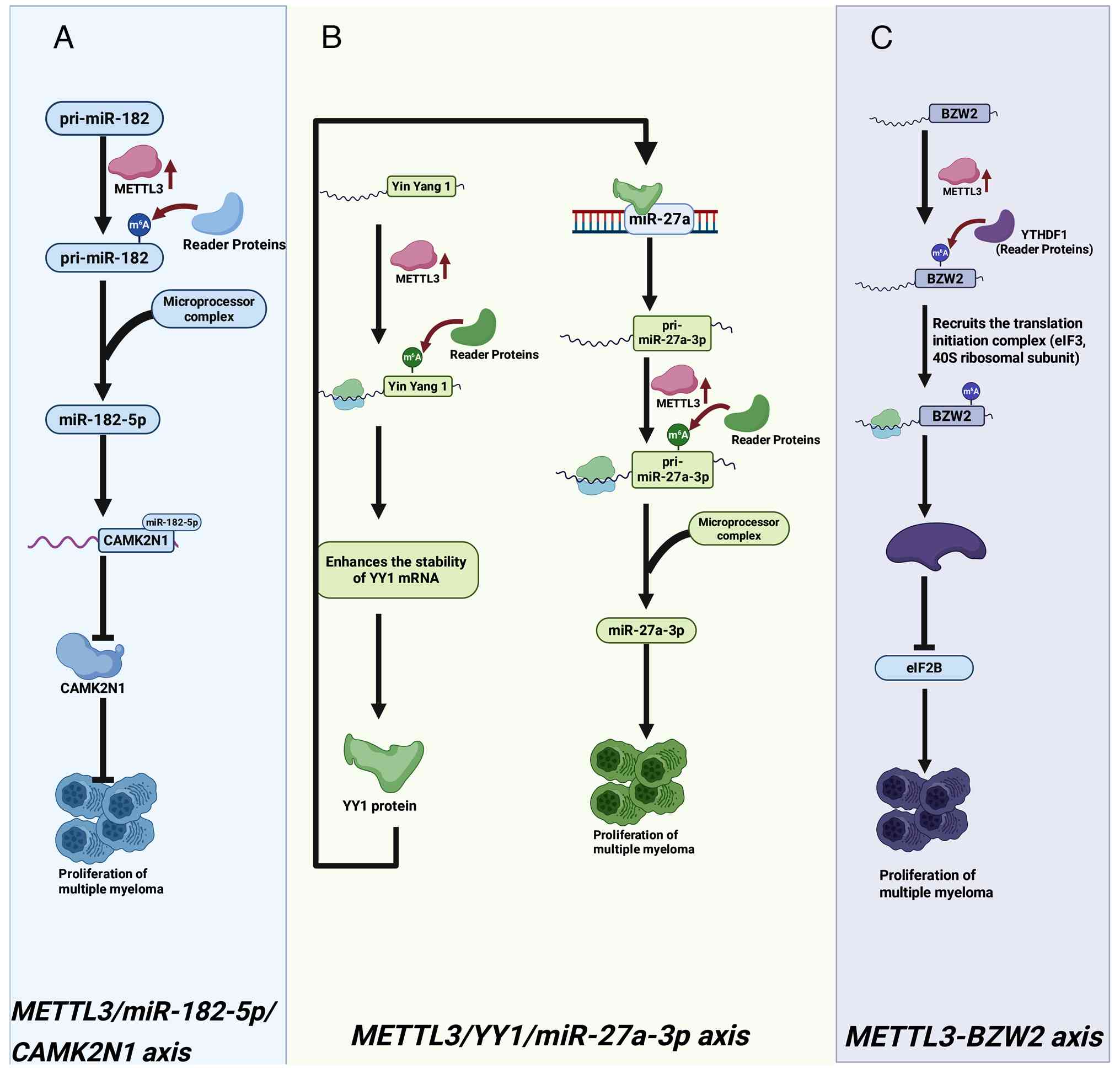
then deubiquitinates and stabilizes the ferroptosis defense protein
solute carrier family 7 member 11 (SLC7A11), protecting MM cells.
The core writer complex component WTAP also appears to function as
a critical oncogene. A recent study identified WTAP as
overexpressed in MM patients and associated with poor survival
(124). Mechanistically, this
study reported that WTAP installs m6A modifications on
microtubule-associated protein 6 domain containing 1 mRNA, thereby
regulating the Hippo signaling pathway to drive MM cell
proliferation. Notably, the writer complex can be regulated
post-translationally; research indicates protein arginine
methyltransferase 1 methylates and stabilizes WTAP, which in turn
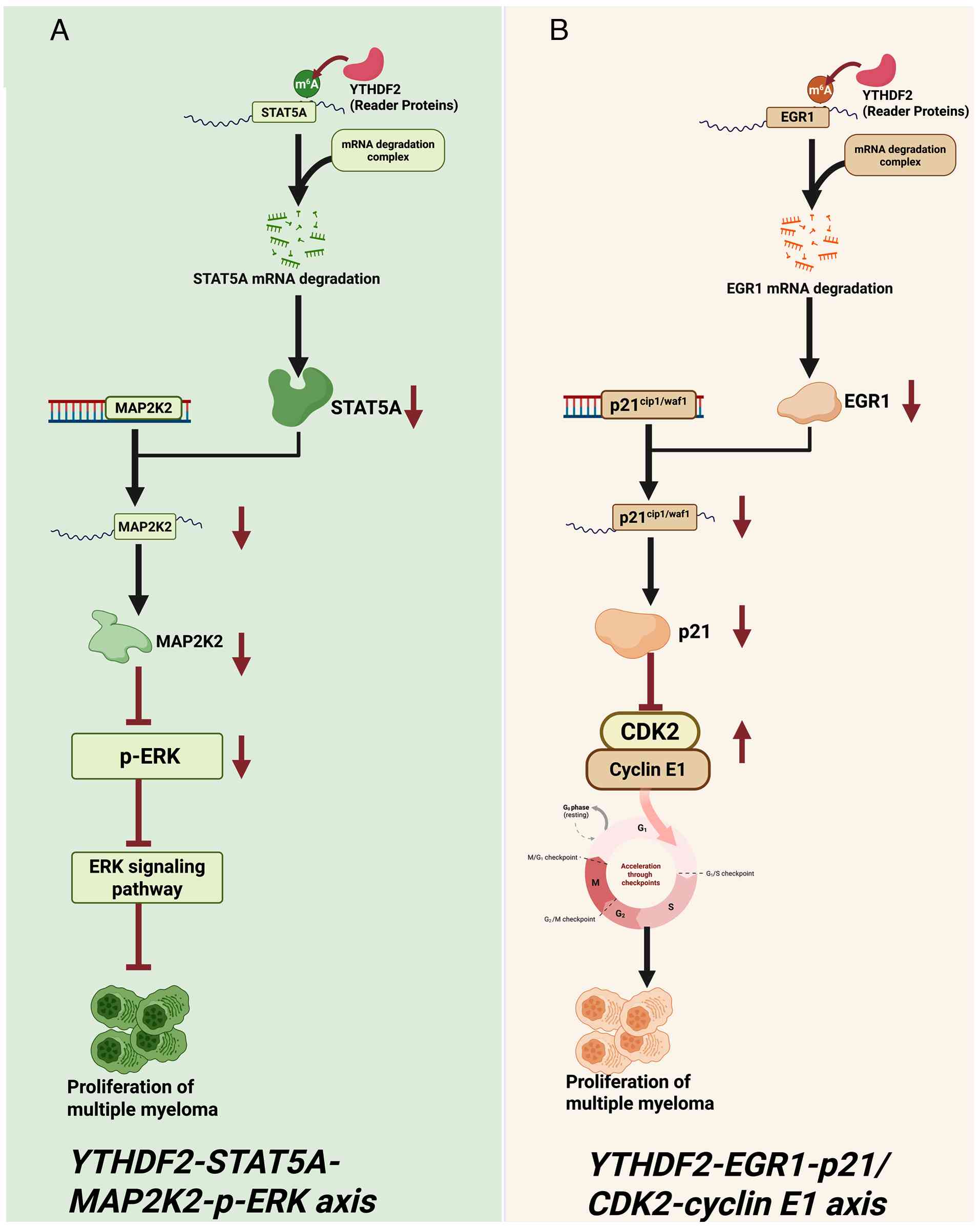
enhances m6A modification of NADH:ubiquinone
oxidoreductase core subunit S6 mRNA, boosting its expression and
activating oxidative phosphorylation to promote tumorigenesis
(125).
The oncogenic role of readers extends to
genetically defined MM subgroups. IGF2BP1 is markedly overexpressed
in patients with chromosome 1q gain, where it predicts an inferior
clinical outcome (128).
Mechanistically, IGF2BP1 directly binds to m6A sites
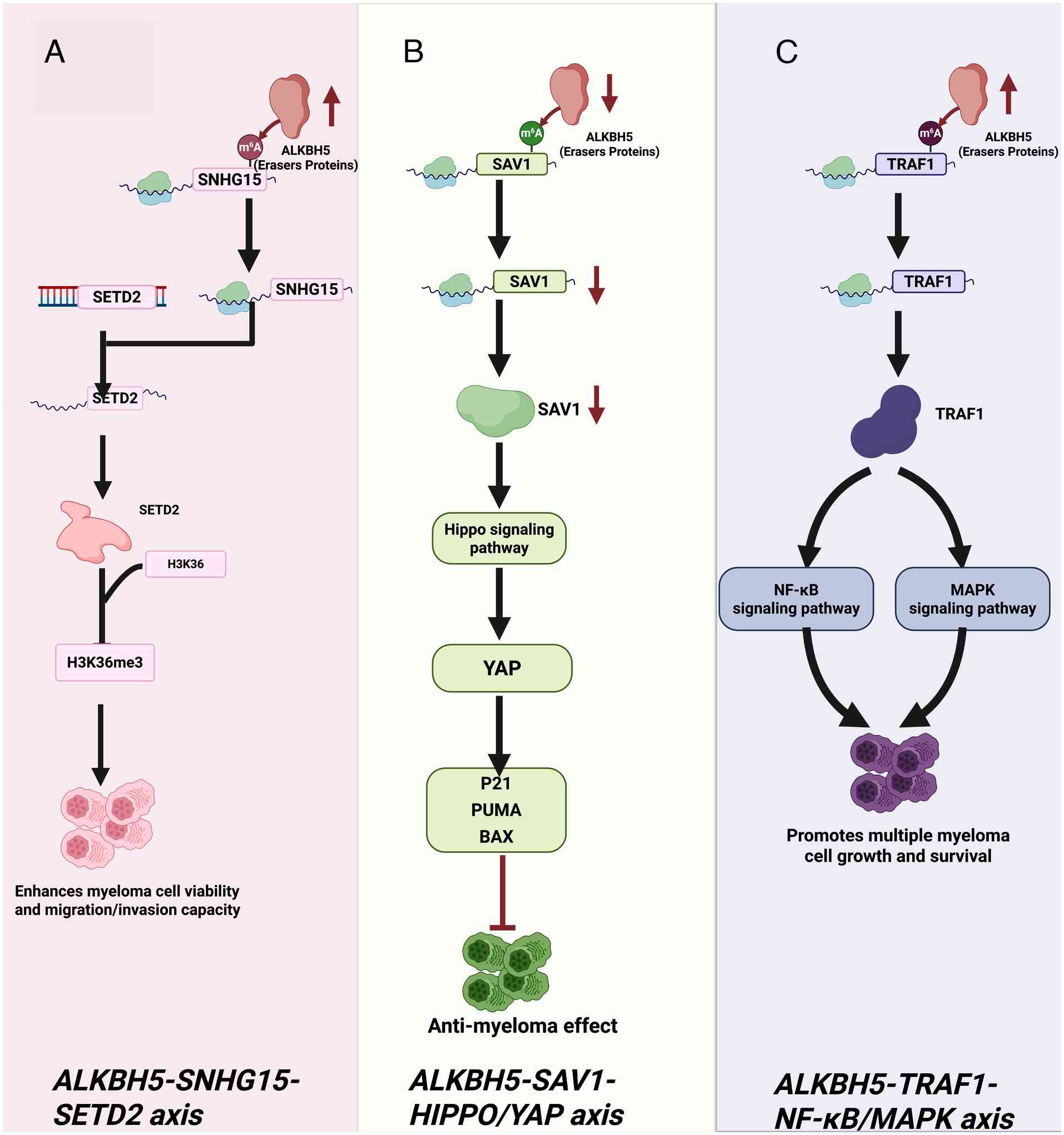
within the cell division cycle 5-Like (CDC5L) mRNA,
enhancing its stability and translation. The functional dependency
of this axis was confirmed by the fact that genetic or
pharmacological inhibition of IGF2BP1 suppressed the pro-tumor
effects, revealing the IGF2BP1-CDC5L axis as a vulnerability in
this high-risk population (128).
The other major eraser, FTO, exerts its oncogenic
role by establishing a hypomethylated transcriptome (138). FTO is upregulated in MM and
promotes tumor growth and metastasis. It specifically demethylates
heat shock factor 1 (HSF1) mRNA, shielding it from
YTHDF2-mediated decay and thus augmenting the heat shock response,
a critical survival pathway under proteotoxic stress (139). Importantly, FTO activity is
metabolically coupled. Isocitrate dehydrogenase 2 (IDH2), which
generates the essential FTO cofactor α-ketoglutarate, is highly
expressed in progressive MM. IDH2 depletion increased global
m6A levels and impaired growth, mechanistically through
suppressing FTO-mediated hypomethylation and stabilization of Wnt
family member 7B mRNA, which activates pro-tumorigenic Wnt
signaling (140,141). The translational promise of
targeting m6A erasers is underscored by the finding that
FTO inhibition synergizes with the frontline therapeutic drug
bortezomib to suppress myeloma growth in vivo, presenting a
compelling combinatorial strategy (139).
Taken together, the ALKBH5-SNHG15-SETD2,
ALKBH5-SAV1-YAP and ALKBH5-TRAF1 axes, along with FTO-mediated
regulation of HSF1 and Wnt signaling, represent important emerging
mechanistic insights. However, the current evidence for these
oncogenic roles of m6A erasers is predominantly derived
from in vitro and mouse xenograft models. Their overarching
significance, interdependence and clinical relevance as therapeutic
targets in primary MM require independent replication and further
validation in patient cohorts.
Collectively, these findings from recent
pre-clinical and bioinformatic studies illuminate an expanding
epitranscriptomic network in MM. While m6A remains the
most extensively characterized, the reported contributions of
ac4C and m5C underscore the broader
regulatory potential of RNA modifications. Targeting these pathways
represents a promising but early-stage frontier for developing
novel therapeutic strategies aimed at overcoming drug resistance
and improving patient outcomes.
The development of treatment resistance, a major
driver of relapse and mortality in MM, is thought to be closely
linked to dynamic adaptations in the epitranscriptome, particularly
m6A modification, which may coordinate both
cell-intrinsic survival pathways and extrinsic communication with
the bone marrow niche (147-150). Beyond directly regulating
oncogenic transcripts, m6A-mediated mechanisms are
implicated in drug resistance by reshaping the chromatin landscape
through crosstalk with other epigenetic modifiers and by modulating
the expression of drug efflux pumps and metabolic enzymes, thereby
reducing intracellular drug exposure and therapeutic efficacy
(83,151-153). A comprehensive dissection of
these epitranscriptomic pathways is therefore a current research
priority to develop strategies to overcome resistance and prevent
disease progression.
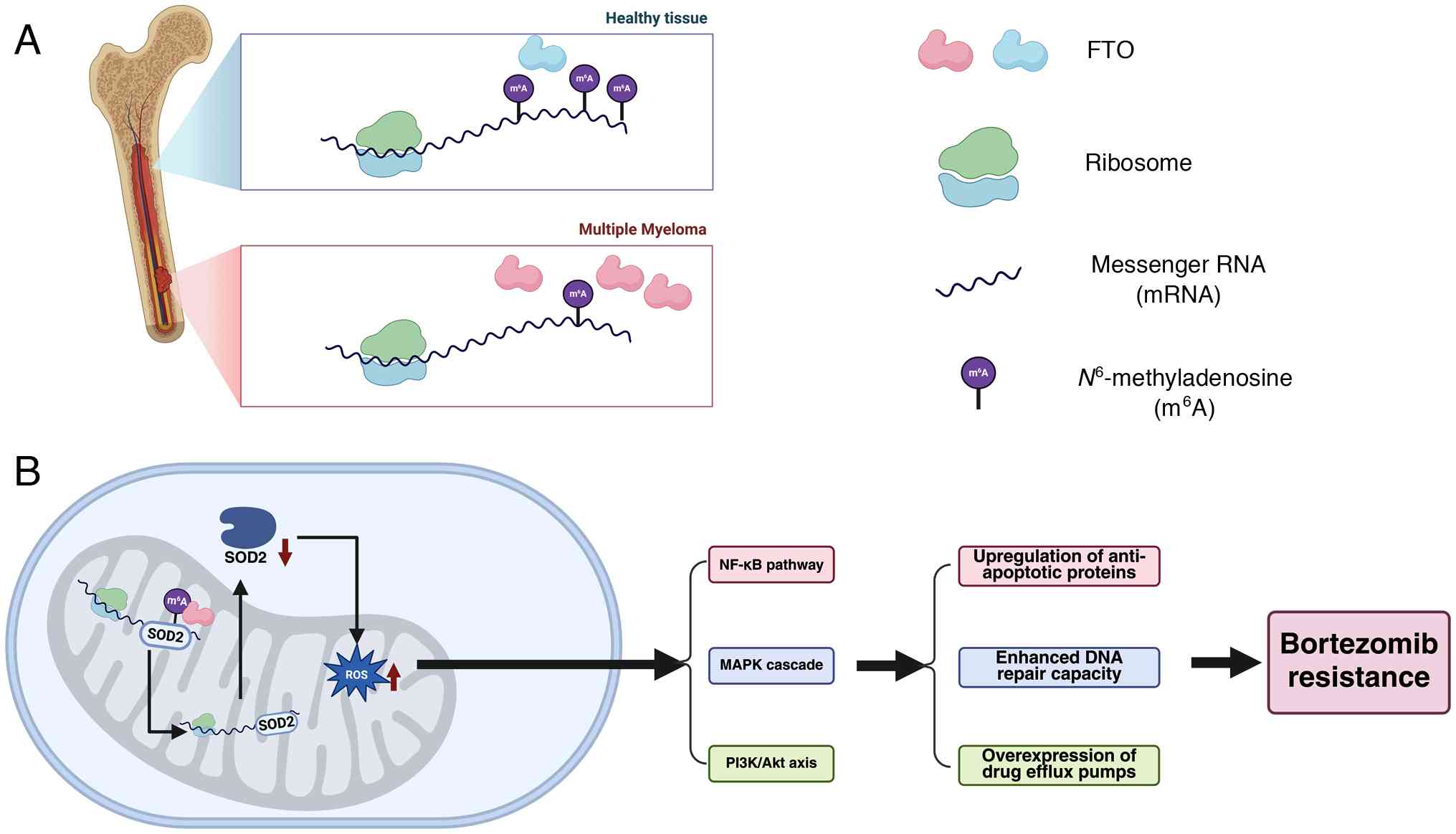
Notably, the regulation of oxidative stress in MM
reveals a complex picture. On one hand, METTL5 upregulates SEPHS2
expression, thereby enhances antioxidant defense (121). By contrast, FTO reduces SOD2
expression, diminishing antioxidant defense yet being reported to
contribute to bortezomib resistance (155). This paradox may be explained by
the dual role of reactive oxygen species (ROS), which at optimal
levels can drive pro-tumorigenic signaling (161,162). Thus, cancer cells may manipulate
different antioxidant pathways to maintain ROS within a
survival-favorable range. Future studies are warranted to validate
this effect and to determine how redox homeostasis can be
therapeutically targeted (163,164).
ALKBH5 has also been implicated in resistance to
targeted therapies. A recent study demonstrated that ALKBH5
overexpression attenuates the efficacy of the histone deacetylase
inhibitor romidepsin by reducing m6A methylation on
FOXM1 mRNA, thereby stabilizing it (165). Conversely, romidepsin treatment
or ALKBH5 knockout increases FOXM1 m6A
modification, accelerating its decay and synergistically enhancing
apoptosis (165). This positions
ALKBH5 as a key modulator of drug response in pre-clinical
models.
This concept extends to predicting response to
combination therapies. In a cohort of patients relapsing after
bortezomib-based regimens, a high m6A risk score (with
an Area Under the Curve of 0.9) was associated with non-response to
salvage therapy with daratumumab, carfilzomib, lenalidomide and
dexamethasone (DARA-KRD) (113).
Although mechanisms such as FTO-SOD2 have been demonstrated to
mediate resistance in preclinical models, these findings are
primarily based on single studies or limited cohorts. The
prevalence of these pathways in patient populations, particularly
among those with different types of resistance and their
interactions with classical resistance mechanisms remain
insufficiently elucidated and warrant validation in large-scale,
prospective clinical cohorts.
The bone marrow adipocyte niche has emerged as a
proposed source of exosome-mediated transfer of chemoresistance, a
process suggested to be governed by epitranscriptomic regulation.
Adipocyte-derived exosomes are reported to enrich MM cells with the
lncRNAs LOC606724 and SNHG1, which inhibit apoptosis
(171). Furthermore, the
methyltransferase METTL7A, whose activity is potentiated by EZH2 in
these models, promotes the m6A modification of these
lncRNAs, thereby enhancing their packaging into exosomes. Upon
delivery to MM cells, these m6A-modified lncRNAs drive
c-Myc expression (171).
This paradigm highlights a potential mechanism of niche-induced
drug resistance that warrants further investigation.
FTO is another high-priority target. MM-specific
evidence shows that FTO is upregulated in patients and promotes
bortezomib resistance through mechanisms including the disruption
of the oxidative stress modulator SOD2 (Fig. 5) (155). The chemical tractability of FTO
is supported by several inhibitors (such as FB23-2, CS1/CS2), which
have demonstrated anti-tumor effects in leukemia models (173,176-178). The potential for integration is
highly compelling. Synthetic small-molecule inhibitors like FB23-2
can selectively block FTO activity, leading to enhanced decay of
oncogenic transcripts such as MYC. Accordingly, FTO inhibition
warrants MM-specific validation as a resensitization strategy,
including testing in acquired bortezomib-resistant models and in
combination regimens with proteasome inhibitors.
The eraser ALKBH5 also presents a viable target.
Its MM-specific validation is underscored by its role in driving
tumorigenesis and mediating resistance to agents such as the
histone deacetylase inhibitor romidepsin by stabilizing FOXM1 mRNA
(165). However, available
ALKBH5 inhibitors (such as IOX1) are generally considered tool
compounds and improved potency/selectivity and MM-specific
pharmacological validation will be required. Modulating ALKBH5 may
still be attractive for combination strategies if on-target
activity and tolerability can be established (194).
Collectively, the inhibitors for METTL3 and FTO
possess the strongest immediate translational rationale. However,
it is critical to note that the current assessment of their
efficacy largely draws on research from other hematologic
malignancies such as AML. Specific pharmacodynamic data in MM,
optimal combination regimens and potential resistance mechanisms
await elucidation through future systematic preclinical studies
dedicated to MM.
Beyond small molecules, genetic and targeted
protein degradation technologies offer alternative paths for
precise and potent modulation of the m6A machinery
(Table II) (199-209). CRISPR-Cas9-mediated knockout of
METTL3 or ALKBH5 has proven effective in impairing cancer
progression in various models, though in vivo delivery to MM
cells in the bone marrow remains a challenge (194,210). More sophisticated epigenetic
editing tools, such as dCas13b-ALKBH5 fusion systems, enable
site-specific m6A demethylation without altering the DNA
sequence, offering the potential to reversibly silence specific
driver genes such as IRF4 while sparing normal hematopoiesis
(208,211).
Proteolysis-targeting chimeras (PROTACs) represent
a groundbreaking modality to achieve complete and selective
degradation of target proteins. PROTACs such as WD6305, which
recruit E3 ubiquitin ligases to degrade METTL3, have demonstrated
potent anti-leukemic effects in vivo (199,212,213). This strategy could be
particularly effective against MM cells that rely on METTL3 for
survival, potentially overcoming resistance mechanisms that arise
with catalytic inhibitors. Similarly, antisense oligonucleotides
(ASOs) designed to silence METTL3 mRNA expression provide
high specificity and could mitigate off-target effects associated
with small molecules (214).
These next-generation approaches are poised to
address the issues of tumor heterogeneity and acquired resistance
that often plague conventional therapies. The future clinical
application of these modalities will hinge on overcoming the
critical challenge of delivery, efficiently and specifically
targeting MM cells within the bone marrow niche. Advances in lipid
nanoparticles, viral vectors, or cell-specific targeting moieties
will be essential to unlock the full potential of genetic and
degradation-based epitranscriptomic therapy for MM.
Notably, for targets such as ALKBH5 and YTHDF2, the
rationale for targeted therapies primarily relies on genetic
approaches (such as knockdown/knockout experiments) and the
efficacy of tool compounds in non-MM models. Conclusive evidence of
'chemical tractability', such as highly selective and potent lead
compounds, remains lacking in the context of MM. Furthermore,
direct single-cell epitranscriptomic mapping of m6A
marks (rather than regulator expression) in MM subtypes remains
limited, representing a key technical and conceptual gap.
The burgeoning field of epitranscriptomics has
fundamentally expanded our understanding of gene regulation in
cancer. The present review consolidated compelling evidence that
the m6A modification machinery is an active regulatory
layer linked to key aspects of MM pathogenesis, progression and
therapy resistance. Through the dysregulated activity of writers,
erasers and readers, m6A modifications fine-tune the
expression of oncogenic networks, remodel the bone marrow
microenvironment and confer resilience to current therapies,
positioning the epitranscriptome as a rich source of novel
prognostic biomarkers and therapeutic vulnerabilities.
Major gaps remain regarding heterogeneity and
clinical translation. MM is a heterogeneous disease characterized
by varying molecular subtypes [such as hyperdiploid, t(11;14) and t(4;14)]
(215,216), yet subtype-resolved
m6A mapping is still limited. Addressing this will
require advanced approaches such as single-cell profiling and
high-resolution mapping (217,218). In parallel, translation must
account for the essential roles of m6A regulators in
normal hematopoiesis and immunity, which raises the risk of
on-target toxicity and underscores the need for selective
inhibitors/degraders, biomarker-guided patient selection and dosing
or delivery strategies that maximize the therapeutic window.
Not applicable.
YM and MM performed study conception and design. YM
and SH wrote the initial draft of the manuscript. MM, DB, YG, NB
and WH reviewed and edited the manuscript. MM and WH conducted
proofreading and further revisions. YM contributed to the
acquisition of funds. Data authentication is not applicable. All
authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by grants from the National
Natural Science Foundation of China (grant no. 82460714), Natural
Science Foundation of Xinjiang Uygur Autonomous Region (grant no.
2024D01E17), Xinjiang Uygur Autonomous Region Tianchi Talent
Introduction Plan ('TianChi Excellent Award for Young Doctoral
Talents'), the Xinjiang Medical University National Young Talent
Cultivation Program (grant no. XYD2024GR05) and the Undergraduate
Innovation Training Program of Xinjiang Medical University (project
no. X202410760050).
|
1
|
de Jong MME, Kellermayer Z, Papazian N,
Tahri S, Hofste Op Bruinink D, Hoogenboezem R, Sanders MA, van de
Woestijne PC, Bos PK, Khandanpour C, et al: The multiple myeloma
microenvironment is defined by an inflammatory stromal cell
landscape. Nat Immunol. 22:769–780. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ramberger E, Sapozhnikova V, Ng YLD,
Dolnik A, Ziehm M, Popp O, Sträng E, Kull M, Grünschläger F, Krüger
J, et al: The proteogenomic landscape of multiple myeloma reveals
insights into disease biology and therapeutic opportunities. Nat
Cancer. 5:1267–1284. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guan L, Su W, Zhong J and Qiu L: M-protein
detection by mass spectrometry for minimal residual disease in
multiple myeloma. Clin Chim Acta. 552:1176232024. View Article : Google Scholar
|
|
4
|
Kumar SK, Rajkumar V, Kyle RA, van Duin M,
Sonneveld P, Mateos MV, Gay F and Anderson KC: Multiple myeloma.
Nat Rev Dis Prim. 3:170462017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mikhael J, Bhutani M and Cole CE: Multiple
myeloma for the primary care provider: A practical review to
promote earlier diagnosis among diverse populations. Am J Med.
136:33–41. 2023. View Article : Google Scholar
|
|
6
|
Rajkumar SV: Multiple myeloma: 2022 update
on diagnosis, risk stratification, and management. Am J Hematol.
97:10862022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang J, Chan SC, Lok V, Zhang L,
Lucero-Prisno DE III, Xu W, Zheng ZJ, Elcarte E, Withers M, Wong
MCS, et al: The epidemiological landscape of multiple myeloma: A
global cancer registry estimate of disease burden, risk factors,
and temporal trends. Lancet Haematol. 9:e670–e677. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Das S, Juliana N, Yazit NAA, Azmani S and
Abu IF: Multiple myeloma: Challenges encountered and future options
for better treatment. Int J Mol Sci. 23:16492022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohty M, Facon T, Malard F and Harousseau
JL: A roadmap towards improving outcomes in multiple myeloma. Blood
Cancer J. 14:1352024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu S, Xu J, Cui W, Jin H, Wang X and
Maimaitiyiming Y: Post-translational modifications in multiple
myeloma: Mechanisms of drug resistance and therapeutic
opportunities. Biomolecules. 15:7022025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rajkumar SV and Kumar S: Multiple myeloma
current treatment algorithms. Blood Cancer J. 10:942020. View Article : Google Scholar :
|
|
12
|
Bhatt P, Kloock C and Comenzo R:
Relapsed/refractory multiple myeloma: A review of available
therapies and clinical scenarios encountered in myeloma relapse.
Curr Oncol. 30:2322–2347. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zeng L, Huang H, Qirong C, Ruan C, Liu Y,
An W, Guo Q and Zhou J: Multiple myeloma patients undergoing
chemotherapy: Which symptom clusters impact quality of life? J Clin
Nurs. 32:7247–7259. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goodman RS, Johnson DB and Balko JM:
Corticosteroids and cancer immunotherapy. Clin Cancer Res.
29:2580–2587. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo H, Feng Y, Wang F, Lin Z, Huang J, Li
Q, Wang X, Liu X, Zhai X, Gao Q, et al: Combinations of ivermectin
with proteasome inhibitors induce synergistic lethality in multiple
myeloma. Cancer Lett. 565:2162182023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Minařík J and Ševčíková S:
Immunomodulatory agents for multiple myeloma. Cancers (Basel).
14:57592022. View Article : Google Scholar
|
|
17
|
Koniarczyk HL, Ferraro C and Miceli T:
Hematopoietic stem cell transplantation for multiple myeloma. Semin
Oncol Nurs. 33:265–278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dima D, Jiang D, Singh DJ, Hasipek M, Shah
HS, Ullah F, Khouri J, Maciejewski JP and Jha BK: Multiple myeloma
therapy: Emerging trends and challenges. Cancers (Basel).
14:40822022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheykhhasan M, Ahmadieh-Yazdi A,
Vicidomini R, Poondla N, Tanzadehpanah H, Dirbaziyan A, Mahaki H,
Manoochehri H, Kalhor N and Dama P: CAR T therapies in multiple
myeloma: Unleashing the future. Cancer Gene Ther. 31:667–686. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Neri P, Leblay N, Lee H, Gulla A, Bahlis
NJ and Anderson KC: Just scratching the surface: novel treatment
approaches for multiple myeloma targeting cell membrane proteins.
Nat Rev Clin Oncol. 21:590–609. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tam T, Smith E, Lozoya E, Heers H and
Andrew Allred P: Roadmap for new practitioners to navigate the
multiple myeloma landscape. Heliyon. 8:e105862022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu Y, Jian X, Chen S, An G, Jiang D, Yang
Q, Zhang J, Hu J, Qiu Y, Feng X, et al: Targeting gut microbial
nitrogen recycling and cellular uptake of ammonium to improve
bortezomib resistance in multiple myeloma. Cell Metab.
36:159–175.e8. 2024. View Article : Google Scholar
|
|
23
|
Neri P, Barwick BG, Jung D, Patton JC,
Maity R, Tagoug I, Stein CK, Tilmont R, Leblay N, Ahn S, et al:
ETV4-Dependent transcriptional plasticity maintains MYC expression
and results in IMiD resistance in multiple myeloma. Blood Cancer
Discov. 5:56–73. 2024. View Article : Google Scholar :
|
|
24
|
Zhang L, Peng X, Ma T, Liu J, Yi Z, Bai J,
Li Y, Li L and Zhang L: Natural killer cells affect the natural
course, drug resistance, and prognosis of multiple myeloma. Front
Cell Dev Biol. 12:13590842024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Malard F, Neri P, Bahlis NJ, Terpos E,
Moukalled N, Hungria VTM, Manier S and Mohty M: Multiple myeloma.
Nat Rev Dis Prim. 10:452024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Negrete-Rodríguez P, Gallardo-Pérez MM,
Lira-Lara O, Melgar-de-la-Paz M, Hamilton-Avilés LE, Ocaña-Ramm G,
Robles-Nasta M, Sánchez-Bonilla D, Olivares-Gazca JC, Mateos MV, et
al: Prevalence and consequences of a delayed diagnosis in multiple
myeloma: A single institution experience. Clin Lymphoma Myeloma
Leuk. 24:478–483. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang P, Qu Y, Wang M, Chu B, Chen W, Zheng
Y, Niu T and Qian Z: Pathogenesis and treatment of multiple
myeloma. MedComm (2020). 3:e1462022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dimopoulos K, Gimsing P and Grønbæk K: The
role of epigenetics in the biology of multiple myeloma. Blood
Cancer J. 4:e2072014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song H, Feng X, Zhang H, Luo Y, Huang J,
Lin M, Jin J, Ding X, Wu S, Huang H, et al: METTL3 and ALKBH5
oppositely regulate m6A modification of TFEB mRNA, which dictates
the fate of hypoxia/reoxygenation-treated cardiomyocytes.
Autophagy. 15:1419–1437. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu R, Gao Q, Foltz SM, Fowles JS, Yao L,
Wang JT, Cao S, Sun H, Wendl MC, Sethuraman S, et al: Co-evolution
of tumor and immune cells during progression of multiple myeloma.
Nat Commun. 12:25592021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vo JN, Wu YM, Mishler J, Hall S, Mannan R,
Wang L, Ning Y, Zhou J, Hopkins AC, Estill JC, et al: The genetic
heterogeneity and drug resistance mechanisms of relapsed refractory
multiple myeloma. Nat Commun. 13:37502022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu L, Wen C, Xia J, Zhang H, Liang Y and
Xu X: Targeted immunotherapy: Harnessing the immune system to
battle multiple myeloma. Cell Death Discov. 10:552024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Skerget S, Penaherrera D, Chari A,
Jagannath S, Siegel DS, Vij R, Orloff G, Jakubowiak A, Niesvizky R,
Liles D, et al: Comprehensive molecular profiling of multiple
myeloma identifies refined copy number and expression subtypes. Nat
Genet. 56:1878–1889. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rosselló-Tortella M, Ferrer G and Esteller
M: Epitranscriptomics in hematopoiesis and hematologic
malignancies. Blood Cancer Discov. 1:26–31. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frye M, Jaffrey SR, Pan T, Rechavi G and
Suzuki T: RNA modifications: What have we learned and where are we
headed? Nat Rev Genet. 17:365–372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang C, Han H and Lin S: RNA
epitranscriptomics: A promising new avenue for cancer therapy. Mol
Ther. 30:2–3. 2022. View Article : Google Scholar
|
|
37
|
Yang L, Tang L, Min Q, Tian H, Li L, Zhao
Y, Wu X, Li M, Du F, Chen Y, et al: Emerging role of RNA
modification and long noncoding RNA interaction in cancer. Cancer
Gene Ther. 31:816–830. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pham CT, Rangan L and Schlenner S: RNA
modifications-a regulatory dimension yet to be deciphered in
immunity. Genes Immun. 24:281–282. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xue C, Chu Q, Zheng Q, Jiang S, Bao Z, Su
Y, Lu J and Li L: Role of main RNA modifications in cancer:
N6-methyladenosine, 5-methylcytosine, and pseudouridine. Signal
Transduct Target Ther. 7:1422022. View Article : Google Scholar
|
|
40
|
Deng X, Qing Y, Horne D, Huang H and Chen
J: The roles and implications of RNA m6A modification in cancer.
Nat Rev Clin Oncol. 20:507–526. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cui L, Ma R, Cai J, Guo C, Chen Z, Yao L,
Wang Y, Fan R, Wang X and Shi Y: RNA modifications: importance in
immune cell biology and related diseases. Signal Transduct Target
Ther. 7:3342022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yao L, Yin H, Hong M, Wang Y, Yu T, Teng
Y, Li T and Wu Q: RNA methylation in hematological malignancies and
its interactions with other epigenetic modifications. Leukemia.
35:1243–1257. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qing Y, Su R and Chen J: RNA modifications
in hematopoietic malignancies: A new research frontier. Blood.
138:637–648. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chao CT, Kuo FC and Lin SH: Epigenetically
regulated inflammation in vascular senescence and renal progression
of chronic kidney disease. Semin Cell Dev Biol. 154:305–315. 2024.
View Article : Google Scholar
|
|
45
|
Kan RL, Chen J and Sallam T: Crosstalk
between epitranscriptomic and epigenetic mechanisms in gene
regulation. Trends Genet. 38:182–193. 2022. View Article : Google Scholar :
|
|
46
|
Ye Z, Mayila M, Bu N, Hao W and
Maimaitiyiming Y: Epigenetic and epitranscriptomic landscape of
phthalate toxicity: Implications for human health and disease.
Environ Pollut. 391:1275592026. View Article : Google Scholar
|
|
47
|
Frye M, Harada BT, Behm M and He C: RNA
modifications modulate gene expression during development. Science.
361:1346–1349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Qiu L, Jing Q, Li Y and Han J: RNA
modification: Mechanisms and therapeutic targets. Mol Biomed.
4:252023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun H, Li K, Liu C and Yi C: Regulation
and functions of non-m6A mRNA modifications. Nat Rev Mol Cell Biol.
24:714–731. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Martinez De La Cruz B, Darsinou M and
Riccio A: From form to function: m6A methylation links mRNA
structure to metabolism. Adv Biol Regul. 87:1009262023. View Article : Google Scholar
|
|
51
|
Deng LJ, Deng WQ, Fan SR, Chen MF, Qi M,
Lyu WY, Qi Q, Tiwari AK, Chen JX, Zhang DM and Chen ZS: m6A
modification: Recent advances, anticancer targeted drug discovery
and beyond. Mol Cancer. 21:522022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Su Y, Maimaitiyiming Y, Wang L, Cheng X
and Hsu CH: Modulation of phase separation by RNA: A glimpse on
N6-Methyladenosine modification. Front Cell Dev Biol. 9:7864542021.
View Article : Google Scholar
|
|
53
|
Mendel M, Delaney K, Pandey RR, Chen KM,
Wenda JM, Vågbø CB, Steiner FA, Homolka D and Pillai RS: Splice
site m6A methylation prevents binding of U2AF35 to inhibit RNA
splicing. Cell. 184:3125–3142.e25. 2021. View Article : Google Scholar
|
|
54
|
Aufgebauer CJ, Bland KM and Horner SM:
Modifying the antiviral innate immune response by selective
writing, erasing, and reading of m6A on viral and cellular RNA.
Cell Chem Biol. 31:100–109. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee SY, Kim JJ and Miller KM: Emerging
roles of RNA modifications in genome integrity. Brief Funct
Genomics. 20:106–112. 2021. View Article : Google Scholar :
|
|
56
|
Zaccara S, Ries RJ and Jaffrey SR:
Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell
Biol. 20:608–624. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Esteva-Socias M and Aguilo F: METTL3 as a
master regulator of translation in cancer: Mechanisms and
implications. NAR Cancer. 6:zcae0092024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yan X, Liu F, Yan J, Hou M, Sun M, Zhang
D, Gong Z, Dong X, Tang C and Yin P: WTAP-VIRMA counteracts dsDNA
binding of the m(6)A writer METTL3-METTL14 complex and maintains
N(6)-adenosine methylation activity. Cell Discov. 9:1002023.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang Z, Zhang S, Xiong J, Xia T, Zhu R,
Miao M, Li K, Chen W, Zhang L, You Y and You B: The m6A
demethylases FTO and ALKBH5 aggravate the malignant progression of
nasopharyngeal carcinoma by coregulating ARHGAP35. Cell Death
Discov. 10:432024. View Article : Google Scholar
|
|
60
|
Zou Z, Sepich-Poore C, Zhou X, Wei J and
He C: The mechanism underlying redundant functions of the YTHDF
proteins. Genome Biol. 24:172023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiao W, Adhikari S, Dahal U, Chen YS, Hao
YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al: Nuclear m(6)A
Reader YTHDC1 Regulates mRNA splicing. Mol Cell. 61:507–519. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Huang H, Weng H, Sun W, Qin X, Shi H, Wu
H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al: Recognition of RNA N
6 -methyladenosine by IGF2BP proteins enhances mRNA stability and
translation. Nat Cell Biol. 20:285–295. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Alarcón CR, Goodarzi H, Lee H, Liu X,
Tavazoie S and Tavazoie SF: HNRNPA2B1 is a mediator of
m6A-dependent nuclear RNA processing events. Cell. 162:1299–1308.
2015. View Article : Google Scholar
|
|
64
|
Xu W, Huang Z, Xiao Y, Li W, Xu M, Zhao Q
and Yi P: HNRNPC promotes estrogen receptor-positive breast cancer
cell cycle by stabilizing WDR77 mRNA in an m6A-dependent manner.
Mol Carcinog. 63:859–873. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Meyer KD, Patil DP, Zhou J, Zinoviev A,
Skabkin MA, Elemento O, Pestova TV, Qian SB and Jaffrey SR: 5' UTR
m6A promotes cap-independent translation. Cell. 163:999–1010. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang
W, Song H, Wu H, Shu Q and Jin P: Fragile X mental retardation
protein modulates the stability of its m6A-marked messenger RNA
targets. Hum Mol Genet. 27:3936–3950. 2018.PubMed/NCBI
|
|
67
|
Mao-Mao, Zhang JJ, Xu YP, Shao MM and Wang
MC: Regulatory effects of natural products on N6-methyladenosine
modification: A novel therapeutic strategy for cancer. Drug Discov
Today. 29:1038752024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhu ZM, Huo FC, Zhang J, Shan HJ and Pei
DS: Crosstalk between m6A modification and alternative splicing
during cancer progression. Clin Transl Med. 13:e14602023.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jain S, Koziej L, Poulis P, Kaczmarczyk I,
Gaik M, Rawski M, Ranjan N, Glatt S and Rodnina MV: Modulation of
translational decoding by m6A modification of mRNA. Nat Commun.
14:47842023. View Article : Google Scholar
|
|
70
|
Qiao Y, Sun Q, Chen X, He L, Wang D, Su R,
Xue Y, Sun H and Wang H: Nuclear m6A Reader YTHDC1 promotes muscle
stem cell activation/proliferation by regulating mRNA splicing and
nuclear export. Elife. 12:e827032023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Boulias K and Greer EL: Biological roles
of adenine methylation in RNA. Nat Rev Genet. 24:143–160. 2023.
View Article : Google Scholar
|
|
72
|
Wang Y, Li Y, Skuland T, Zhou C, Li A,
Hashim A, Jermstad I, Khan S, Dalen KT, Greggains GD, et al: The
RNA m6A landscape of mouse oocytes and preimplantation embryos. Nat
Struct Mol Biol. 30:703–709. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wen J, Lv R, Ma H, Shen H, He C, Wang J,
Jiao F, Liu H, Yang P, Tan L, et al: Zc3h13 regulates nuclear RNA
m6A methylation and mouse embryonic stem cell self-renewal. Mol
Cell. 69:1028–1038.e6. 2018. View Article : Google Scholar
|
|
74
|
Yoon KJ, Ringeling FR, Vissers C, Jacob F,
Pokrass M, Jimenez-Cyrus D, Su Y, Kim NS, Zhu Y, Zheng L, et al:
Temporal control of mammalian cortical neurogenesis by m6A
methylation. Cell. 171:877–889.e17. 2017. View Article : Google Scholar
|
|
75
|
Wang L, Maimaitiyiming Y, Su K and Hsu CH:
RNA m6A Modification: The Mediator Between Cellular Stresses and
Biological Effects. RNA Technologies. 12:353–390. 2021. View Article : Google Scholar
|
|
76
|
Xiang Y, Laurent B, Hsu CH, Nachtergaele
S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al: RNA m(6)A
methylation regulates the ultraviolet-induced DNA damage response.
Nature. 543:573–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang L, Zhan G, Maimaitiyiming Y, Su Y,
Lin S, Liu J, Su K, Lin J, Shen S, He W, et al: m6A modification
confers thermal vulnerability to HPV E7 oncotranscripts via reverse
regulation of its reader protein IGF2BP1 upon heat stress. Cell
Rep. 41:1115462022. View Article : Google Scholar
|
|
78
|
Engel M, Eggert C, Kaplick PM, Eder M, Röh
S, Tietze L, Namendorf C, Arloth J, Weber P, Rex-Haffner M, et al:
The role of m6A/m-RNA methylation in stress response regulation.
Neuron. 99:389–403.e9. 2018. View Article : Google Scholar
|
|
79
|
Chuong NN, Doan PPT, Wang L, Kim JH and
Kim J: Current insights into m6A RNA methylation and its emerging
role in plant circadian clock. Plants (Basel). 12:6242023.
|
|
80
|
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang
Y, Jin Y, Yang Y, Chen LL, Wang Y, et al: Extensive translation of
circular RNAs driven by N 6 -methyladenosine. Cell Res. 27:626–641.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Alarcón CR, Lee H, Goodarzi H, Halberg N
and Tavazoie SF: N6-methyladenosine marks primary microRNAs for
processing. Nature. 519:482–485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH,
Wang F, Wang TT, Xu QG, Zhou WP and Sun SH: METTL14 suppresses the
metastatic potential of hepatocellular carcinoma by modulating
N6-methyladenosine-dependent primary MicroRNA processing.
Hepatology. 65:529–543. 2017. View Article : Google Scholar
|
|
83
|
Deng S, Zhang J, Su J, Zuo Z, Zeng L, Liu
K, Zheng Y, Huang X, Bai R, Zhuang L, et al: RNA m6A regulates
transcription via DNA demethylation and chromatin accessibility.
Nat Genet. 54:1427–1437. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Höfler S and Duss O: Interconnections
between m6 A RNA modification, RNA structure, and protein-RNA
complex assembly. Life Sci Alliance. 7:e2023022402024. View Article : Google Scholar
|
|
85
|
Vaid R, Thombare K, Mendez A,
Burgos-Panadero R, Djos A, Jachimowicz D, Lundberg KI, Bartenhagen
C, Kumar N, Tümmler C, et al: MET TL3 drives telomere targ eting of
TERRA lncRNA through m 6 A-dependent R-loop formation: A
therapeutic target for ALT-positive neuroblastoma. Nucleic Acids
Res. 52:2648–2671. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lee SY, Lee SH, Kwak MJ, Kim JY, Perren
JO, Miller KM and Kim JJ: Depletion of BRD9-mediated R-loop
accumulation inhibits leukemia cell growth via
transcription-replication conflict. Nucleic Acids Res.
53:gkaf6132025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Verghese M, Wilkinson E and He YY: Role of
RNA modifications in carcinogenesis and carcinogen damage response.
Mol Carcinog. 62:24–37. 2023. View Article : Google Scholar :
|
|
88
|
Yang J, Xu J, Wang W, Zhang B, Yu X and
Shi S: Epigenetic regulation in the tumor microenvironment:
Molecular mechanisms and therapeutic targets. Signal Transduct
Target Ther. 8:2102023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lin S and Kuang M: RNA
modification-mediated mRNA translation regulation in liver cancer:
Mechanisms and clinical perspectives. Nat Rev Gastroenterol
Hepatol. 21:267–281. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Dong L, Cao Y, Hou Y and Liu G:
N6-methyladenosine RNA methylation: A novel regulator of the
development and function of immune cells. J Cell Physiol.
237:329–345. 2022. View Article : Google Scholar
|
|
91
|
Delaunay S, Helm M and Frye M: RNA
modifications in physiology and disease: Towards clinical
applications. Nat Rev Genet. 25:104–122. 2024. View Article : Google Scholar
|
|
92
|
Berdasco M and Esteller M: Towards a
druggable epitranscriptome: Compounds that target RNA modifications
in cancer. Br J Pharmacol. 179:2868–2889. 2022. View Article : Google Scholar
|
|
93
|
Zheng J, Lu Y, Lin Y, Si S, Guo B, Zhao X
and Cui L: Epitranscriptomic modifications in mesenchymal stem cell
differentiation: Advances, mechanistic insights, and beyond. Cell
Death Differ. 31:9–27. 2024. View Article : Google Scholar
|
|
94
|
Delaunay S and Frye M: RNA modifications
regulating cell fate in cancer. Nat Cell Biol. 21:552–559. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shi J, Zhang Q, Yin X, Ye J, Gao S, Chen
C, Yang Y, Wu B, Fu Y, Zhang H, et al: Stabilization of IGF2BP1 by
USP10 promotes breast cancer metastasis via CPT1A in an
m6A-dependent manner. Int J Biol Sci. 19:449–464. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lv D, Gimple RC, Zhong C, Zhong C, Wu Q,
Yang K, Prager BC, Godugu B, Qiu Z, Zhao L, et al: PDGF signaling
inhibits mitophagy in glioblastoma stem cells through
N6-methyladenosine. Dev Cell. 57:1466–1481.e6. 2022. View Article : Google Scholar
|
|
97
|
Feng Y, Yuan P, Guo H, Gu L, Yang Z, Wang
J, Zhu W, Zhang Q, Cao J, Wang L and Jiao Y: METTL3 mediates
epithelial-mesenchymal transition by modulating FOXO1 mRNA
N6-Methyladenosine-Dependent YTHDF2 Binding: A novel mechanism of
radiation-induced lung injury. Adv Sci (Weinh). 10:e22047842023.
View Article : Google Scholar
|
|
98
|
Geula S, Moshitch-Moshkovitz S,
Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V,
Peer E, Mor N, Manor YS, et al: Stem Cells m6A mRNA methylation
facilitates resolution of naïve pluripotency toward
differentiation. Science. 347:1002–1006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S,
Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al: m6A demethylase
ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by
sustaining FOXM1 expression and cell proliferation program. Cancer
Cell. 31:591–606.e6. 2017. View Article : Google Scholar
|
|
101
|
Xiao S, Ma S, Sun B, Pu W, Duan S, Han J,
Hong Y, Zhang J, Peng Y, He C, et al: The tumor-intrinsic role of
the m6A reader YTHDF2 in regulating immune evasion. Sci Immunol.
9:eadl21712024. View Article : Google Scholar
|
|
102
|
Wang X, Zhao BS, Roundtree IA, Lu Z, Han
D, Ma H, Weng X, Chen K, Shi H and He C: N6-methyladenosine
modulates messenger RNA translation efficiency. Cell.
161:1388–1399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zeng L, Huang X, Zhang J, Lin D and Zheng
J: Roles and implications of mRNA N6-methyladenosine in cancer.
Cancer Commun (Lond). 43:729–748. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhuang H, Yu B, Tao D, Xu X, Xu Y, Wang J,
Jiao Y and Wang L: The role of m6A methylation in therapy
resistance in cancer. Mol Cancer. 22:912023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jin Z, MacPherson K, Liu Z and Vu LP: RNA
modifications in hematological malignancies. Int J Hematol.
117:807–820. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li
W, Yang YG, Zhou Q and Liu F: Endothelial-specific m6A modulates
mouse hematopoietic stem and progenitor cell development via Notch
signaling. Cell Res. 28:249–252. 2018. View Article : Google Scholar
|
|
107
|
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C,
Huang H, Nachtergaele S, Dong L, Hu C, et al: FTO plays an
oncogenic role in acute myeloid leukemia as a N6-Methyladenosine
RNA demethylase. Cancer Cell. 31:127–141. 2017. View Article : Google Scholar
|
|
108
|
Vu LP, Pickering BF, Cheng Y, Zaccara S,
Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al:
The N 6 -methyl-adenosine (m 6 A)-forming enzyme METTL3 controls
myeloid differentiation of normal hematopoietic and leukemia cells.
Nat Med. 23:1369–1376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Barbieri I, Tzelepis K, Pandolfini L, Shi
J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister
AJ, Han N, et al: Promoter-bound METTL3 maintains myeloid leukaemia
by m6A-dependent translation control. Nature. 552:126–131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhang N, Shen Y, Li H, Chen Y, Zhang P,
Lou S and Deng J: The m6A reader IGF2BP3 promotes acute myeloid
leukemia progression by enhancing RCC2 stability. Exp Mol Med.
54:194–205. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wilkinson E, Cui YH and He YY:
Context-dependent roles of RNA modifications in stress responses
and diseases. Int J Mol Sci. 22:19492021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wang J, Zuo Y, Lv C, Zhou M and Wan Y:
N6-methyladenosine regulators are potential prognostic biomarkers
for multiple myeloma. IUBMB Life. 75:137–148. 2023. View Article : Google Scholar :
|
|
113
|
Deng Y, Zhu H and Peng H: Enhancing
staging in multiple myeloma using an m6A regulatory gene-pairing
model. Clin Exp Med. 25:402025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Bao J, Xu T, Wang W, Xu H, Chen X and Xia
R: N6-methyladenosine-induced miR-182-5p promotes multiple myeloma
tumorigenesis by regulating CAMK2N1. Mol Cell Biochem.
479:3077–3089. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Che F, Ye X, Wang Y, Wang X, Ma S, Tan Y,
Mao Y and Luo Z: METTL3 facilitates multiple myeloma tumorigenesis
by enhancing YY1 stability and pri-microRNA-27 maturation in
m6A-dependent manner. Cell Biol Toxicol. 39:2033–2050. 2023.
View Article : Google Scholar
|
|
116
|
Huang X, Yang Z, Li Y and Long X: m6A
methyltransferase METTL3 facilitates multiple myeloma cell growth
through the m6A modification of BZW2. Ann Hematol. 102:1801–1810.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lu X, Li Y, Li R, Zhang J, Peng J and
Zhang Y: Regulatory role of the METTL3/MALAT1 axis in multiple
myeloma progression. J Bone Oncol. 53:1006952025. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhao Y, Zhang E, Lv N, Ma L, Yao S, Yan M,
Zi F, Deng G, Liu X, He J, et al: Metformin and FTY720
synergistically induce apoptosis in multiple myeloma cells. Cell
Physiol Biochem. 48:785–800. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Gao L, Li L, Hu J, Li G, Zhang Y, Dai X,
De Z and Xu F: Metformin inhibits multiple myeloma serum-induced
endothelial cell thrombosis by down-regulating miR-532. Ann Vasc
Surg. 85:347–357.e2. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chen CJ, Huang JY, Huang JQ, Deng JY,
Shangguan XH, Chen AZ, Chen LT and Wu WH: Metformin attenuates
multiple myeloma cell proliferation and encourages apoptosis by
suppressing METTL3-mediated m6A methylation of THRAP3, RBM25, and
USP4. Cell Cycle. 22:986–1004. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Jiang J, Zhong F, Xiao Z, Yao F, Liu J, Li
M, Zeng H, Qiu Y, Zhang J, Zhang H, et al: METTL5 regulates
SEPHS2-mediated selenoprotein synthesis to promote multiple myeloma
survival and progression. Cell Death Dis. 16:5852025. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wu Y, Luo Y, Yao X, Shi X, Xu Z, Re J, Shi
M, Li M, Liu J, He Y and Du X: KIAA1429 increases FOXM1 expression
through YTHDF1-mediated m6A modification to promote aerobic
glycolysis and tumorigenesis in multiple myeloma. Cell Biol
Toxicol. 40:582024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Su Q, Liu W, Wang P and Wang M: Long
non-coding RNA FEZF1-AS1 suppresses ferroptosis in multiple myeloma
cells through KIAA1429-mediated m6A modification. Hum Cell.
38:1782025. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Xu H, Xu M, Ding J and Bao J: WTAP
promotes the proliferation of multiple myeloma by regulating the
hippo pathway through m(6)A modification of MAP6D1. Leuk Lymphoma.
67:148–163. 2026. View Article : Google Scholar
|
|
125
|
Jia Y, Yu X, Liu R, Shi L, Jin H, Yang D,
Zhang X, Shen Y, Feng Y, Zhang P, et al: PRMT1 methylation of WTAP
promotes multiple myeloma tumorigenesis by activating oxidative
phosphorylation via m6A modification of NDUFS6. Cell Death Dis.
14:5122023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Hua Z, Wei R, Guo M, Lin Z, Yu X, Li X, Gu
C and Yang Y: YTHDF2 promotes multiple myeloma cell proliferation
via STAT5A/MAP2K2/p-ERK axis. Oncogene. 41:1482–1491. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liu R, Miao J, Jia Y, Kong G, Hong F, Li
F, Zhai M, Zhang R, Liu J, Xu X, et al: N6-methyladenosine reader
YTHDF2 promotes multiple myeloma cell proliferation through
EGR1/p21cip1/waf1/CDK2-Cyclin E1 axis-mediated cell cycle
transition. Oncogene. 42:1607–1619. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Xu J, Wang Y, Ren L, Li P and Liu P:
IGF2BP1 promotes multiple myeloma with chromosome 1q gain via
increasing CDC5L expression in an m6A-dependent manner. Genes Dis.
12:1012142024. View Article : Google Scholar
|
|
129
|
Bernstein ZS, Kim EB and Raje N: Bone
disease in multiple myeloma: Biologic and clinical implications.
Cells. 11:23082022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Terpos E, Ntanasis-Stathopoulos I,
Gavriatopoulou M and Dimopoulos MA: Pathogenesis of bone disease in
multiple myeloma: From bench to bedside. Blood Cancer J. 8:72018.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Liu R, Zhong Y, Chen R, Chu C, Liu G, Zhou
Y, Huang Y, Fang Z and Liu H: m6A reader hnRNPA2B1 drives multiple
myeloma osteolytic bone disease. Theranostics. 12:7760–7774. 2022.
View Article : Google Scholar
|
|
132
|
Jiang F, Tang X, Tang C, Hua Z, Ke M, Wang
C, Zhao J, Gao S, Jurczyszyn A, Janz S, et al: HNRNPA2B1 promotes
multiple myeloma progression by increasing AKT3 expression via
m6A-dependent stabilization of ILF3 mRNA. J Hematol Oncol.
14:542021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Guo Y, Jia C, Wang X, Luo K, Chi L, Xu Q,
Gong T and Quan L: HNRNPA2B1 promotes the progression of multiple
myeloma via endoplasmic reticulum stress and autophagy mediated by
CK2 Kinase. J Proteome Res. 24:5921–5931. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Tang J, Li J, Qin S, Xiao Y, Liu J, Chen X
and Zhang Y: Identification and validation of the m6A-binding
protein LRPPRC to promote tumorigenesis in multiple myeloma.
Hematology. 30:25230822025. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Yao L, Li T, Teng Y, Guo J, Zhang H, Xia L
and Wu Q: ALKHB5-demethylated lncRNA SNHG15 promotes myeloma
tumorigenicity by increasing chromatin accessibility and recruiting
H3K36me3 modifier SETD2. Am J Physiol Cell Physiol. 326:C684–C697.
2024. View Article : Google Scholar :
|
|
136
|
Yu T, Yao L, Yin H, Teng Y, Hong M and Wu
Q: ALKBH5 promotes multiple myeloma tumorigenicity through inducing
m6A-demethylation of SAV1 mRNA and myeloma stem cell phenotype. Int
J Biol Sci. 18:2235–2248. 2022. View Article : Google Scholar
|
|
137
|
Qu J, Hou Y, Chen Q, Chen J, Li Y, Zhang
E, Gu H, Xu R, Liu Y, Cao W, et al: RNA demethylase ALKBH5 promotes
tumorigenesis in multiple myeloma via TRAF1-mediated activation of
NF-κB and MAPK signaling pathways. Oncogene. 41:400–413. 2022.
View Article : Google Scholar :
|
|
138
|
Badraldin SQ, Alfarttoosi KH, Sameer HN,
Bishoyi AK, Ganesan S, Shankhyan A, Ray S, Nathiya D, Yaseen A,
Athab ZH and Adil M: Mechanistic role of FTO in cancer
pathogenesis, immune evasion, chemotherapy resistance, and
immunotherapy response. Semin Oncol. 52:1523682025. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Xu A, Zhang J, Zuo L, Yan H, Chen L, Zhao
F, Fan F, Xu J, Zhang B, Zhang Y, et al: FTO promotes multiple
myeloma progression by posttranscriptional activation of HSF1 in an
m6A-YTHDF2-dependent manner. Mol Ther. 30:1104–1118. 2022.
View Article : Google Scholar
|
|
140
|
Li JJ, Yu T, Zeng P, Tian J, Liu P, Qiao
S, Wen S, Hu Y, Liu Q, Lu W, et al: Wild-type IDH2 is a therapeutic
target for triple-negative breast cancer. Nat Commun. 15:34452024.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Song S, Fan G, Li Q, Su Q, Zhang X, Xue X,
Wang Z, Qian C, Jin Z, Li B and Zhuang W: IDH2 contributes to
tumorigenesis and poor prognosis by regulating m6A RNA methylation
in multiple myeloma. Oncogene. 40:5393–5402. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Zhang Y, Deng Z, Sun S, Xie S, Jiang M,
Chen B, Gu C and Yang Y: NAT10 acetylates BCL-XL mRNA to promote
the proliferation of multiple myeloma cells through PI3K-AKT
pathway. Front Oncol. 12:9678112022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Liu H, Zhang X, Lu Q and Zhang H: NAT10
contributes to the progression of multiple myeloma through ac4C
modification of GPR37. Hematology. 30:25557792025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Ren H, Liu C, Wu H, Wang Z, Chen S, Zhang
X, Ren J, Qiu H and Zhou L: m5C Regulator-mediated methylation
modification clusters contribute to the immune microenvironment
regulation of multiple myeloma. Front Genet. 13:9201642022.
View Article : Google Scholar
|
|
145
|
Jiang Y, Sun J, Chen Y, Cheng L, Feng S,
Wang Y and Sun C: NSUN2-mediated RNA m(5)C modification drives
multiple myeloma progression by enhancing the stability of HIP1
mRNA. Sci Rep. 15:278882025. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Fu J, Han X, Gao W, Yu M and Cui X: m1A
regulator-mediated methylation modifications and gene signatures
and their prognostic value in multiple myeloma. Exp Ther Med.
29:182025. View Article : Google Scholar
|
|
147
|
Cohen YC, Zada M, Wang SY, Bornstein C,
David E, Moshe A, Li B, Shlomi-Loubaton S, Gatt ME, Gur C, et al:
Identification of resistance pathways and therapeutic targets in
relapsed multiple myeloma patients through single-cell sequencing.
Nat Med. 27:491–503. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Ferguson ID, Patiño-Escobar B, Tuomivaara
ST, Lin YT, Nix MA, Leung KK, Kasap C, Ramos E, Nieves Vasquez W,
Talbot A, et al: The surfaceome of multiple myeloma cells suggests
potential immunotherapeutic strategies and protein markers of drug
resistance. Nat Commun. 13:41212022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Sun J, Corradini S, Azab F, Shokeen M, Muz
B, Miari KE, Maksimos M, Diedrich C, Asare O, Alhallak K, et al:
IL-10R inhibition reprograms tumor-associated macrophages and
reverses drug resistance in multiple myeloma. Leukemia.
38:2355–2365. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Bird S and Pawlyn C: IMiD resistance in
multiple myeloma: Current understanding of the underpinning biology
and clinical impact. Blood. 142:131–140. 2023.PubMed/NCBI
|
|
151
|
Tzelepis K, Rausch O and Kouzarides T:
RNA-modifying enzymes and their function in a chromatin context.
Nat Struct Mol Biol. 26:858–862. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Chen H, Jia B, Zhang Q and Zhang Y:
Meclofenamic acid restores gefinitib sensitivity by downregulating
breast cancer resistance protein and multidrug resistance protein 7
via FTO/m6A-Demethylation/c-Myc in non-small cell lung cancer.
Front Oncol. 12:8706362022. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Yuan J, Guan W, Li X, Wang F, Liu H and Xu
G: RBM15-mediating MDR1 mRNA m6A methylation regulated by the TGF-β
signaling pathway in paclitaxel-resistant ovarian cancer. Int J
Oncol. 63:1122023. View Article : Google Scholar
|
|
154
|
Liu R, Shen Y, Hu J, Wang X, Wu D, Zhai M,
Bai J and He A: Comprehensive Analysis of m6A RNA methylation
regulators in the prognosis and immune microenvironment of multiple
myeloma. Front Oncol. 11:7319572021. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wang C, Li L, Li M, Wang W and Jiang Z:
FTO promotes Bortezomib resistance via m6A-dependent
destabilization of SOD2 expression in multiple myeloma. Cancer Gene
Ther. 30:622–628. 2023. View Article : Google Scholar
|
|
156
|
Prabhu KS, Ahmad F, Kuttikrishnan S, Leo
R, Ali TA, Izadi M, Mateo JM, Alam M, Ahmad A, Al-Shabeeb Akil AS,
et al: Bortezomib exerts its anti-cancer activity through the
regulation of Skp2/p53 axis in non-melanoma skin cancer cells and
C. elegans. Cell Death Discov. 10:2252024. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Sogbein O, Paul P, Umar M, Chaari A,
Batuman V and Upadhyay R: Bortezomib in cancer therapy: Mechanisms,
side effects, and future proteasome inhibitors. Life Sci.
358:1231252024. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Hurt EM, Thomas SB, Peng B and Farrar WL:
Integrated molecular profiling of SOD2 expression in multiple
myeloma. Blood. 109:3953–3962. 2007. View Article : Google Scholar
|
|
159
|
Hodge DR, Peng B, Pompeia C, Thomas S, Cho
E, Clausen PA, Marquez VE and Farrar WL: Epigenetic silencing of
manganese superoxide dismutase (SOD-2) in KAS 6/1 human multiple
myeloma cells increases cell proliferation. Cancer Biol Ther.
4:585–592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Song IS, Kim HK, Lee SR, Jeong SH, Kim N,
Ko KS, Rhee BD and Han J: Mitochondrial modulation decreases the
bortezomib-resistance in multiple myeloma cells. Int J Cancer.
133:1357–1367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Jomova K, Alomar SY, Alwasel SH,
Nepovimova E, Kuca K and Valko M: Several lines of antioxidant
defense against oxidative stress: antioxidant enzymes,
nanomaterials with multiple enzyme-mimicking activities, and
low-molecular-weight antioxidants. Arch Toxicol. 98:1323–1367.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Huang R, Chen H, Liang J, Li Y, Yang J,
Luo C, Tang Y, Ding Y, Liu X, Yuan Q, et al: Dual role of reactive
oxygen species and their application in cancer therapy. J Cancer.
12:5543–5561. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Jahankhani K, Taghipour N, Nikoonezhad M,
Behboudi H, Mehdizadeh M, Kadkhoda D, Hajifathali A and Mosaffa N:
Adjuvant therapy with zinc supplementation; anti-inflammatory and
anti-oxidative role in multiple myeloma patients receiving
autologous hematopoietic stem cell transplantation: A randomized
controlled clinical trial. Biometals. 37:1609–1627. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Yu W, Cao D, Zhou H, Hu Y and Guo T:
PGC-1α is responsible for survival of multiple myeloma cells under
hyperglycemia and chemotherapy. Oncol Rep. 33:2086–2092. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Zhang Y, Cao X, Li W, Cui Z, Mao J, Yao R
and Liu L: ALKBH5 reverses romidepsin-mediated anti-multiple
myeloma activity via regulation of m6A modification of FOXM1.
Biochem Pharmacol. 239:1169982025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Quan L, Jia C, Guo Y, Chen Y, Wang X, Xu Q
and Zhang Y: HNRNPA2B1-mediated m6A modification of TLR4 mRNA
promotes progression of multiple myeloma. J Transl Med. 20:5372022.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Giallongo C, Tibullo D, Puglisi F, Barbato
A, Vicario N, Cambria D, Parrinello NL, Romano A, Conticello C,
Forte S, et al: Inhibition of TLR4 signaling affects mitochondrial
fitness and overcomes bortezomib resistance in myeloma plasma
cells. Cancers (Basel). 12:19992020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Bagratuni T, Sklirou AD, Kastritis E,
Liacos CI, Spilioti C, Eleutherakis-Papaiakovou E, Kanellias N,
Gavriatopoulou M, Terpos E, Trougakos IP and Dimopoulos MA:
Toll-like receptor 4 activation promotes multiple myeloma cell
growth and survival via suppression of the endoplasmic reticulum
stress factor chop. Sci Rep. 9:32452019. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Jiang S, Gao L, Li J, Zhang F, Zhang Y and
Liu J: N6-methyladenosine-modified circ_0000337 sustains bortezomib
resistance in multiple myeloma by regulating DNA repair. Front Cell
Dev Biol. 12:13832322024. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Wang G and Wu W, He D, Wang J, Kong H and
Wu W: N6-methyladenosine-mediated upregulation of H19 promotes
resistance to bortezomib by modulating the miR-184/CARM1 axis in
multiple myeloma. Clin Exp Med. 25:1022025. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Wang Z, He J, Bach DH, Huang YH, Li Z, Liu
H, Lin P and Yang J: Induction of m6A methylation in adipocyte
exosomal LncRNAs mediates myeloma drug resistance. J Exp Clin
Cancer Res. 41:42022. View Article : Google Scholar
|
|
172
|
Sun X, Zhou Y, Zhu W and Chen H: Research
progress on N6-methyladenosine and non-coding RNA in multiple
myeloma. Discov Oncol. 16:6152025. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Huang Y, Xia W, Dong Z and Yang CG:
Chemical inhibitors targeting the oncogenic m6A Modifying Proteins.
Acc Chem Res. 56:3010–3022. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
He B, Hu Y, Wu Y, Wang C, Gao L, Gong C,
Li Z, Gao N, Yang H, Xiao Y and Yang S: Helicobacter pylori CagA
elevates FTO to induce gastric cancer progression via a
'hit-and-run' paradigm. Cancer Commun (Lond). 45:608–631. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Xiao L, Li X, Mu Z, Zhou J, Zhou P, Xie C
and Jiang S: FTO inhibition enhances the antitumor effect of
temozolomide by targeting MYC-miR-155/23a cluster-MXI1 feedback
circuit in glioma. Cancer Res. 80:3945–3958. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Xu Y, Zhou J, Li L, Yang W, Zhang Z, Zhang
K, Ma K, Xie H, Zhang Z, Cai L, et al: FTO-mediated autophagy
promotes progression of clear cell renal cell carcinoma via
regulating SIK2 mRNA stability. Int J Biol Sci. 18:5943–5962. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Jiang L, Liang R, Luo Q, Chen Z and Song
G: Targeting FTO suppresses hepatocellular carcinoma by inhibiting
ERBB3 and TUBB4A expression. Biochem Pharmacol. 226:1163752024.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Zhang J, Li G, Wu R, Shi L, Tian C, Jiang
H, Che H, Jiang Y, Jin Z, Yu R, et al: The m6A RNA demethylase FTO
promotes radioresistance and stemness maintenance of glioma stem
cells. Cell Signal. 132:1117822025. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Yang Q and Al-Hendy A: The functional role
and regulatory mechanism of FTO m6A RNA demethylase in human
uterine leiomyosarcoma. Int J Mol Sci. 24:79572023. View Article : Google Scholar
|
|
180
|
Su R, Dong L, Li Y, Gao M, Han L,
Wunderlich M, Deng X, Li H, Huang Y, Gao L, et al: Targeting FTO
suppresses cancer stem cell maintenance and immune evasion. Cancer
Cell. 38:79–96.e11. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Huff S, Kummetha IR, Zhang L, Wang L, Bray
W, Yin J, Kelley V, Wang Y and Rana TM: Rational design and
optimization of m6A-RNA Demethylase FTO inhibitors as anticancer
agents. J Med Chem. 65:10920–10937. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Q,
Wang Y, Zhao H, Gao C, Zhang S, et al: Identification of entacapone
as a chemical inhibitor of FTO mediating metabolic regulation
through FOXO1. Sci Transl Med. 11:eaau71162019. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Ramedani F, Jafari SM, Saghaeian Jazi M,
Mohammadi Z and Asadi J: Anti-cancer effect of entacaponeon
esophageal cancer cells via apoptosis induction and cell cycle
modulation. Cancer Rep (Hoboken). 6:e17592023.
|
|
184
|
Yankova E, Blackaby W, Albertella M, Rak
J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D,
Hendrick AG, et al: Small-molecule inhibition of METTL3 as a
strategy against myeloid leukaemia. Nature. 593:597–601. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Sun Y, Shen W, Hu S, Lyu Q, Wang Q, Wei T,
Zhu W and Zhang J: METTL3 promotes chemoresistance in small cell
lung cancer by inducing mitophagy. J Exp Clin Cancer Res.
42:652023. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Jin X, Lv Y, Bie F, Duan J, Ma C, Dai M,
Chen J, Lu L, Xu S, Zhou J, et al: METTL3 confers oxaliplatin
resistance through the activation of G6PD-enhanced pentose
phosphate pathway in hepatocellular carcinoma. Cell Death Differ.
32:466–479. 2025. View Article : Google Scholar :
|
|
187
|
Hao S, Sun H, Sun H, Zhang B, Ji K, Liu P,
Nie F and Han W: STM2457 Inhibits the invasion and metastasis of
pancreatic cancer by down-regulating BRAF-Activated Noncoding RNA
N6-Methyladenosine modification. Curr Issues Mol Biol.
45:8852–8863. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Tang H, Zhang R and Zhang A:
Small-molecule inhibitors targeting RNA m(6)A modifiers for cancer
therapeutics : Latest advances and future perspectives. J Med Chem.
68:18114–18142. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Du Y, Yuan Y, Xu L, Zhao F, Wang W, Xu Y
and Tian X: Discovery of METTL3 small molecule inhibitors by
virtual screening of natural products. Front Pharmacol.
13:8781352022. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Dolbois A, Bedi RK, Bochenkova E, Müller
A, Moroz-Omori EV, Huang D and Caflisch A:
1,4,9-Triazaspiro[5.5]undecan-2-one derivatives as potent and
selective METTL3 Inhibitors. J Med Chem. 64:127382021. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Li J and Gregory RI: Mining for METTL3
inhibitors to suppress cancer. Nat Struct Mol Biol. 28:460–462.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Malacrida A, Di Domizio A, Bentivegna A,
Cislaghi G, Messuti E, Tabano SM, Giussani C, Zuliani V, Rivara M
and Nicolini G: MV1035 overcomes temozolomide resistance in
patient-derived glioblastoma stem cell lines. Biology (Basel).
11:702022.PubMed/NCBI
|
|
193
|
Li N, Kang Y, Wang L, Huff S, Tang R, Hui
H, Agrawal K, Gonzalez GM, Wang Y, Patel SP and Rana TM: ALKBH5
regulates anti-PD-1 therapy response by modulating lactate and
suppressive immune cell accumulation in tumor microenvironment.
Proc Natl Acad Sci USA. 117:20159–20170. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Tang W, Xu N, Zhou J, He Z, Lenahan C,
Wang C, Ji H, Liu B, Zou Y, Zeng H and Guo H: ALKBH5 promotes
PD-L1-mediated immune escape through m6A modification of ZDHHC3 in
glioma. Cell Death Discov. 8:4972022. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Schott A, Simon T, Müller S, Rausch A,
Busch B, Glaß M, Misiak D, Dipto M, Elrewany H, Peters LM, et al:
The IGF2BP1 oncogene is a druggable m6A-dependent enhancer of
YAP1-driven gene expression in ovarian cancer. NAR Cancer.
7:zcaf0062025. View Article : Google Scholar
|
|
196
|
Singh A, Singh V, Wallis N, Abis G,
Oberman F, Wood T, Dhamdhere M, Gershon T, Ramos A, Yisraeli J, et
al: Development of a specific and potent IGF2BP1 inhibitor: A
promising therapeutic agent for IGF2BP1-expressing cancers. Eur J
Med Chem. 263:1159402024. View Article : Google Scholar
|
|
197
|
Feng P, Chen D, Wang X, Li Y, Li Z, Li B,
Zhang Y, Li W, Zhang J, Ye J, et al: Inhibition of the m6A reader
IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia.
Leukemia. 36:2180–2188. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Qian L, Ji Z, Mei L and Zhao J: IGF2BP2
promotes lung adenocarcinoma progression by regulating LOX1 and
tumor-associated neutrophils. Immunol Res. 73:162024. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Du W, Huang Y, Chen X, Deng Y, Sun Y, Yang
H, Shi Q, Wu F, Liu G, Huang H, et al: Discovery of a PROTAC
degrader for METTL3-METTL14 complex. Cell Chem Biol.
31:177–183.e17. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Rauff R, Abedeera SM, Schmocker S, Xie J
and Abeysirigunawardena SC: Peptides targeting RNA m6A methylations
influence the viability of cancer cells. ChemMedChem.
18:e2022005492023. View Article : Google Scholar
|
|
201
|
Li Z, Feng Y, Han H, Jiang X, Chen W, Ma
X, Mei Y, Yuan D, Zhang D and Shi J: A stapled peptide inhibitor
targeting the binding interface of N6-Adenosine-Methyltransferase
Subunits METTL3 and METTL14 for cancer therapy. Angew Chem Int Ed
Engl. 63:e2024026112024. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Huang CS, Zhu YQ, Xu QC, Chen S, Huang Y,
Zhao G, Ni X, Liu B, Zhao W and Yin XY: YTHDF2 promotes
intrahepatic cholangiocarcinoma progression and desensitises
cisplatin treatment by increasing CDKN1B mRNA degradation. Clin
Transl Med. 12:e8482022. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Hua Z, Gong B and Li Z: Silencing YTHDF2
induces apoptosis of neuroblastoma cells in a cell line-dependent
manner via regulating the expression of DLK1. Mol Neurobiol.
62:8121–8134. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Paris J, Morgan M, Campos J, Spencer GJ,
Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA,
Sepulveda C, et al: Targeting the RNA m6A Reader YTHDF2 selectively
compromises cancer stem cells in acute myeloid leukemia. Cell Stem
Cell. 25:137–148.e6. 2019. View Article : Google Scholar
|
|
205
|
Bao Y, Zhai J, Chen H, Wong CC, Liang C,
Ding Y, Huang D, Gou H, Chen D, Pan Y, et al: Targeting m 6 A
reader YTHDF1 augments antitumour immunity and boosts anti-PD-1
efficacy in colorectal cancer. Gut. 72:1497–1509. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Wang L, Zhu L, Liang C, Huang X, Liu Z,
Huo J, Zhang Y, Zhang Y, Chen L, Xu H, et al: Targeting
N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor
immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J Hepatol.
79:1185–1200. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Xie LJ, Yang XT, Wang RL, Cheng HP, Li ZY,
Liu L, Mao L, Wang M and Cheng L: Identification of flavin
mononucleotide as a cell-active artificial N6-Methyladenosine RNA
Demethylase. Angew Chem Int Ed Engl. 58:5028–5032. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Wilson C, Chen PJ, Miao Z and Liu DR:
Programmable m6A modification of cellular RNAs with a
Cas13-directed methyltransferase. Nat Biotechnol. 38:1431–1440.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Li J, Chen Z, Chen F, Xie G, Ling Y, Peng
Y, Lin Y, Luo N, Chiang CM and Wang H: Targeted mRNA demethylation
using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids
Res. 48:5684–5694. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Sun Y, Gong W and Zhang S: METTL3 promotes
colorectal cancer progression through activating JAK1/STAT3
signaling pathway. Cell Death Dis. 14:7652023. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Sun X, Wang DO and Wang J: Targeted
manipulation of m6A RNA modification through CRISPR-Cas-based
strategies. Methods. 203:56–61. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Nar R, Wu Z, Li Y, Smith A, Zhang Y, Wang
J, Yu F, Gao S, Yu C, Huo Z, et al: Targeting METTL3 protein by
proteolysis-targeting chimeras: A novel therapeutic approach for
acute myeloid leukemia. Genes Dis. 12:1014522024. View Article : Google Scholar
|
|
213
|
Kim S, Hwang I, Kim YK, Kim DS, Choi YJ
and Jeung EB: Treatment of dexamethasone and lenalidomide-resistant
multiple myeloma via RAD51 degradation using PROTAC and synergistic
effects with chemotherapy. J Physiol Pharmacol. 75:2024.
|
|
214
|
Li Y, Zhu S, Chen Y, Ma Q, Kan D, Yu W,
Zhang B, Chen X, Wei W, Shao Y, et al: Post-transcriptional
modification of m6A methylase METTL3 regulates ERK-induced
androgen-deprived treatment resistance prostate cancer. Cell Death
Dis. 14:2892023. View Article : Google Scholar
|
|
215
|
Kamiya T, Oshima M, Koide S,
Nakajima-Takagi Y, Aoyama K, Itokawa N, Yamashita M, Doki N,
Kataoka K and Iwama A: Unraveling the heterogeneity of multiple
myeloma cells by single-cell RNA sequencing analysis. Blood.
140:9939–9940. 2022. View Article : Google Scholar
|
|
216
|
Wang Y, Peng Y, Yang C, Xiong D, Wang Z,
Peng H, Wu X, Xiao X and Liu J: Single-cell sequencing analysis of
multiple myeloma heterogeneity and identification of new
theranostic targets. Cell Death Dis. 15:6722024. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Liu C, Liang H, Wan AH, Xiao M, Sun L, Yu
Y, Yan S, Deng Y, Liu R, Fang J, et al: Decoding the m6A
epitranscriptomic landscape for biotechnological applications using
a direct RNA sequencing approach. Nat Commun. 16:7982025.
View Article : Google Scholar
|
|
218
|
Jin R, Zou Q and Luo X: From detection to
prediction: Advances in m6A methylation analysis through machine
learning and deep learning with implications in cancer. Int J Mol
Sci. 26:67012025. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Li Q, Liu J, Guo L, Zhang Y, Chen Y, Liu
H, Cheng H, Deng L, Qiu J, Zhang K, et al: Decoding the interplay
between m6A modification and stress granule stability by live-cell
imaging. Sci Adv. 10:eadp56892024. View Article : Google Scholar
|
|
220
|
Xi JF, Liu BD, Tang GR, Ren ZH, Chen HX,
Lan YL, Yin F, Li Z, Cheng WS, Wang J, et al: m6A modification
regulates cell proliferation via reprogramming the balance between
glycolysis and pentose phosphate pathway. Commun Biol. 8:4962025.
View Article : Google Scholar
|
|
221
|
Zhang L, Wei J, Zou Z and He C: RNA
modification systems as therapeutic targets. Nat Rev Drug Discov.
25:59–78. 2026. View Article : Google Scholar
|