Introduction
The anticancer efficacy of tumor therapeutics is
impaired by inherent or acquired tumor cell resistance. In
addition, adverse effects on normal tissue limit the maximum
possible cumulative dose of the anticancer drug that can be
applied. Against this background, alternative and well-tolerated
therapeutic options are needed. Anthracyclines are conventional
(that is, genotoxic) anticancer therapeutics (cAT) which are used
for the treatment of numerous malignancies, including hematological
disorders, sarcomas and breast cancer (1). They impair the genetic integrity and
thus the malignancy of tumor cells by inhibiting topoisomerase II
(Topo II), which is essential for DNA replication. As a consequence
of Topo II poisoning, DNA double-strand breaks (DSB) are formed,
which effectively trigger mechanisms of cell death (2,3).
DNA intercalation, inhibition of DNA helicases, disruption of
mitochondrial functions and formation of reactive oxygen species
(ROS) (4) also contribute to the
antitumor effect of anthracyclines. Tumor cell resistance
mechanisms are often agent-specific and were classified into pre-,
on- and post-target mechanisms (5). Pre-target resistance mechanisms,
such as mechanisms of transport or detoxification, eventually
reduce the level of drug-induced primary DNA damage and, in
consequence, DNA damage-triggered cell death. With Doxo,
overexpression of the drug exporter protein p-glycoprotein
(P-gp/MDR1) is considered as an important mechanism of acquired
Doxo resistance (6,7). However, as cells express a variety
of different transporters (importers and exporters) for Doxo and
other cAT (7,8), the outcome of anticancer drug
treatment is ultimately determined by the combined
activity/expression of multiple importers and exporters. Against
this background and having in mind that cellular mechanisms
contributing to acquired drug resistance in a genetically
heterogenous tumor cell population are probably manifold, it would
be desirable to effectively target transport dependent (that is,
pre-target mechanisms) and/or transport-independent (that is, on-
or post-target) mechanisms that contribute to drug resistance.
Since cAT-induced DNA damage induces a complex stress response
termed DNA damage response (DDR), which regulates mechanisms of
cell cycle progression, DNA repair and, finally, survival- and
death-related pathways (3,9),
factors of the DDR are considered as particular promising
pharmacological targets to overcome inherent or acquired tumor cell
resistance (10-12).
The DDR becomes fine-tuned by the PI3-like kinase
Ataxia telangiectasia mutated (ATM) and the ATM and Rad3-related
kinase (ATR) (13-15), with ATM being of particular
relevance for the regulation of DSB-induced stress responses and
ATR for replicative stress responses (16-18). By coordinating the activation of
cell cycle checkpoints, DNA repair and cell death-related pathways,
the ATM/ATR-regulated network represents the major molecular switch
that defines the balance between survival and death (3). In line with this, ATM- and
ATR-regulated pathways contribute to tumor cell resistance
(19) and DDR modulating
compounds have been proved as useful to improve anticancer therapy
(20-23). In the case of oncogene-driven
replicative stress, tumor cells are often particular sensitive to
compounds that impair a coordinated replicative stress response,
thereby eventually enforcing replication fork collapse and death
(21,22,24-27). Alterations in DNA repair provides
another Achilles' heel for personalized anticancer therapy as
reflected by synthetic lethality (28-30). Here, defective DSB repair by
homologous recombination, for example due to hereditary breast
cancer associated gene 1/2 deficiency (BRCAness), predicts the
hypersensitivity of malignant cells to inhibition of PARP-related
backup DNA repair pathways by PARP inhibitors (such as olaparib or
niraparib) (31,32).
The present study used ovarian carcinoma cells
(A2780ADR) as in vitro model of acquired Doxo resistance
(33). It comparatively
characterized i) the stress responses of wild-type A2780 and drug
resistant A2780ADR cells to Doxo treatment, ii) the
cross-resistance of A2780ADR variants to other anticancer drugs
(Eto and CisPt) as well as to a set of candidate compounds
interfering with DDR/DNA repair, RAC1 GTPase signaling or drug
transport and iii) the outcome of a combined treatment of A2780ADR
cells with Doxo plus the aforementioned inhibitors. Thereby, the
present study aimed to identify compounds that are particularly
effective to overcome acquired Doxo resistance of malignant
cells.
Materials and methods
Materials
Chemicals were obtained from the following
providers: Entinostat (MS-275) was obtained from Selleck Chemicals,
Doxo from STADA Consumer Health & STADAPHARM GmbH, etoposide,
Ehop16, prexasertib (AZD-7762) and dexrazoxane were from
MilliporeSigma, cisplatin from Accord Healthcare GmbH, olaparib
from APeXBIO Technology LLC, niraparib from MedChemExpress, EHT1864
was purchased from Tocris Bioscience, rabusertib (LY2603618) and
ricolinostat (ACY-1215) from MedChemExpress and verapamil (Ver)
from Thermo Fisher Scientific, Inc. The following primary
antibodies were used: Copper transporting ATPase (ATP7A),
extracellular regulated kinase 2 (ERK2), phosphorylated (p)-histone
H3 (Ser10) from Thermo Fisher Scientific Inc., cleaved caspase-7
(Asp198), p-Chk1 (Ser 345), cyclin B1, galactosidase β (E2U2I),
GAPDH (14C10), MDR1/ABCB1 (D3H1Q), p-P53 (S15), PARP,
TopBP1(D8G4L), topoisomerase IIa (D10G9), 53BP1 and Ki67 were from
Cell Signaling Technology Inc., pChk2 (T68) [Y171], copper uptake
protein 1 (CTR1/SLC31A1) [EPR7936] and Rad51 from Abcam, γH2AX (Ser
139) clone JBW301, p-KAP-1 (S824) and p-RPA32 (S4/S8) from Bethyl
Laboratories Inc., organic cation transporter-2 (OCT2) from Biozol
Diagnostics Vertrieb GmbH, p16 (F-12) and p21 (C-19) from Santa
Cruz Biotechnology, Inc. As secondary antibodies, horseradish
peroxidase-conjugated secondary antibodies goat anti-mouse IgG and
mouse anti-rabbit IgG were used (Rockland Immunochemicals
Inc.).
Cell culture and treatment of cells
Parental A2780 ovarian carcinoma cells (A2780) as
well as a doxorubicin resistant variant (A2780ADR) were from the
European Collection of Authenticated Cell Cultures and were
cultured in RPMI-1640 medium (MilliporeSigma) containing 10% fetal
calf serum, 1% glutamine and 1% Pen/Strep at 37°C in a humidified
atmosphere containing 5% CO2. Cells were authenticated
by STR profiling during the last three years. All experiments were
performed with mycoplasma free cells. Immortalized HL-1
cardiomyocytes were provided by Professor W.C. Claycomb (Louisiana
State University, New Orleans, LA, USA) (34) and were grown on gelatin (2
mg/ml)/fibronectin (1 mg/ml) (Sigma Aldrich; Merck KGaA) coated
dishes and maintained in Claycomb medium, supplemented with 10% FBS
and 100 μM norepinephrine (Sigma Aldrich; Merck KGaA). Mouse
embryonic stem cells (ESC; LF2) were isolated from the mouse strain
129J (35) and were from
Professor A. Smith (University of Oxford, UK). They were cultivated
under feeder-free conditions on 0,1% gelatine-coating using
knock-out Dulbecco's Modified Eagle's Medium (KO-DMEM) (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with knock-out serum
replacement (15%) (Gibco; Thermo Fisher Scientific, Inc.),
penicillin/streptomycin (1%), glutamax (1%), β-mercaptoethanol
(5×10−5 M) (Invitrogen; Thermo Fisher Scientific, Inc.)
and leukemia inhibitory factor (LIF; MilliporeSigma) (1,000 U/ml)
at 37°C in an atmosphere containing 5% CO2. b4-hiPSC
were generated from human foreskin fibroblasts as previously
described (36). Cells were
cultured on plates coated with reduced growth factor basement
membrane matrix (Gibco; Thermo Fisher Scientific, Inc.) in StemMacs
medium (Miltenyi Biotec GmbH) supplemented with 10 mM Y-27632
dihydrochloride (MilliporeSigma).
Determination of cell viability
Cell viability was determined using the Alamar blue
assay (37). Viable cells are
characterized by an effective mitochondrial metabolization of the
non-fluorescent dye resazurin (Sigma-Aldrich; Merck KGaA) to
fluorescent resorufin (excitation: 535 nm, emission: 590 nm).
Relative viability in the untreated control was set to 100%. If not
stated otherwise, data are shown as the mean ± standard deviation
(SD) of ≥3 independent experiments, each performed in biological
quadruplicates. The combination index (CI) was determined (38) for the calculation of additive (CI
>0.8<1.2), synergistic (CI≤0.8) or antagonistic (CI≥1.2) drug
interactions in the co-treatment experiments.
Analysis of doxorubicin import and
export
Doxo import and export were measured by exploiting
the inherent red fluorescence of Doxo as previously described
(39). Briefly, following 2 h of
pulse-treatment with different Doxo concentrations, the
fluorescence of the cells was measured as a surrogate marker of
drug uptake by flow cytometry (excitation: 450 nm, emission: 560
nm). After a post-incubation period of up to 6 h in the absence of
Doxo, the residual intracellular fluorescence was again measured by
flow cytometry. The time dependent decrease in Doxo fluorescence
was calculated as a surrogate marker of Doxo export.
Cell cycle analysis by flow
cytometry
For flow cytometry-based analysis of cell cycle
distribution, cells were trypsinized and combined with floating
cells present in the medium. Cells were pelleted (1,000 × g, 10
min, 4°C) and suspended in PBS. DNase-free RNase A (SERVA
Electrophoresis GmbH) was added (2 μg/ml, 1 h at room
temperature). DNA was stained with propidium iodide (PI)
(Sigma-Aldrich; Merck KGaA) for 20 min in the dark. Cell number was
adjusted to 106 cells/ml with PBS and analysis was
performed using BD Accuri C6 flow cytometer (BD Biosciences).
Analysis of apoptosis and senescence
Cell death by apoptosis was monitored by flow
cytometry-based quantitation of the subG1 fraction,
which represents the apoptotic cell fraction. In addition, cleavage
of PARP protein and pro-caspase-7 was monitored by western
blotting. To monitor senescence β-GAL expression was analyzed by
western blotting.
Analysis of proliferation
To monitor proliferation, the incorporation of the
nucleoside analogue 5-ethynyl-2'-deoxyuridine (EdU) into S-phase
cells as well as the percentage of pH3 (Ser10) positive cells
(mitotic index) and Ki-67 positive cells were determined. To this
end, cells were seeded on cover slips and cultivated for the
indicated time period. EdU-incorporation was analyzed using the
EdU-Click 488 Kit (baseclick GmbH), which is based on a
pulse-labeling of S-phase cells with 10 μM EdU according to
the manufacturer's protocol. To determine the mitotic index, cells
were fixed with 4% formaldehyde/PBS followed by incubation PBS
−0.3% TritonX-100 (5 min; room temperature). After blockage of
unspecific binding (5% BSA in 0.3% Triton X-100/PBS (1 h; room
temperature), anti-Ser10 phosphorylated histone H3 antibody (pH3;
Thermo Fisher Scientific Inc.; dilution 1:1,000; 16 h; 4°C) and
Ki-67 antibody (Cell Signaling Technology Inc.; dilution 1:500; 16
h; 4°C) were added. Incubation with Alexa Fluor® 488
labeled goat anti-rabbit and Alexa Fluor 555 labeled goat
anti-mouse secondary antibody was performed for 120 min at room
temperature. pH3- and Ki-67-positive cells were counterstained with
DAPI-containing Vectashield (Vector Laboratories, Inc.) and
analyzed by Olympus BX43 microscope (40× objective) (Olympus
Corporation).
Immunocytochemistry-based analysis of DSB
formation
The frequency of nuclear foci formed by S139
phosphorylated H2AX (γH2AX foci) is a commonly used surrogate
marker of DSBs (40,41) and was assayed by
immunocytochemistry-based method. The appearance of nuclear 53BP1
foci, which is another marker of DSBs (42,43), was also determined by
immunocytochemistry. Upon treatment of cells grown on cover slips,
cells were fixed with 4% formaldehyde in phosphate-buffered saline
(PBS; Merck KGaA; 15 min; room temperature) followed by
permeabilization by incubation in PBS −0.3% TritonX-100 (5 min;
room temperature). After blocking [1 h; room temperature; blocking
solution: 5% BSA (Merck KGaA) in PBS/0.3% Triton X-100
(Sigma-Aldrich; Merck KGaA)], incubation with primary γH2AX
antibody (1:2,000; cat. no. 07-727; MilliporeSigma) and 53BP1
antibody (1:500; cat. no. 4937S; Invitrogen; Thermo Fisher
Scientific, Inc.) was performed overnight (4°C), followed by
incubation with the secondary fluorescence-labelled antibody [Alexa
Fluor 488 goat-anti-rabbit IgG (H+L); cat. no. A11008; Invitrogen;
Thermo Fisher Scientific, Inc.] (1:500, 2 h; room temperature, in
the dark). Cells were mounted in Vectashield Mounting medium
(anti-fading; Vector Laboratories, Inc.) containing the blue
fluorescent DNA stain 4',6-Diamidin-2-phenylindol (DAPI; cat. no.
H-1200; Biozol Diagnostics Vertrieb GmbH) at room temperature and
the number of nuclear γH2AX foci and 53BP1 foci was scored using an
Olympus BX43 fluorescence microscope (Olympus Corporation). Only
nuclei with distinct foci were evaluated. γH2AX foci pan-stained
nuclei, which are indicative of apoptotic cells, were excluded from
the analyses. If not stated otherwise, data are shown as the mean ±
SD from three independent experiments with each ≥50 nuclei analyzed
per experimental condition.
Western blotting
The activation of the DDR was investigated by
western blotting using total cell extracts obtained by lysing an
equal number of cells in 150 μl RIPA buffer (20 min on ice).
RCDC-Protein Assay (cat. no. 500-0120; Bio-Rad Laboratories, Inc.)
was used for protein determination. After sonication (6 KHz; 5×2
sec; on ice) (EpiShear Probe sonicator; Active Motif, Inc.) and
centrifugation (10,000 × g; 4°C; 10 min), Roti®-Load
buffer (5 min; room temperature) was added to the supernatant and
proteins were denatured by heating (5 min, 95°C). Afterwards, 20
μg protein was loaded per lane and proteins were separated
by SDS-PAGE (6 or 12.5% gel) and transferred onto a nitrocellulose
membrane (Cytiva) via the Protean Mini Cell System. After blocking
[5% non-fat milk in TBS/0.1% Tween 20 (Merck KGaA; 2 h; room
temperature)], the membrane was incubated with the corresponding
primary antibody (1:1,000; overnight; 4°C). The following primary
antibodies were used: ATP7A (cat. no. PA5-103110; Invitrogen;
Thermo Fisher Scientific; Inc.), Caspase-7 cleaved (Asp198; cat.
no. 9491S; Cell Signaling Technology, Inc.), Chk1phospho (Ser345;
cat. no. 2341, Cell Signaling Technology, Inc.), Chk2phosphoT68
(Y171; cat. no. ab32148; Abcam), CTR1/SLC31A1 (EPR7936; cat. no.
ab129067; Abcam), Cyclin B1 (cat. no. 4138, Cell Signaling
Technology, Inc.), ERK2 [PA5-32396, Invitrogen/Thermo Fisher
Scientific; Carlsbad, USA], Galactosidase beta (E2U2I) (cat. no.
27198, Cell Signaling Technology, Inc.), GAPDH (14C10; cat. no.
2118S; Cell Signaling Technology, Inc.), H2AX phospho (Ser139,
cloneJBW301; cat. no. 05-636, Merck KGaA), MDR1/ABCB1 (D3H1Q; cat.
no. 12683; Cell Signaling Technology, Inc.), OCT2 (cat. no.
MBS9600162, Biozol Diagnostics Vertrieb GmbH), P16 (F-12; cat. no.
sc-1661; Santa Cruz Biotechnology, Inc.), p21 (C-19; cat. no.
sc-397; Santa Cruz Biotechnology, Inc.), P53 phospho (S15; cat. no.
9284S; Cell Signaling Technology, Inc.), PARP (cat. no. 9542S, Cell
Signaling Technology, Inc.), Rad51 (cat. no. ab63801; Abcam), RPA32
phospho (S4/S8; cat. no. ICH-00422; Bethyl Laboratories Inc.),
TopBP1 (D8G4L; cat. no. 14342, Cell Signaling Technology, Inc.),
Topoisomerase II alpha (D10G9; cat. no. 12286, Cell Signaling
Technology, Inc.). After washing (TBS/0.1% Tween 20), the secondary
(peroxidase-conjugated) antibody was added (1:2,000; 2 h; room
temperature) [Anti-mouse IgG (H&L; goat) Antibody Peroxidase
Conjugated (cat. no. 610-1302, Rockland Immunochemicals, Inc.) or
Anti-rabbit IgG (H&L; goat) Antibody Peroxidase Conjugated
(cat. no. 611-1302, Rockland Immunochemicals, Inc.] The ChemiDoc
Touch Imaging System (Bio-Rad Laboratories, Inc.) was used for
visualization of the bound antibodies. Image Lab 6.1.0 build 7
(Bio-Rad Laboratories, Inc.) was used for densitometrical analyses.
Changes in protein expression of factors of interest were
identified by normalization to the expression of a housekeeping
protein (such as ERK2).
Reverse transcription-quantitative (RT-q)
PCR
Total RNA was purified from up to 5×106
cells per preparation using the RNeasy Mini Kit (Qiagen, Hilden,
Germany), followed by reverse transcriptase reaction with High
Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific,
Inc.). RNA extraction, cDNA synthesis and qPCR were performed
according the manufacturer's protocols. For each PCR reaction 40 ng
of cDNA and 0.25 μM of the corresponding primers (Eurofins
MWG Synthesis GmbH) were used. Quantitative real-time PCR analysis
was performed in triplicates using the SensiMix SYBR Hi-ROX Kit
(Meridian Bioscience, Inc.) and a CFX96 Real-Time System (Bio-Rad
Laboratories, Inc.) with the Bio-Rad CFX Manager 3.1 software. PCR
runs (35-40 cycles) were performed as follows: 95°C, 10 min; 95°C,
15 sec; 60°C, 30 sec; 72°C, 40 sec; 72°C, 10 min. Melting curves
were recorded to ensure the specificity of the amplification
reaction. mRNA levels were normalized by geometric averaging using
β-actin and GAPDH as internal control genes as reported (44). Unless stated otherwise, relative
mRNA expression of untreated control cells was set to 1.0. Primer
sequences used for RT-qPCR analyses are depicted in Table SI.
Statistical analyses
Student's t-test and the One-way ANOVA with
Dunnett's post-hoc test were employed to confirm statistically
significant differences between different experimental groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Doxorubicin-resistant (A2780ADR) ovarian
cancer cells are cross-resistant to the anticancer drugs etoposide
and cisplatin
The present study employed parental ovarian cancer
cells (A2780) and thereof derived Doxo resistant variants
(A2780ADR). Comparative analysis of their viability following Doxo
treatment for 72 h revealed IC50 of 0.04 and 0.38
μM for A2780 and A2780ADR, respectively (Fig. 1A). A2780ADR cells revealed a
profound cross-resistance to the Topo II inhibitor etoposide (Eto;
IC50 A2780: 0.14 μM; IC50 A2780ADR:
0.92 μM; Fig. 1B), while
being only weakly cross-resistant to the platinating agent
cisplatin (CisPt; IC50 A2780: 0.62 μM;
IC50 A2780ADR: 1.91 μM; Fig. 1C). Based on their corresponding
IC50, A2780ADR cells are characterized by a ~10, ~7 and
~3-fold higher resistance to Doxo, Eto and CisPt, respectively, as
compared with the parental A2780 cells. Analyzing viability
following a 24 h treatment period, no major differences were
observed as concluded from the calculation of the corresponding
IC50 values (Fig.
S1).
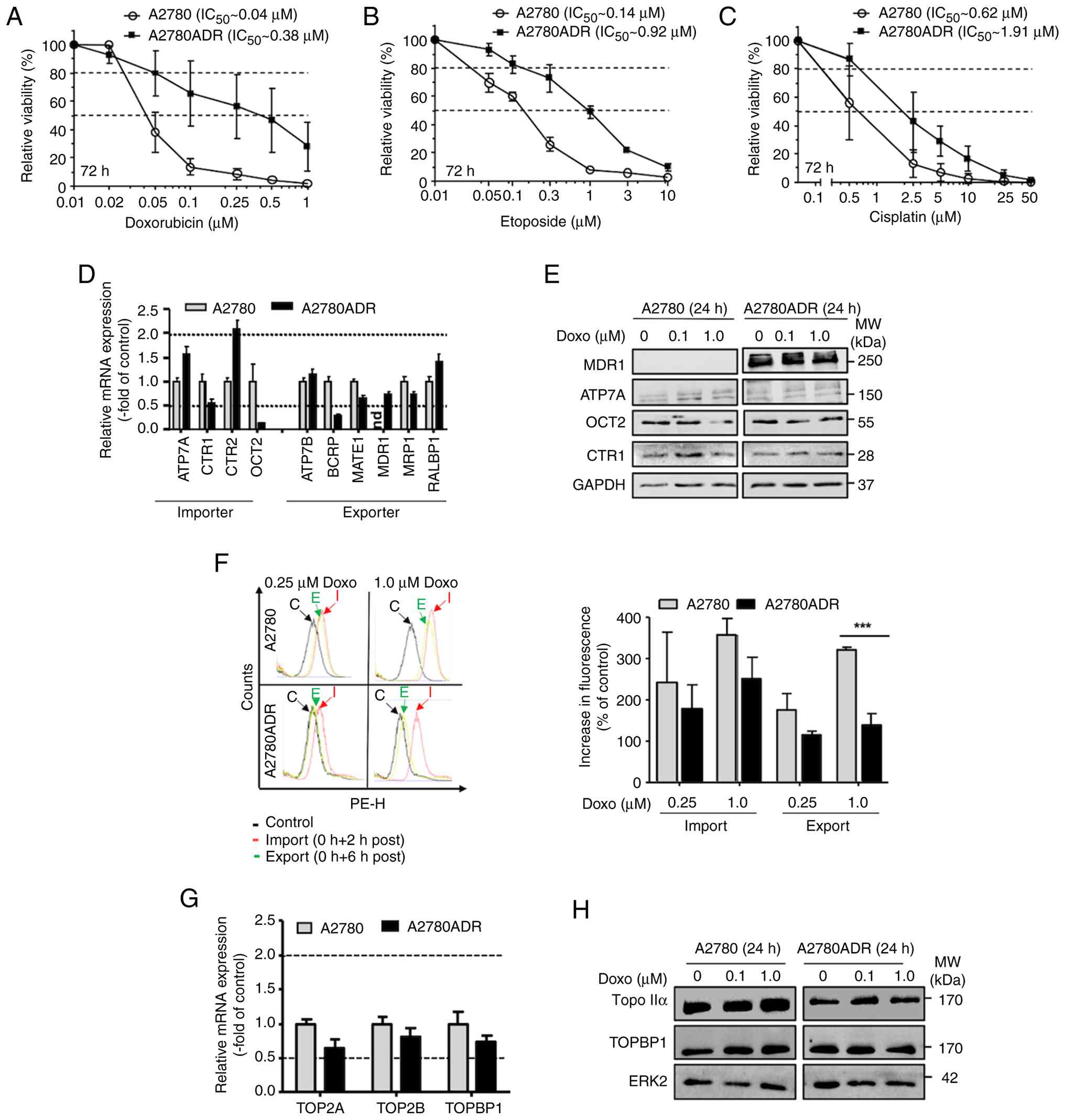 | Figure 1Comparative analysis of the response
of A2780 and A2780ADR cells to treatment with anticancer drugs
(Doxo, Eto and CisPt) and effect of mechanisms of drug transport.
Logarithmically growing parental A2780 and A2780ADR variant cells
were treated with the anticancer drugs (A) Doxo, (B) Eto and (C)
CisPt at the indicated concentrations. At 72 h after drug addition,
viability was monitored by use of the AlamarBlue assay as described
in methods. Data shown are the mean ± SD from three independent
experiments each performed in biological quadruplicates (n=3; n=4).
Dashed lines indicate inhibitory concentrations (IC20
and IC50). For viability data (IC50) after 24
and 72 h of treatment also see Fig.
S1 and Table SII. (D)
Comparative analysis of the mRNA expression of selected
transporters in A2780 and A2780ADR. Data shown are the mean ± SD
from triplicate determinations. mRNA expression of transporters was
normalized to GAPDH mRNA levels and set to 1.0 in the parental
A2780 cells. The dashed lines indicate changes in mRNA levels of
≥2.0 and ≤0.5, which are considered as biologically relevant. (E)
Comparative analysis of the protein expression of representative
drug transporters under basal situation and after 24 h treatment
with Doxo (0.1 and 1.0 μM). Data shown are from a
representative western blotting using ERK2 protein levels as
loading control. Data obtained after 72 h are presented in Fig. S2 (left panel). (F) Intracellular
Doxo fluorescence was measured by flow cytometry-based methods
after 2 h Doxo pulse-treatment (0.25 and 1.0 μM) and was
taken as indicative of drug import. To measure drug export, Doxo
pulse-treated cells were post-incubated for 6 h in the absence of
the drug before fluorescence was monitored. Data shown in the left
panel are representative results obtained from flow cytometry
analyses. C, control; I, import, E, export. The histogram in the
right panel depicts quantitative data obtained from n=3 independent
experiments each performed in biological triplicates (n=3).
***P≤0.001. (G) Analysis of basal mRNA expression of
topoisomerase II isoforms TOP2A, TOP2B and TopBP1. Data shown are
the mean ± SD from triplicate determinations. Relative mRNA level
in A2780 cells was set to 1.0. The dashed lines indicate changes in
mRNA levels of ≥2.0 and ≤0.5, which are considered as biologically
relevant. (H) Comparative analysis of the protein expression of
TOP2A and TopBP1 under basal situation and after 24 h treatment
with Doxo (0.1 μM, 1.0 μM). Data shown are from a
representative western blotting using ERK2 as protein loading
control. Data obtained after 72 h Dox treatment are presented in
Fig. S2 (right panel). Doxo,
doxorubicin; Eto, etoposide; CisPt, cisplatin; SD, standard
deviation; Nd, not detectable TOP2A, topoisomerase IIα; TOP2B,
topoisomerase IIβ; TOPBP1, topoisomerase binding protein 1; MDR1,
multi-drug resistance gene 1; ATP7A, copper transporting ATPase;
OCT2, organic cation transporter-2; CTR1, copper uptake protein 1;
Topo IIa, topoisomerase IIα; ERK2, extracellular regulated kinase
2. |
Doxorubicin-resistant (A2780ADR) ovarian
cancer cells are characterized by altered expression of drug
transporters and enhanced doxorubicin export
Altered drug transport, as mediated for instance by
p-glycoprotein, is one possible mechanism contributing to acquired
drug resistance of cancer cells (45,46). Analyzing the mRNA expression of
various drug importers and exporters, an elevated mRNA expression
of the importer CTR2 and a reduced mRNA expression of the exporter
BCRP was observed in Doxo resistant A2780ADR cells as compared with
the wild-type cells (Fig. 1D).
Notably, while the mRNA expression of MDR1 was clearly detectable
in A2780ADR cells, it was below detection limit in A2780 cells
(Fig. 1D). Lack of MDR1
expression in the parental cells and high expression in A2780ADR
was confirmed on the protein level (Figs. 1E and S2). To monitor the cells' activity of
drug import and export, the present study took advantage of the
inherent reddish fluorescence of Doxo and comparatively analyzed
alterations in the intracellular fluorescence of A2780 and A2780ADR
cells following Doxo treatment. The data show a similar increase in
fluorescence in both cell lines after 2 h of Doxo pulse-treatment
(Fig. 1F), indicating that both
cell lines have comparable Doxo uptake capacity (i.e. import).
However, analyzing the remaining intracellular Doxo concentration
after a subsequent 6 h post-incubation period in the absence of
Doxo, residual fluorescence was markedly lower (~50%) in A2780ADR
as compared with parental A2780 cells (Fig. 1F). This finding showed that
A2780ADR cells were characterized by an about twice as fast drug
export as A2780 parental cells, which is very probably due to their
elevated MDR1 expression as concluded from the results of the mRNA
and protein expression analyses. Thus, increased drug export
probably contributes to the high Doxo resistance of A2780ADR cells.
However, having in mind the ~10-fold higher Doxo resistance of
A2780ADR cells as compared with A2780 cells, it was hypothesized
that mechanisms other than just drug export additionally
contributed to their pronounced drug resistant phenotype. Since
Topo IIα and IIβ are relevant primary targets for the anticancer
efficacy of Doxo, we additionally investigated their mRNA and
protein expression. A2780 and A2780ADR cells revealed similar mRNA
levels of Topo IIα, Topo IIβ and Topoisomerase IIβ binding protein
(TopBP1; Fig. 1G). On the protein
level, A2780ADR were characterized by a reduced expression of Topo
IIα protein under basal situation and 24-72 h following Doxo
treatment (Figs. 1H and S2). Overall, these data indicated that
both alterations in drug transport catalyzed by MDR1 and the
protein expression of Topo IIα contributed to the strongly enhanced
Doxo resistance of A2780ADR cells as well as to their profound
cross-resistance to etoposide.
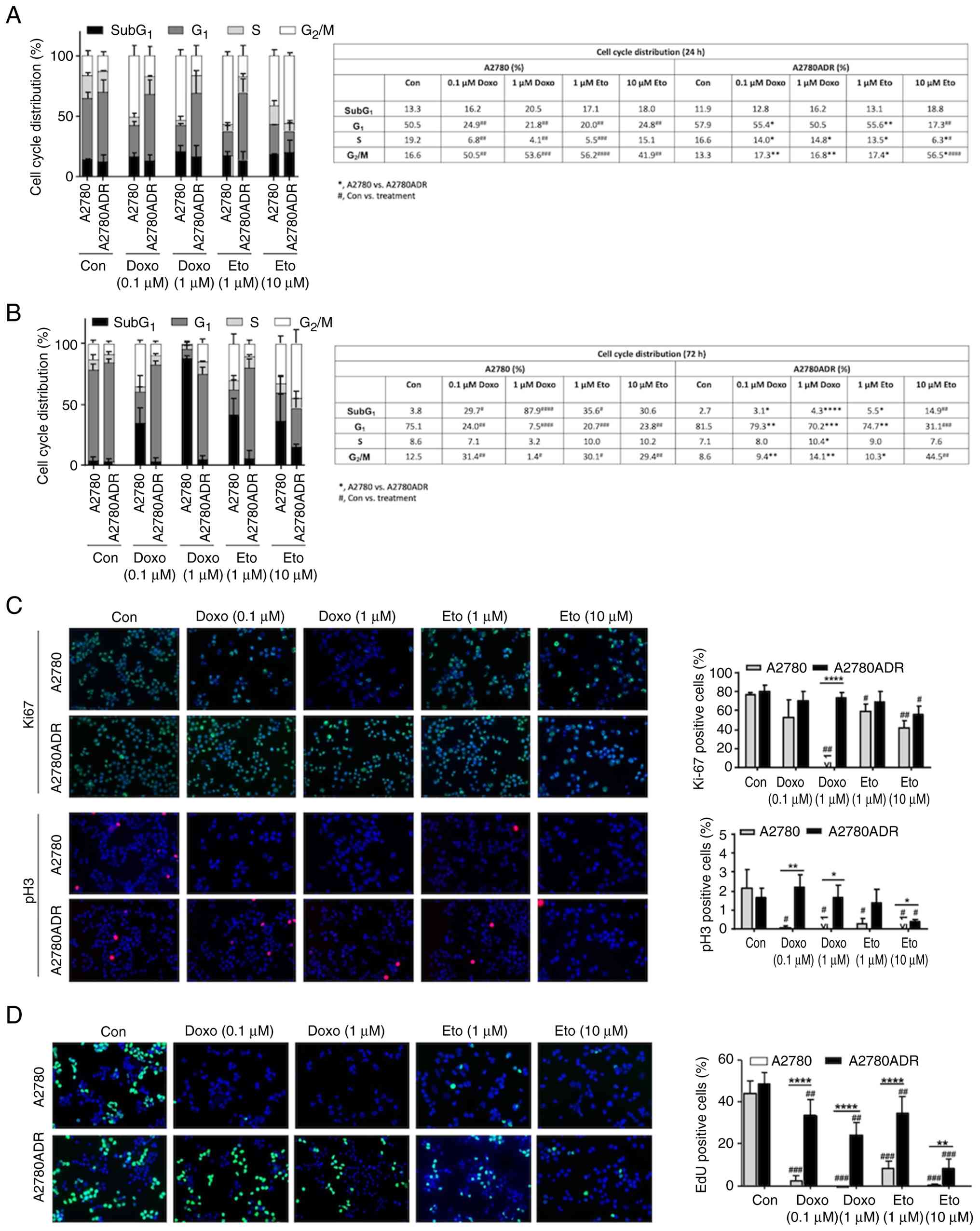
Analyses of proliferation and cell cycle
progression of A2780 and A2780ADR cells following treatment with
topoisomerase II inhibitors
To further analyze the outcome of altered drug
export in A2780 vs. A2780ADR cells, cell cycle progression was
analyzed after 24-72 h of Doxo and Eto treatment. At early time
point of analysis (that is, 24 h), A2780 revealed a more pronounced
accumulation of cells in G2/M phase compared with
A2780ADR (Fig. 2A). This effect
was seen both upon both Doxo and Eto treatment (Fig. 2A). Notably, SubG1
fraction was not yet enhanced following Doxo or Eto treatment in
A2780 cells at this early time point of analysis. Under basal
situation, the percentage of SubG1 phase cells was
slightly higher at the 24 h time point of analysis as compared with
72 h. It was hypothesized that this minor effect reflected stress
that resulted from the reseeding procedure. After an extended
treatment period of 72 h, parental cells revealed a clear increase
in the percentage of Doxo- and Eto-treated cells present in the
SubG1 fraction, which was not observed in the drug
resistant A2780ADR variants (Fig.
2B). Taken together, these data showed that parental A2780
cells were hypersensitive to G2/M blocking activity and
apoptosis induction triggered by Topo II poisoning anticancer
drugs.
To monitor proliferative activity, the percentage of
Ki-67 positive cells was analyzed in both cell variants. In
addition, mitotic index was calculated by determining the
percentage of pH3 positive cells. The data obtained show a marked
decrease in the percentage of Ki-67 positive A2780 parental cells
following treatment with Doxo but not Eto (Fig. 2C). By contrast, measuring the
mitotic index, a marked decrease in the frequency of pH3 positive
cells was found in both parental cells and resistant variants
following Doxo or Eto treatment (Fig.
2C). Measuring S-phase activity by analyzing the incorporation
of EdU, a very strong reduction in the percentage of EdU positive
parental cells upon both Doxo and Eto treatment it was again
observed, which was much weaker in the resistant A2780ADR cells
compared with the wild-type cells (Fig. 2D). Summarizing, the data show that
A2780ADR cells are highly resistant to the antiproliferative
activity of Topo II inhibitory compounds.
Comparative analyses of Doxo-induced
formation of DSB and activation of DDR-related mechanisms in A2780
and A2780ADR cells
Inhibition of Topo II isoforms leading to the
formation of DSB is considered as a major molecular mechanism
underlying the anticancer activity of Doxo (47,48). Measuring the formation of nuclear
γH2AX foci as well as 53BP1 foci and co-localized γH2AX/53BP1 foci
as surrogate markers of DNA damage (that is, DSB), a markedly lower
number of DNA damage-associated foci in the A2780ADR cells as
compared with the parental A2780 cells was observed, both after
treatment with Doxo and Eto (Fig.
3A). Following high-dose treatment with Doxo, a high percentage
of γH2AX pan-stained A2780 cells was detectable, which was not
observed in A2780ADR cells (Fig.
3A). These data show that the level of Doxo- and Eto-induced
DSB was substantially reduced in the drug resistant variant
compared with the parental cells. As DSBs are a potent trigger of
mechanisms of the DDR, the present study next investigated the
activation status of prototypical markers of the DDR by western
blotting. An excessive increase in the DDR-related protein levels
of γH2AX, p-Kap1, p-Chk2 and p-p53 in Doxo treated A2780 parental
cells only, both after Doxo treatment period of 24 and 72 h. By
contrast activation of these DDR-related factors was not found in
A2780ADR cells (Fig. 3B).
Notably, parental cells were also characterized by a great increase
in the protein expression of the senescence marker p21, which was
not detectable in the Doxo resistant A2780ADR cells (Fig. 3B). Eto treatment of A2780 cells
also caused a distinct increase in γH2AX, p-P53 and p21 protein
expression (Fig. S3).
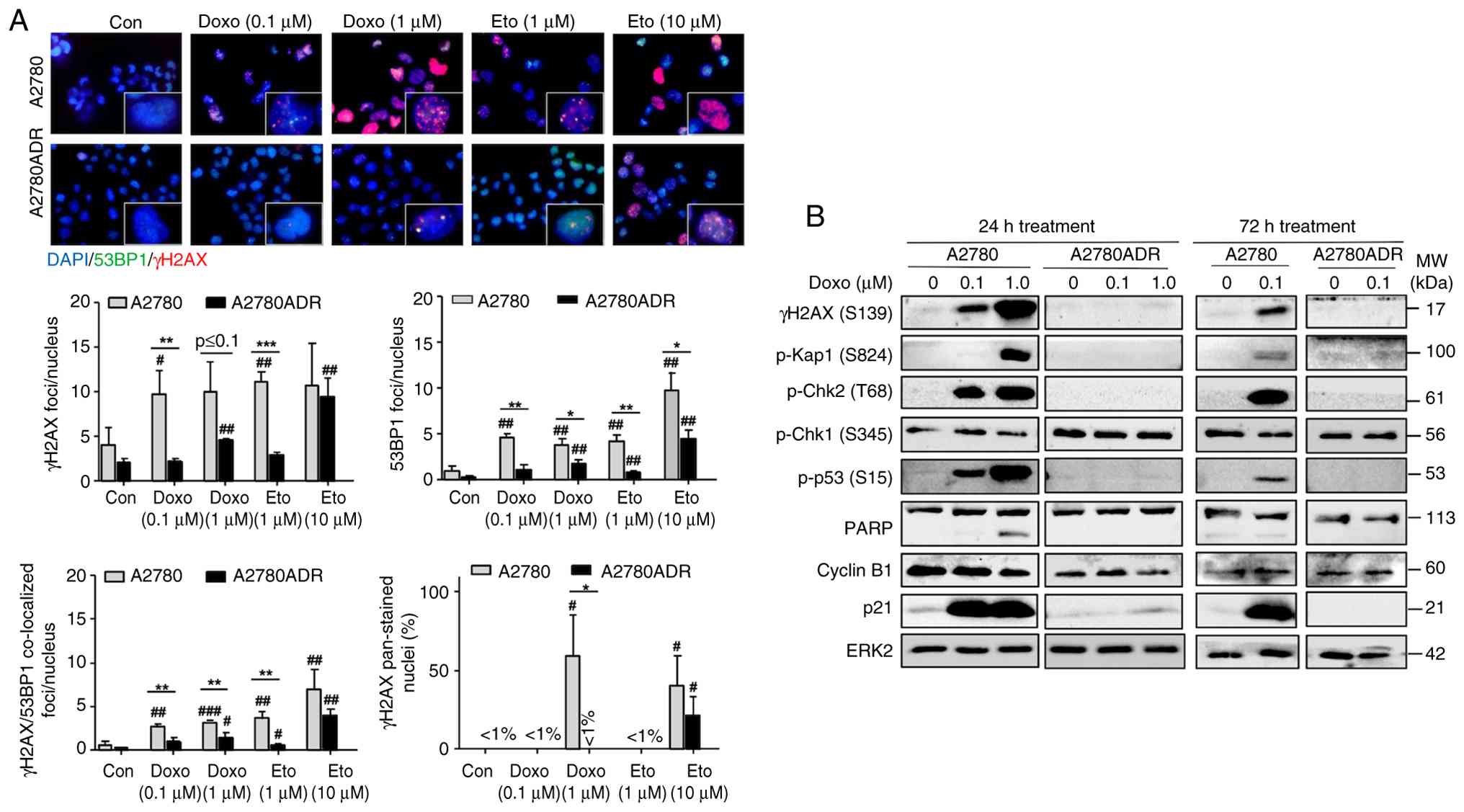 | Figure 3Effect of Topo II inhibitors on DNA
damage formation and activation of DDR-related mechanisms in A2780
and A2780ADR cells. (A) At 24 h after treatment of logarithmically
growing cells with the indicated concentrations of Doxo or Eto, the
number of nuclear γH2AX-foci, 53BP1-foci, γH2AX/53BP1 co-localized
foci and γH2AX pan-stained cells was analyzed. The upper part of
the figure shows representative images (total magnification,
×1,000). Quantitative data depicted in the histogram are the mean ±
SD from n=3 independent experiments with each five images being
analyzed per experimental condition. *P≤0.05;
**P≤0.01; ***P≤0.001 (A2780 vs. A2780ADR);
#P≤0.05; ##P≤0.01; ###P≤0.001
(treated vs. untreated control). Control experiments performed by
use of 1st or 2nd antibody only or no antibody at all did not
interfere with the signal of main interest (that is, nuclear foci;
data not shown). (B) Logarithmically growing cells were treated
with the indicated concentrations of Doxo for 24 or 72 h.
Afterwards, the protein expression of DDR-related factors was
analyzed by western blotting using EKR2 protein expression as
loading control. Doxo, doxorubicin; Eto, etoposide; p-,
phosphorylated; Chk1/2, checkpoint kinase 1/2; ERK2, extracellular
regulated kinase; γH2AX, Ser139 phosphorylated histone H2AX; 2;
Kap1, KRAB-associated protein 1; PARP, poly (ADP-ribose)
polymerase; p21, cyclin-dependent kinase inhibitor 1; p53, tumor
suppressor p53. |
Cross-sensitivity of parental (A2780) and
doxorubicin-resistant (A2780ADR) ovarian carcinoma cells to various
anticancer drugs and inhibitors of DNA repair and DDR
Aiming to overcome the Doxo resistance of A2780ADR
cells, the present study investigated their response to selected
inhibitors of DDR- and DNA repair-related mechanisms, which are
considered as promising targets to improve anticancer therapy and
to overcome acquired drug resistance of tumor cells (49-52). To this end, the present study
comparatively investigated the outcome of a 72 h treatment period
of A2780 parental and A2780ADR cells with Poly (ADP-ribose)
polymerase (PARP) inhibitors (olaparib and niraparib), checkpoint
kinase 1/2 (Chk1/2) inhibitors (prexasertib and rabusertib) and
histone deacetylase (HDAC) inhibitors (entinostat and
ricolinostat). In addition, the calcium channel blocker Ver, which
is reported to interfere with drug transport (53), the catalytic topo II inhibitor
dexrazoxane, which is able to protect the heart from Doxo-induced
cardiotoxicity by depleting both topo II isoforms independent of
metal chelation (54-56) and Rac1 GTPase inhibitors (EHT1864
and Ehop16), which are reported to interfere with Doxo-induced DNA
damage formation and DDR activation (57,58), were included into the study. From
the IC50 it was concluded that the data revealed a clear
cross-resistance of A2780ADR cells to the poly (ADP-ribose)
polymerase inhibitor (PARPi) olaparib (>10-fold) and niraparib
(>5-fold), the Chk1/2i prexasertib (~4-fold) and rabusertib
(~2.5-fold) and the HDAC class I inhibitor entinostat (~3-fold;
Fig. 4). A2780ADR cells were
neither appreciably cross-sensitive nor hypersensitive to any of
the other pharmacological modulators (such as ricolinostat, Ver,
EHT1864, EHOP16 and dexrazoxane) employed. Measuring cell viability
24 h after drug treatment, no major differences were observed
(Fig. S4). The IC50
and cross-sensitivities are summarized in Table SII.
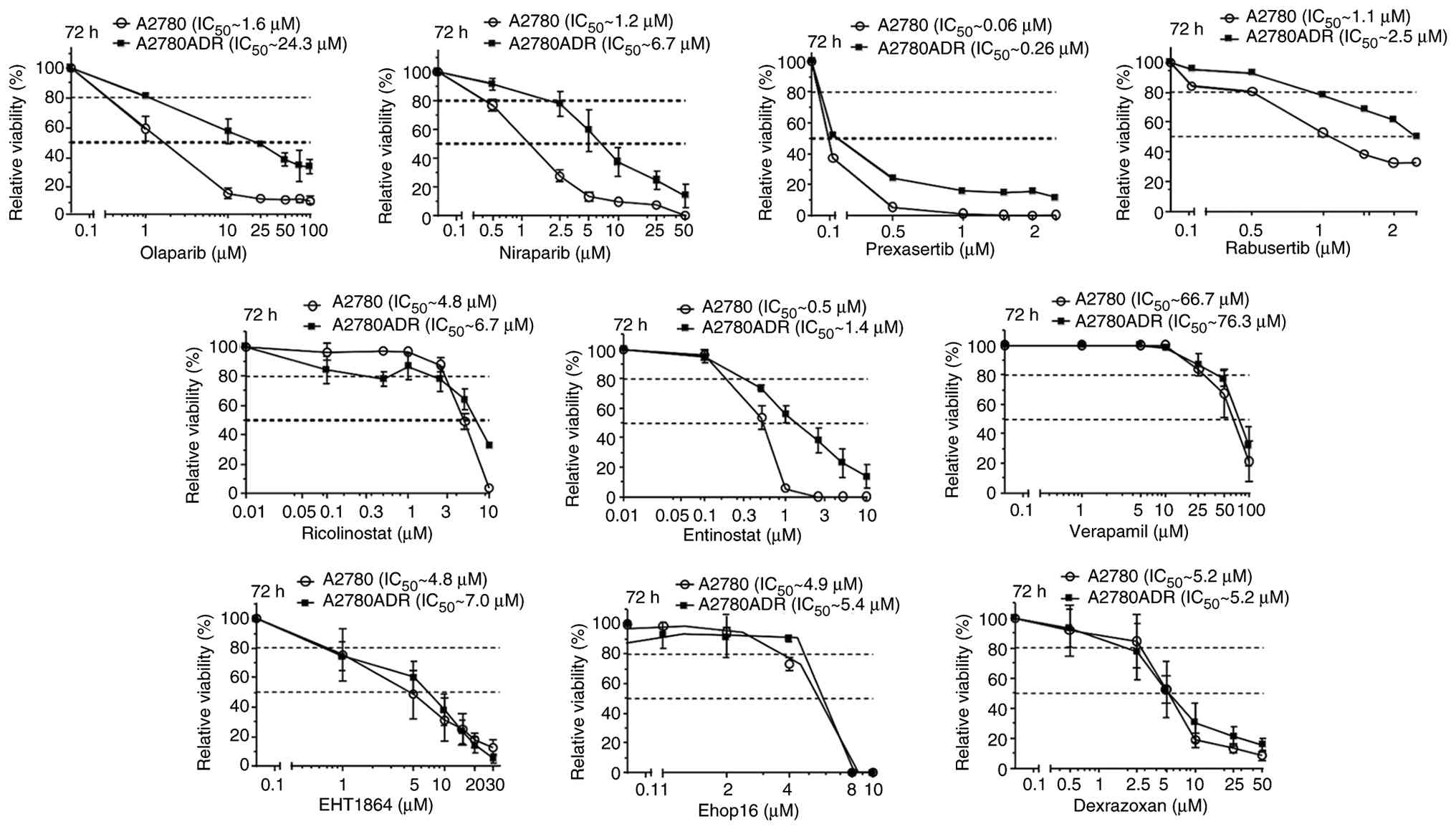 | Figure 4Analysis of cross-sensitivity of
parental A2780 and Doxo-resistant A2780ADR cells to selected
inhibitors of DDR- and DNA repair-related mechanisms.
Logarithmically growing parental A2780 and A2780ADR variant cells
were treated with selected pharmacological inhibitors of DNA repair
(olaparib and niraparib), DDR (prexasertib and rabusertib), HDAC
(ricolinistat and entinostat), Rac1 GTPase (EHT1864 and Ehop16),
drug transport (verapamil) and Topo II (dexrazoxane) at the
indicated concentrations. At 72 h after drug addition, viability
was monitored by use of the AlamarBlue assay as described in
methods. Data shown are the mean ± SD from three independent
experiments each performed in biological quadruplicates (n=3; n=4).
Dashed lines indicate inhibitory concentrations (IC20
and IC50). Data obtained from treatment period of 24 h
are presented in Fig. S4. For
IC50 after 24 h and 72 h see Table SII. Doxo, doxorubicin; DDR, DNA
damage response; HDAC, histone deacetylase; SD, standard deviation;
EHT, Rac1 inhibitor EHT1864. |
Co-treatment of doxorubicin resistant
A2780ADR with DDR modifiers overcomes Doxo resistance by inducing
synergistic toxicity
Based on the results of these extensive studies, the
present study addressed the question whether the pharmacological
inhibitors under investigation were able to overcome the acquired
Doxo resistance of A2780ADR cells. To this end, entinostat,
EHT1864, rabusertib, dexrazoxane and Ver were selected for Doxo
co-treatment analyses. Each of the selected inhibitors
synergistically increased the cytotoxicity of Doxo in the drug
resistant A2780ADR cells as concluded from the calculated CI
(Fig. 5A). Based on the CI, the
most pronounced synergistic toxicity was observed when Doxo was
combined with the transport inhibitor Ver or the Rac1 inhibitor
EHT1864 (Fig. 5A). The high
efficacy of Ver was associated with increased intracellular steady
state levels of Doxo under situation of co-treatment (Fig. 5B). It was not found if EHT or EST
were used for Doxo co-treatment (Fig.
5B). These data indicated that synergistic toxicity resulting
from combined treatment of A2780ADR cells with Doxo and Ver was at
least partially due to Ver-mediated inhibition of drug export
mechanisms leading to higher concentration of intracellular Doxo.
By contrast, synergism observed upon use of EST and EHT1864 is
suggested to be independent of drug transport. Notably, the data
were in line with published data showing that Rac1 is involved in
chemoresistance (59,60) and class I HDACi are useful of
overcome acquired anticancer drug resistance of malignant cells
(61-64).
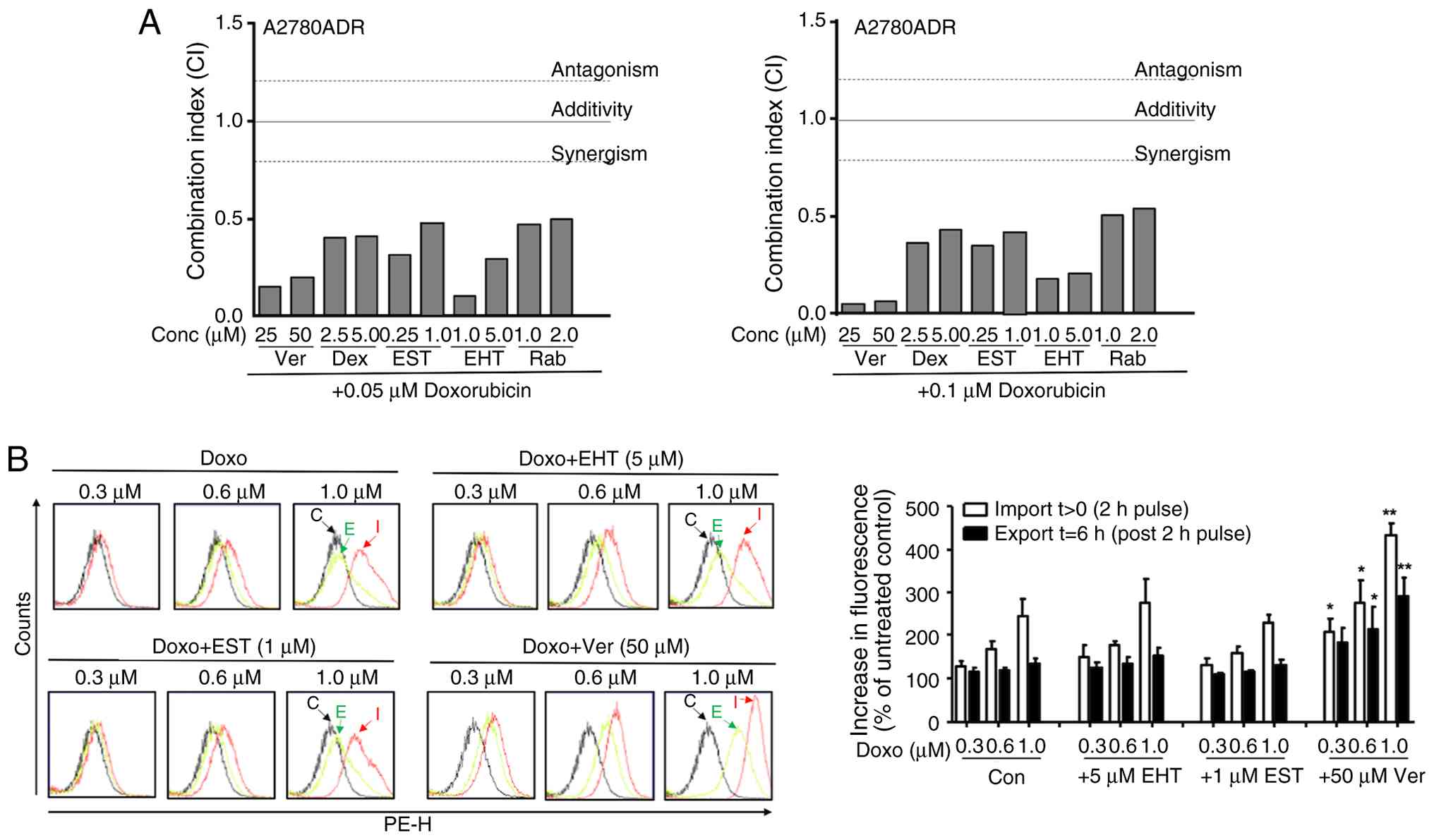 | Figure 5Combined treatment of A2780ADR with
Doxo and selected inhibitors causes synergistic toxicity. (A)
Logarithmically growing Doxo resistant A2780ADR cells were
co-treated with Doxo and selected pharmacological inhibitors at the
indicated concentrations. At 72 h after drug addition, viability
was monitored by use of the AlamarBlue assay and CI was calculated
as described in methods. Data shown are the mean ± SD from three
independent experiments each performed in biological quadruplicates
(n=3; n=4). (B) Intracellular Doxo fluorescence was measured by
flow cytometry-based method after co-treatment with Doxo and
selected pharmacological inhibitors as described in methods. To
measure drug export, Doxo pulse-treated cells were post-incubated
for 6 h in the absence of the drug before fluorescence was
monitored. Data shown in the left panel are representative results
obtained from flow cytometry analyses. C, control; I, import, E,
export. The histogram in the right panel depicts quantitative data
obtained from n=3 independent experiments each performed in
biological triplicates (n=3). Statistical significance:
*P≤0.05; **P≤0.001. Doxo, doxorubicin; CI,
combination index; SD, standard deviation; EST, entinostat; EHT,
Rac1 inhibitor EHT1864; Dex, dexrazoxane; Rab, rabusertib; Ver,
verapamil. |
Notably, Ver also synergistically increased the
cytotoxicity of Doxo in A2780 parental cells (Fig. S5A), showing that the Ver effect
is not restricted to cells with acquired Doxo resistance but also
pertains to parental tumor cells. The pharmacological inhibitors
entinotat (EST), EHT1864 and rabusertib (Rab) were also effective
in combination with Doxo whereas dexrazoxane (Dex) was not
(Fig. S5A). In addition,
verapamil (Ver) also conferred synergistic toxicity in A2780ADR
cells in combination with etoposide (Fig. S5B), showing that the Ver effect
is not limited to Doxo but also comprises other anticancer drugs.
Other inhibitors also conferred synergistic toxicity in combination
with Eto, with EHT being most effective (Fig. S5B). To further investigate
whether Ver is also able to overcome acquired resistance to
anticancer drugs others than Topo II inhibitors, the present study
investigated the influence of Ver on the CisPt sensitivity of
CisPt-resistant A2780CisR cells, including EHT and EST for control.
Data obtained show that all three modulators promoted CisPt-induced
cytotoxicity in A2780CisR cells, with Ver and EST being most
effective (Fig. S6).
In order to investigate whether the synergistic
cytotoxicity evoked by combined treatment with Ver and Doxo was
related to an elevated DNA damage induction, the present study
measured the outcome of the co-treatments on the formation of DSB.
To this end, the number of nuclear γH2AX and 53BP1 foci, which are
indicative of DSB, was analyzed. Data obtained show that Ver most
markedly increased the number of both nuclear γH2AX and 53BP1 foci
as well as γH2AX/53BP1 co-localized foci compared with Doxo
mono-treatment (Fig. 6A). In
addition, the percentage of γH2AX pan-stained cells was also
markedly enhanced upon co-treatment with Ver and Doxo as compared
with the mono-treatments (Fig.
6A). Apparently, Ver was able to markedly stimulate the
formation of DSB if used in combination with Doxo, demonstrating
that Ver potentiates the genotoxic effects of Doxo in A2780ADR
cells. Analyzing proliferation by monitoring the percentage of EdU
positive cells, the most substantial antiproliferative effects were
also observed upon combining Doxo with Ver (Fig. 6B). In addition, Ver also markedly
increased the percentage of PI positive cells if used in
combination with Doxo (Fig. 6C),
showing that Ver potentiates Doxo-induced toxicity.
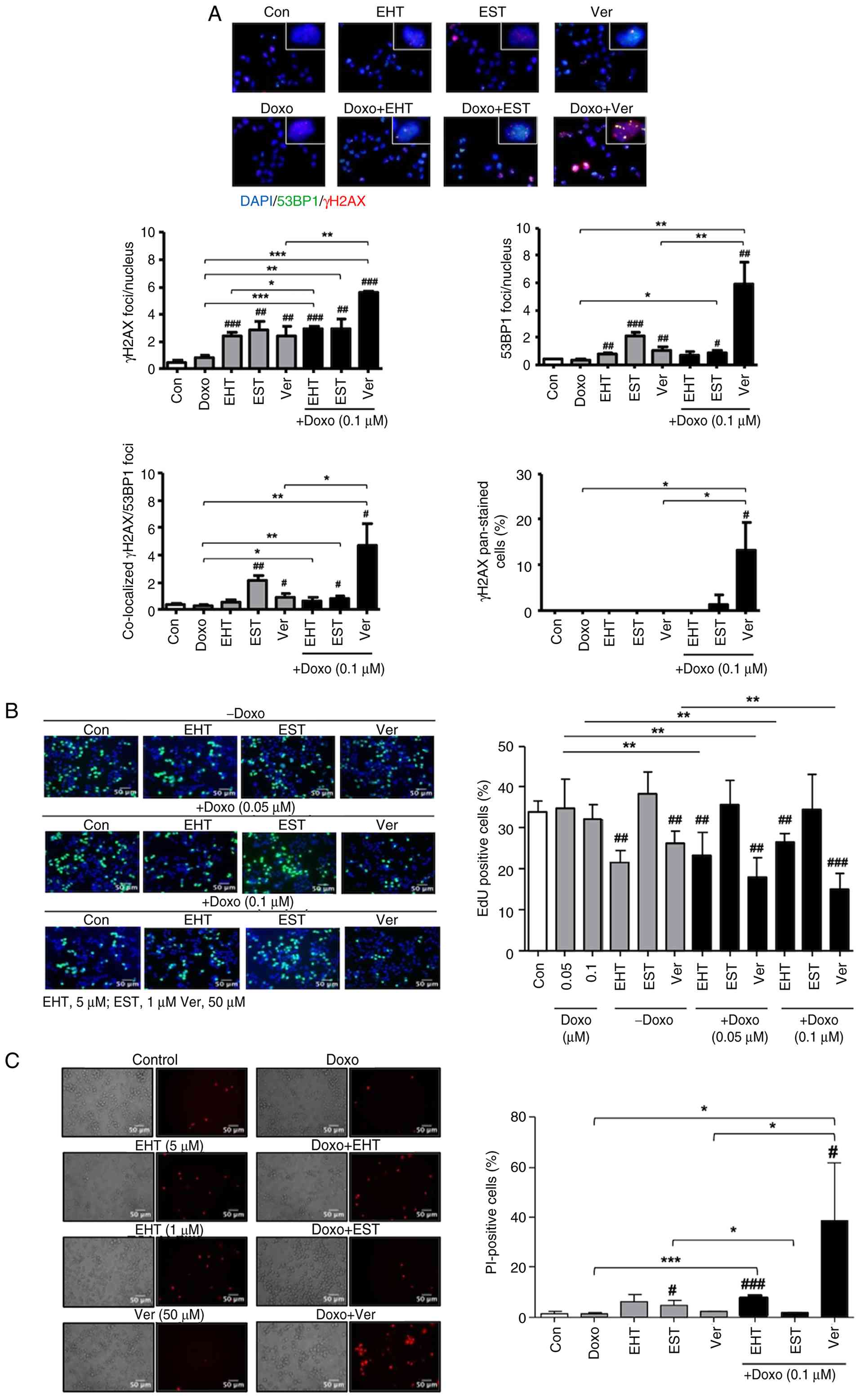 | Figure 6Influence of combined treatment of
A2780ADR with Doxo and selected inhibitors on DNA damage formation,
proliferation and cell death. (A) At 24 h after treatment of
logarithmically growing cells with the indicated concentrations of
Doxo and inhibitors (EHT, 5 μM; EST, 1 μM; Ver, 50
μM), the number of nuclear γH2AX-foci, 53BP1-foci,
γH2AX/53BP1 co-localized foci and γH2AX pan-stained cells was
analyzed as described in methods. The upper part of the figure
shows representative images (total magnification, ×1,000).
Quantitative data depicted in the histogram are the mean ± SD from
n=5 microscopical images analyzed per experimental condition.
*P≤0.05, **P≤0.01, ***P≤0.001
mono-treatment vs. co-treatment; #P≤0.05,
##P≤0.01, ###P≤0.001 untreated vs. treated
group). (B) Logarithmically growing Doxo resistant A2780ADR cells
were co-treated with the indicated concentrations of Doxo and
selected pharmacological inhibitors (EHT, 5 μM; EST, 1
μM; Ver, 50 μM). At 24 h later, cells were
pulse-labeled with EdU to monitor proliferation as described in
methods and the percentage of EdU positive cells was determined
microscopically (total magnification, ×400). Quantitative data
shown in the histogram are the mean ± SD from five replicates.
**P≤0.05; ##P≤0.01; ###P≤0.001
(vs. untreated control). (C) PI staining of mono- and co-treated
A2780ADR cells 72 h after treatment with Doxo (0.1 μM) and
pharmacological inhibitors (EHT, 5 μM; EST, 1 μM;
Ver, 50 μM). Left panel: representative images; right panel:
percentage of PI positive cells (mean ± SD from n=5 microscopical
images analyzed per experimental condition) (40× microscope
objective). *P≤0.05; **P≤0.01. mono-treatment
vs. co-treatment; #P≤0.05; ##P≤0.01, Con vs
treated group. Doxo, doxorubicin; EST, entinostat; EHT, Rac1
inhibitor EHT1864; Ver, verapamil; SD, standard deviation; PI,
propidium iodide. |
Influence of combined treatment of A2780
and A2780ADR cells with Doxo and selected inhibitors on mechanisms
of the DDR and mRNA expression of selected susceptibility-related
genes
Aiming to further elucidate the molecular mechanisms
underlying the observed synergistic toxicity and to substantiate
the assumed increase in DSB formation under situation of Ver + Doxo
co-treatment, the present study investigated the outcome of
combined treatments on the activation status of a subset of
DDR-related factors that are regulating replicative stress
responses and cell death by western blotting. The data obtained
showed that Ver especially was able to potentiate Doxo-mediated
phosphorylation of the DDR-related factors p53, RPA32, Chk2 and
H2AX, especially following 72 h of co-treatment (Fig. 7A). Moreover, if Doxo was combined
with Ver, PAPR cleavage and cleavage of pro-caspase 7, which are
indicative of the activation of apoptotic pathways, was observed
(Fig. 7A). Thus, it was
hypothesized that specific inhibition of drug transport by Ver
conferred a broad re-sensitization of A2780ADR cells by fostering
Doxo-induced formation of DSB and, in consequence, by activating
DDR-dependent mechanisms that stimulated pro-apoptotic pathways. In
addition, co-treatment of A2780ADR cells with Doxo plus Ver
increased the mean mRNA expression of pre-selected factors involved
in the regulation of oxidative stress response, DNA repair and cell
cycle regulation compared with Doxo or Ver mono-treated control
(Fig. 7B).
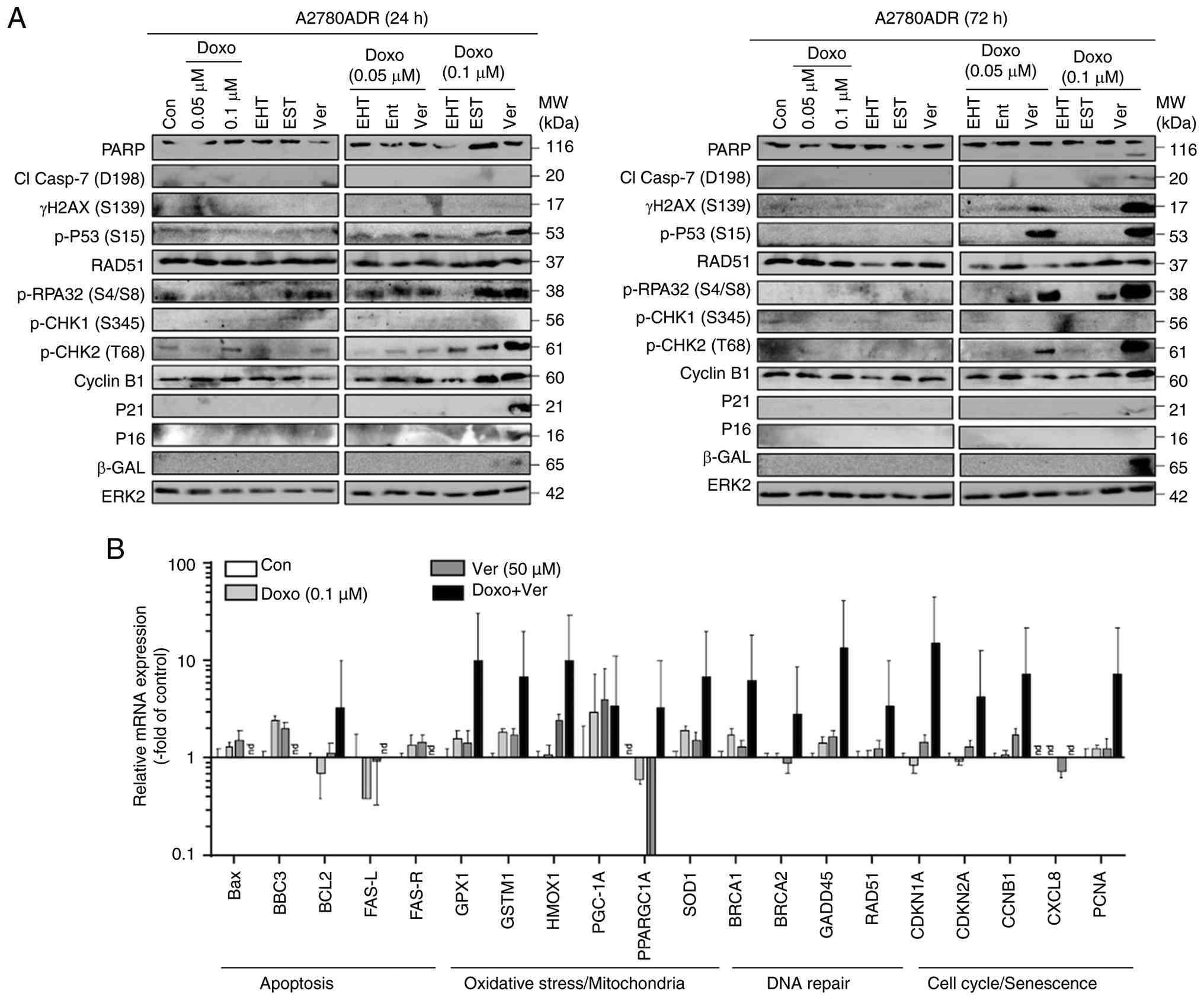 | Figure 7Influence of combined treatment of
A2780ADR cells with Doxo and selected inhibitors on mechanism of
the DDR and mRNA expression of selected susceptibility-related
genes. (A) Logarithmically growing A2780ADR cells were co-treated
with the indicated concentrations of Doxo and selected
pharmacological inhibitors (concentrations see Fig. 5) for 24 or 72 h. Afterwards, the
protein expression of DDR-related factors was analyzed by western
blotting. For loading control, blots were reprobed with ERK2
antibody. (B) Reverse transcription-quantitative PCR of the mRNA
expression of selected factors known to contribute to different
mechanisms of drug sensitivity. Data shown are mean ± SD from
triplicate determinations as described in methods. Relative mRNA
level in untreated A2780ADR cells was set to 1.0. Doxo,
doxorubicin; DDR, DNA damage response; p-, phosphorylated; nd, not
detectable; Bax, Bcl-2 associated protein X; Bcl-2, B-cell
lymphoma; BBC3, Bcl-2 binding component 2; BRCA1, 2, breast cancer
associated gene 1,2; Cl casp-7, cleaved caspase 7; Chk, checkpoint
kinase; CXCL8, chemokine ligand 8 (interleukin 8); p21, CDK
inhibitor 1; p16, CDK inhibitor 2; CDKN1A/2A, cyclin dependent
kinae inhibitor 1A/2A; CCNB1, Cyclin B1; b-Gal, beta-galactosidase;
FASL, FAS ligand; FASR, FAS receptor; GADD, growth arrest and DNA
damage inducible GPX1, glutathione peroxidase 1; GSTM1, glutathione
S-transferase 1; HMOX1, heme oxygenase 1; γH2AX, Ser139
phosphorylated histone H2AX; p53, tumor suppressor p53; PARP, poly
(ADP-ribose) polymerase; PCNA-proliferating cell nuclear antigen;
PGC1A, PPARG coactivator 1; PPARGC1A, peroxisome
proliferator-activated receptor gamma coactivator 1-alpha; RAD51,
radiation damage gene 51; RPAreplication protein A; SOD1,
superoxide dismutase 1; Ver, verapamil. |
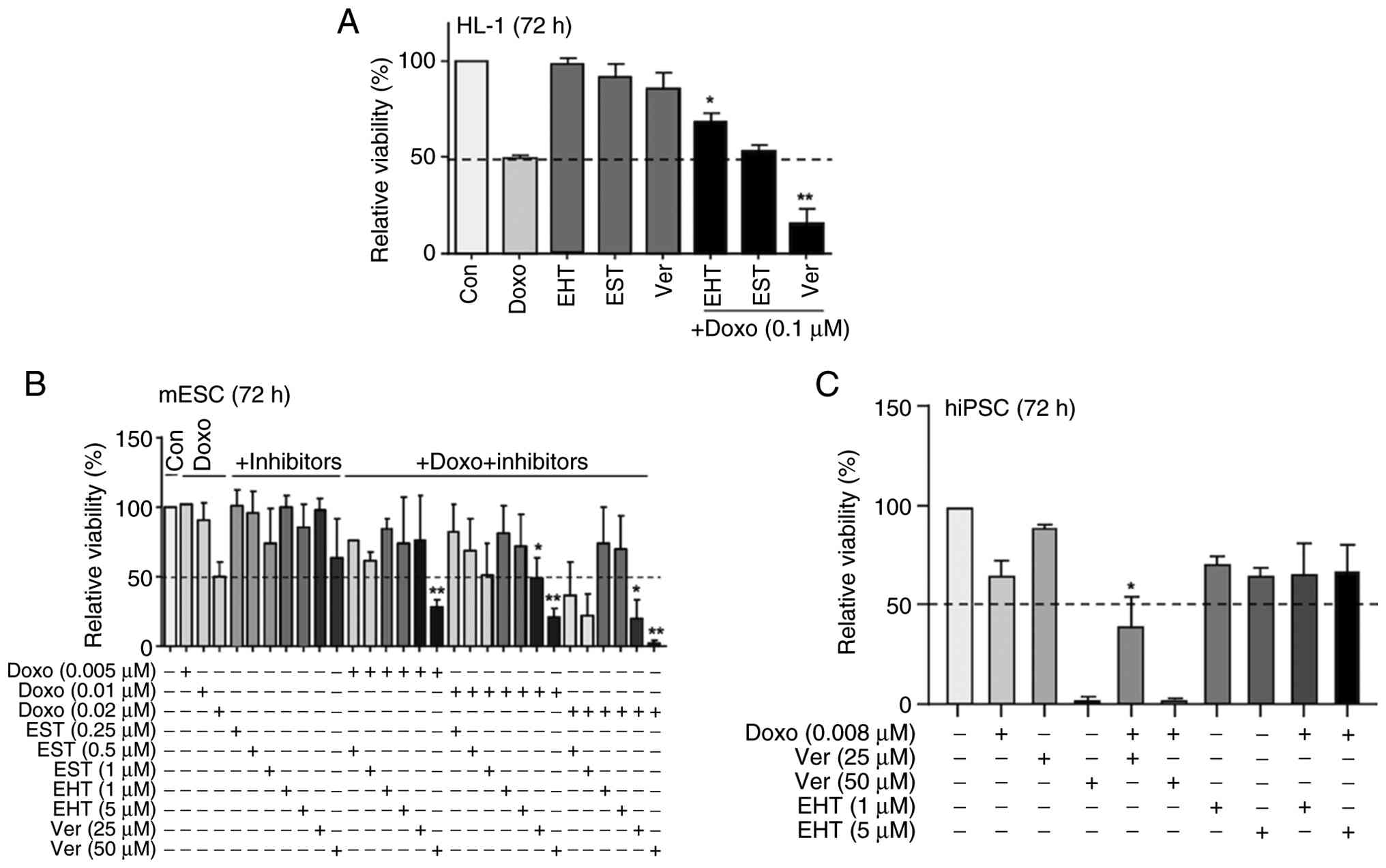
Influence of combined treatment on the
viability of non-malignant cells
Irreversible cardiotoxicity is a dose-limiting
adverse effect of anthracyclines (65,66). Hence, potentiating the anticancer
efficacy of Doxo if combined with pharmacological modifiers brings
up the concern of elevated side effects of the co-treatment,
especially regarding heart damage. To address this aspect, the
present study investigated the effect of combination treatments
using immortalized murine HL-1 cardiomyocyte cells as an in
vitro model. The results revealed more than additive
cytotoxicity if Doxo was combined with Ver (Fig. 8A). By contrast, tendentially
antagonistic effects were observed if Doxo was combined with the
Rac1 inhibitor EHT1864 (Fig. 8A).
This is in line with previous studies showing that pharmacological
inhibition of Rac1 is able to protect cardiac cell types from
Doxo-induced injury in vitro and in vivo (67-70). Moreover, the present study
investigated the cytotoxicity of Doxo in combination with Ver,
EHT1864 and EST using stem cell lines of murine (mESC) and human
(hiPSC) origin. We found that Ver promotes the Doxo sensitivity of
both mESC and hiPSC (Fig. 8B and
C). By contrast Rac1 inhibition by EHT1864 again did not evoke
synergistic toxicity in combination with Doxo in these
non-malignant cells (Fig.
8A-C).
Discussion
In order to characterize the molecular mechanisms
contributing to the Doxo resistant phenotype of A2780ADR cells and,
furthermore, to identify bioactive compounds to overcome their
acquired platin-resistance, extensive cross-resistance analyses
were performed. The results obtained show profound cross-resistance
of A2780ADR cells to Eto and a rather moderate cross-resistance to
CisPt. Having in mind that inhibition of Topo II is a common
mechanism underlying the anticancer efficacy of Dox and Eto, we
assume that the acquired Doxo resistance of A2780DAR cells is
related to Topo II-associated mechanisms, although the contribution
of other mechanisms to Doxo resistance of A2780ADR cells, such as
ROS formation, intercalation or inhibition of helicases (4,71,72), cannot be ruled out. The weak
cross-resistance to CisPt might be based on overlapping drug
transporters for Doxo, Eto and CisPt (7,8) or
DSB as a common type of DNA damage that result from both
Doxo-/Eto-mediated Topo II inhibition and DNA interstrand
cross-links caused by CisPt. Data obtained from the subsequent
analysis of drug export supported the hypothesis that drug export
contributes to the Doxo resistance of A2780ADR cells. Notably, the
export activity of parental A2780 cells is only moderately (~20%)
higher than that of A2780 cells. By contrast, the drug resistant
A2780ADR variants reveal an ~10-fold higher Doxo resistance than
the parental cells as concluded from their IC50.
Therefore, it was assumed that mechanisms others than drug export
additionally contributed to the profound Doxo resistant phenotype
of A2780 cells. One of this hypothesized Doxo-related additional
resistance mechanism of A2780 cells might be a reduced protein
expression of the main Doxo target protein Topo IIα (73).
Other feasible mechanisms of acquired Doxo
resistance may be alteration in DDR and DNA repair, since such
mechanisms are well-known to define the balance between cell death
and survival signaling (3) by
regulating cell cycle progression, cell death and DNA repair.
Analyzing Doxo- and Eto-induced alterations in the activation of
cell cycle checkpoints, the present study observed a pronounced
resistance of A2780ADR cells to both Doxo- and Eto-induced
G2/M arrest as well as apoptosis as reflected by the
percentage of cells present in the SubG1 fraction.
Notably, while Doxo caused a great increase in the SubG1
fraction of A2780 cells, such an effect was not found following Eto
treatment. It is hypothesized that this may be related to the not
fully identical mode of action of both drugs. While both drugs are
poisoning Topo II, Doxo has additional cytotoxic activities, such
as intercalation into DNA, inhibition of DNA helicases and ROS
formation via mitochondrial toxicity (5). Hence, it is feasible that, apart
from TOP2 dependent mechanism, A2780ADR cells also benefit from
additional cytoprotective mechanisms against Doxo, including
mechanisms related to oxidative stress defense. Moreover, the
activation of S-phase checkpoint by Doxo is also mitigated in
A2780ADR as compared with wild-type cells. This is concluded from
their very weak S-phase block following Doxo treatment as measured
by EdU incorporation analyses as well as the analyses of Ki67 and
pH3 positive cells. Summarizing, it is tempting to hypothesize that
acquired drug resistance of A2780ADR cells to Doxo is due to
alterations in their drug-stimulated activation of cell cycle
checkpoints, including S-phase related checkpoints, and
apoptosis-related mechanisms of the DDR. Having in mind that DSB
are potent trigger of the DDR, the present study next assayed the
steady state levels of DSB after 24 h of drug treatment by
monitoring the number of nuclear γH2AX foci, which are
well-accepted surrogate markers of DSB (40). Indeed, both Doxo- and Eto induced
formation of DSB was largely reduced in the A2780ADR cells as
compared with the wild-type. These findings indicated that lower
steady-state levels of drug-induced DSB in the Doxo resistant
variant majorly account for their substantially increased drug
resistance. This hypothesis gains further support by almost
complete lack of activation of DDR-related factors (such as
KRAB-associated protein 1, Chk2, p53 and p21) in A2780ADR compared
with parental A2780 cells following Doxo treatment. As with Doxo,
treatment of A2780 cells with Eto also caused a distinct increase
in the protein expression of γH2AX, pP53 and p21. This is
noteworthy, since the pronounced p21 induction following Doxo
treatment was associated with a strong decrease in the percentage
of Ki67 positive cells, while Eto treatment rather also caused
strong p21 induction but no major reduction in Ki67 positive cells.
So, it appears feasible that the Doxo-induced p21 induction
reflects an early stress response that is associated with
senescence while the response to Eto is not. Discussing the
response of cells regarding Ki67, it should be noted that Ki67 is
considered more than just a proliferation marker (74). It is related to chromatin and
subject to ubiquination-related mechanisms (75) as well as complex transcriptional
control mechanisms (76). Hence,
it was hypothesized that Doxo and Eto differently interfere with
mechanisms of Ki67 regulation. Overall, the present study supported
the hypothesis that the immense Doxo resistance of A2780ADR cells
(that is, ~10-fold as compared with A2780 cells) is also
attributable to an attenuated Doxo-induced formation of DNA damage
(that is, DSB) and largely reduced activation of DDR-related
pro-death mechanisms (51,52,77).
DDR and DNA repair-associated factors are promising
targets to improve anticancer therapy and to overcome acquired drug
resistance of tumor cells (49-52). Therefore, the present study
analyzed the influence of a set of pharmacological inhibitors
interfering with these mechanisms on the viability of A2780ADR
cells. The data showed cross-resistance of the Doxo resistant cells
to PARPi (that is, Olaparib and niraparib) as well as Chk1/2
inhibitors (that is, prexasertib and rabusertib) and the HDACi
entinostat. This cross-resistance data further supported the
hypothesis that mechanisms of DNA repair/DDR contribute to the
Doxo-resistant phenotype of A2780ADR cells. However, the A2780ADR
cells were not hypersensitive to any of the other inhibitors testet
(that is, ricolinostat, Ver, EHT1864, Ehop16 and dexrazoxane),
showing the specificity of the effects observed with PARPi and
Chk1/2i. These data clearly demonstrated that mono-treatment with
DDR modifying drugs is not particularly useful for preferential
killing of drug resistant cells. Therefore, the present study next
investigated whether co-treatment with a selected subset of
inhibitors (that is, entinostat, EHT1864, rabusertib, dexrazoxane
and Ver) was useful to re-sensitize A2780ADR cells to Doxo
treatment. The data obtained showed that EHT and Ver evoked the
most noticeable synergistic toxicity in combination with Doxo.
Notably, Ver and EHT also caused synergistic toxicity in
combination with Eto and, moreover, both Ver and EST were highly
efficient at overcoming acquired CisPt resistance of A2780CisR
cells. In summary, the data demonstrated that Ver is particular
useful to re-sensitize both Doxo-, Eto- and CisPt-resistant A2780
cells, indicating that Ver is able to overcome acquired drug
resistance towards multiple anticancer agents.
As to the molecular mechanisms involved, the present
study found that part of the Ver effect was related to a reduced
drug export, leading to higher intracellular steady-state
concentration of Doxo. Notably, synergistic toxicity observed if
EHT or EST are combined with Doxo is independent of drug transport.
To investigate the influence of the pharmacological inhibitors on
Doxo-induced DNA damage formation, the steady state level of DSB
was monitored after a 24 h treatment period. This experimental
setup was considered as particular appropriate because it takes
into account the complex kinetic processes that define the level of
Doxo-induced DNA damage that is detectable at a certain time point
of analysis and, furthermore, allows the detection of the net
outcome of the pharmacological modifiers on these processes. In
line with the toxicity data, co-treatment with Ver resulted in a
significant increase in Doxo-induced formation of DSB, as measured
by the number of nuclear γH2AX and 53BP1 foci, which was not
observed for EHT and EST. In addition, the percentage of γH2AX
pan-stained and PI positive cells also largely increased under
situation of co-treatment. Surprisingly, mono-treatment with both
EHT, EST and Ver caused a weak increase in DNA damage as reflected
on the level of nuclear γH2AX foci. As to EHT, we have previously
demonstrated DSB-inducing activity also in non-malignant cells
(78). Moreover, Rac1 has been
reported to be expressed in the nucleus (79) and is involved in the regulation of
DDR- and DNA repair-related mechanisms (80,81). These pleiotropic functions of Rac1
are a feasible explanation of a moderate induction of DNA damage
under situation of ETH mono-treatment. Regarding EST, it was
hypothesized that DNA damage resulting from mono-treatment is due
to the well-known interference of this class I HDACi with
mechanisms of DNA repair and DDR (82-84). With respect to Ver it is important
to bear in mind that this Ca2+ channel blocker is
considered to preferentially induce apoptosis in multi-drug
resistant cells via ROS-dependent mechanisms (85). Thus, it is feasible that induction
of oxidative DNA damage is responsible for the induction of DSB
under the situation of Ver mono-treatment. Moreover, in line with
the data of the present study, the reversal of Doxo resistance by
Ver has been associated with drug accumulation and DNA damage
formation (86,87). Taken together, the present study
showed that Ver caused a distinct potentiation of the genotoxic,
antiproliferative and cell-death inducing effects of Doxo. Thus, it
is hypothesized that Ver is able to overcome acquired Doxo
resistance of A2780ADR cells by fostering the Doxo-induced toxicity
via increased formation of DSB, leading to potentiated cytotoxicity
by pronounced inhibition of proliferation and induction of cell
death.
To further characterize the molecular mechanisms
underlying the observed synergistic toxicity and to substantiate
the increase in DSB formation under situation of Ver plus Doxo
co-treatment, the outcome of combined treatments on the activation
status of a subset of DDR-related factors were analyzed by western
blotting. The data demonstrated that specifically Ver, but not EHT
or EST, potentiated the Doxo-stimulated activation of multiple
DDR-related factors (that is, p53, RPA32 and Chk2) as well as
cleavage of PARP and pro-caspase 7, which is indicative of
apoptotic cell death. In addition, complex alterations in the mRNA
expression of susceptibility-related factors involved in the
regulation of oxidative stress responses, cell cycle regulation and
senescence as well as DNA repair were observed. These results
indicated that the synergistic activity of Ver in combination with
Doxo probably relies on an amplification of multiple DDR-related
pro-toxic stress responses of A2780ADR cells. To understand whether
combined treatment may also increase adverse effects of Doxo on
normal cells, combination experiments were performed using
different types of non-malignant cells (that is, HL-1
cardiomyocytes, murine and human stem cells). Unfortunately, data
obtained indicated that Doxo-induced toxicity was also elevated in
non-malignant cells if used in combination with Ver. This finding
raised the concern of a possible increase in adverse effects
resulting from co-treatment regimen in vivo. Thus, while
Doxo plus Ver co-treatment is evoking substantial synergistic
toxicity in Doxo resistant malignant cells, the therapeutic window
of this combination treatment might be narrow. Notably in this
context, Rac1 inhibition by EHT1864 did not cause any synergistic
toxicity in combination with Doxo in non-malignant cells. This
finding is in line with in vitro and in vivo data
(80,88,89) and points to a potentially
favorable therapeutic window of EHT1864 if used in combination with
Doxo.
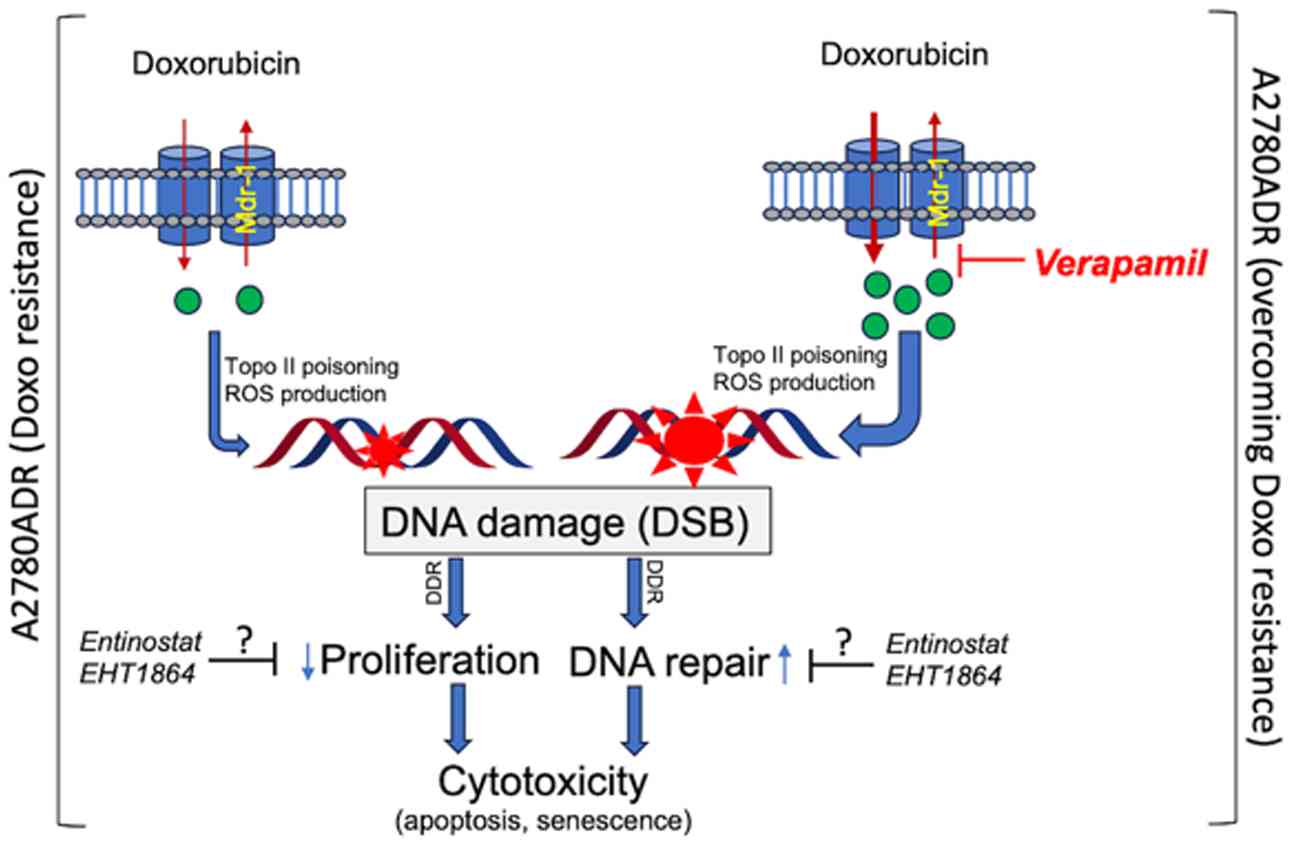
In summary, acquired Doxo resistance of A2780ADR was
associated with a reduced drug uptake, which is probably due to
high MDR-1 expression and a diminished Topo IIα expression.
Furthermore, it was accompanied by an attenuated drug-stimulated
S-phase block, mitigated formation of DSB and reduced DDR
activation as compared with A2780 parental cells. Notably, acquired
Doxo resistance of A2780 cells can be overcome most effectively by
inhibition of drug exporters using Ver (Fig. 9). In consequence of Ver
co-treatment, the intracellular steady-state concentration of Doxo
increased, promoting the formation of DSB, thereby in turn
triggering pro-toxic mechanisms of the DDR that promoted inhibition
of cell proliferation and stimulation of senescence- and cell
death-associated mechanisms. Notably, targeting of Rac1- and/or
HDAC class I-related mechanisms was also highly efficient to
overcome acquired Doxo resistance, yet independent of drug
transport. However, the molecular mechanisms involved here are
still unclear and, hence, will be subject of forthcoming studies.
In conclusion, the present study suggested that Ver is highly
effective to re-sensitize Doxo-resistant tumor cells by promoting
DNA damage-triggered cytotoxicity related to proliferation-, cell
death- and, possibly, senescence-associated mechanisms. However,
combined treatment with Ver may also amplify Doxo-stimulated
adverse outcome pathways in non-malignant cells. Accordingly,
forthcoming pre-clinical in vivo studies are needed for a
meaningful definition of the therapeutic range of a Doxo plus Ver
co-treatment regimen.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
EM was responsible for conceptualization,
methodology, performing experiments, data analysis and
interpretation, visualization and writing. SF and LM were
responsible for performing experiments, data analysis and
visualization. MS was responsible for methodology, data analysis
and supervision. JM was responsible for methodology and data
analysis. LA was responsible for performing experiments, data
analysis and visualization. MK was responsible for resources,
funding acquisition and writing original draft. GF was responsible
for conceptualization, data interpretation, funding acquisition,
writing original draft, resources and supervision. EM, LA, SF, LM
and GF confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ATM
|
Ataxia telangiectasia mutated
|
|
ATR
|
ATM- and Rad-3 related
|
|
cAT
|
conventional anticancer
therapeutics
|
|
Chk1/2
|
checkpoint kinase 1/2
|
|
CisPt
|
cisplatin
|
|
DDR
|
DNA damage response
|
|
Doxo
|
doxorubicin
|
|
DSB
|
DNA double-strand breaks
|
|
EHT
|
Rac1 inhibitor EHT1864
|
|
Eto
|
etoposide
|
|
EST
|
entinostat
|
|
ERK2
|
extracellular regulated kinase 2
|
|
γH2AX
|
Ser139 phosphorylated histone
H2AX
|
|
HDACi
|
histone deacetylase inhibitor
|
|
hiPSC
|
human induced pluripotent stem
cells
|
|
MDR1
|
multi-drug resistance gene 1
|
|
mESC
|
mouse embryonic stem cells
|
|
PARPi
|
poly (ADP-ribose) polymerase
inhibitor
|
|
Ver
|
verapamil.
|
Acknowledgements
The authors would like to thank Dr Christian
Henninger (Institute of Toxicology, HHU Duesseldorf) for discussion
and proofreading of the manuscript.
Funding
The present study was supported by the Deutsche
Forschungsgemeinschaft [DFG Research Training Group (RTG)
417677437/GRK2578 (RG Fritz) and DFG Research Training Group (RTG)
270650915/GRK2158 (RG Fritz)].
References
|
1
|
Minotti G, Menna P, Salvatorelli E, Cairo
G and Gianni L: Anthracyclines: Molecular advances and
pharmacologic developments in antitumor activity and
cardiotoxicity. Pharmacol Rev. 56:185–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roos WP and Kaina B: DNA damage-induced
cell death: From specific DNA lesions to the DNA damage response
and apoptosis. Cancer Lett. 332:237–248. 2013. View Article : Google Scholar
|
|
3
|
Roos WP, Thomas AD and Kaina B: DNA damage
and the balance between survival and death in cancer biology. Nat
Rev Cancer. 16:20–33. 2016. View Article : Google Scholar
|
|
4
|
Rivankar S: An overview of doxorubicin
formulations in cancer therapy. J Cancer Res Ther. 10:853–858.
2014. View Article : Google Scholar
|
|
5
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
6
|
Nielsen D, Maare C and Skovsgaard T:
Cellular resistance to anthracyclines. Gen Pharmacol. 27:251–255.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gillet JP, Efferth T and Remacle J:
Chemotherapy-induced resistance by ATP-binding cassette transporter
genes. Biochim Biophys Acta. 1775:237–262. 2007.PubMed/NCBI
|
|
8
|
Konkimalla VB, Kaina B and Efferth T: Role
of transporter genes in cisplatin resistance. In Vivo. 22:279–283.
2008.PubMed/NCBI
|
|
9
|
Woods D and Turchi JJ: Chemotherapy
induced DNA damage response: Convergence of drugs and pathways.
Cancer Biol Ther. 14:379–389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferri A, Stagni V and Barila D: Targeting
the DNA damage response to overcome cancer drug resistance in
glioblastoma. Int J Mol Sci. 21:49102020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harper JW and Elledge SJ: The DNA damage
response: Ten years after. Mol Cell. 28:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shiloh Y: ATM and ATR: Networking cellular
responses to DNA damage. Curr Opin Genet Dev. 11:71–77. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsuoka S, Ballif BA, Smogorzewska A,
McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini
N, Lerenthal Y, et al: ATM and ATR substrate analysis reveals
extensive protein networks responsive to DNA damage. Science.
316:1160–1166. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Durocher D and Jackson SP: DNA-PK, ATM and
ATR as sensors of DNA damage: Variations on a theme? Curr Opin Cell
Biol. 13:225–231. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kumagai A and Dunphy WG: How cells
activate ATR. Cell Cycle. 5:1265–1268. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Petermann E and Caldecott KW: Evidence
that the ATR/Chk1 pathway maintains normal replication fork
progression during unperturbed S phase. Cell Cycle. 5:2203–2209.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eich M, Roos WP, Nikolova T and Kaina B:
Contribution of ATM and ATR to the resistance of glioblastoma and
malignant melanoma cells to the methylating anticancer drug
temozolomide. Mol Cancer Ther. 12:2529–2540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weber AM and Ryan AJ: ATM and ATR as
therapeutic targets in cancer. Pharmacol Ther. 149:124–138. 2015.
View Article : Google Scholar
|
|
21
|
Carrassa L and Damia G: DNA damage
response inhibitors: Mechanisms and potential applications in
cancer therapy. Cancer Treat Rev. 60:139–151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dobbelstein M and Sorensen CS: Exploiting
replicative stress to treat cancer. Nat Rev Drug Discov.
14:405–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morgan MA and Lawrence TS: Molecular
pathways: Overcoming radiation resistance by targeting DNA damage
response pathways. Clin Cancer Res. 21:2898–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saini P, Li Y and Dobbelstein M: Wee1 is
required to sustain ATR/Chk1 signaling upon replicative stress.
Oncotarget. 6:13072–13087. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Lai J, Du Z, Gao J, Yang S,
Gorityala S, Xiong X, Deng O, Ma Z, Yan C, et al: Targeting
radioresistant breast cancer cells by single agent CHK1 inhibitor
via enhancing replication stress. Oncotarget. 7:34688–34702. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toledo LI, Murga M and Fernandez-Capetillo
O: Targeting ATR and Chk1 kinases for cancer treatment: A new model
for new (and old) drugs. Mol Oncol. 5:368–373. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murga M, Campaner S, Lopez-Contreras AJ,
Toledo LI, Soria R, Montaña MF, Artista L, Schleker T, Guerra C,
Garcia E, et al: Exploiting oncogene-induced replicative stress for
the selective killing of Myc-driven tumors. Nat Struct Mol Biol.
18:1331–1335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gilad O, Nabet BY, Ragland RL, Schoppy DW,
Smith KD, Durham AC and Brown EJ: Combining ATR suppression with
oncogenic Ras synergistically increases genomic instability,
causing synthetic lethality or tumorigenesis in a dosage-dependent
manner. Cancer Res. 70:9693–9702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Helleday T: The underlying mechanism for
the PARP and BRCA synthetic lethality: Clearing up the
misunderstandings. Mol Oncol. 5:387–393. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lord CJ and Ashworth A: PARP inhibitors:
Synthetic lethality in the clinic. Science. 355:1152–1158. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lord CJ, Tutt ANJ and Ashworth A:
Synthetic lethality and cancer therapy: Lessons learned from the
development of PARP inhibitors. Ann Rev Med. 66:455–470. 2015.
View Article : Google Scholar
|
|
32
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sriraman SK, Pan J, Sarisozen C, Luther E
and Torchilin V: Enhanced cytotoxicity of folic Acid-targeted
liposomes Co-loaded with C6 ceramide and doxorubicin: In vitro
evaluation on HeLa, A2780-ADR, and H69-AR cells. Mol Pharm.
13:428–437. 2016. View Article : Google Scholar
|
|
34
|
Claycomb WC, Lanson NA Jr, Stallworth BS,
Egeland DB, Delcarpio JB, Bahinski A and Izzo NJ Jr: HL-1 cells: A
cardiac muscle cell line that contracts and retains phenotypic
characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA.
95:2979–2984. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nichols J, Evans EP and Smith AG:
Establishment of germ-line-competent embryonic stem (ES) cells
using differentiation inhibiting activity. Development.
110:1341–1348. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y and Adjaye J: A cyclic AMP analog,
8-Br-cAMP, enhances the induction of pluripotency in human
fibroblast cells. Stem Cell Rev Rep. 7:331–341. 2011. View Article : Google Scholar
|
|
37
|
O'Brien J, Wilson I, Orton T and Pognan F:
Investigation of the Alamar Blue (resazurin) fluorescent dye for
the assessment of mammalian cell cytotoxicity. Eur J Biochem.
267:5421–5426. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chou TC: Comparison of drug combinations
in vitro, in animals, and in clinics by using the combination index
method via computer simulation. Cancer Res. 692009.
|
|
39
|
Luk CK and Tannock IF: Flow cytometric
analysis of doxorubicin accumulation in cells from human and rodent
cell lines. J Natl Cancer Inst. 81:55–59. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Olive PL: Detection of DNA damage in
individual cells by analysis of histone H2AX phosphorylation.
Methods Cell Biol. 75:355–373. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zimmermann M and de Lange T: 53BP1: Pro
choice in DNA repair. Trends Cell Biol. 24:108–117. 2014.
View Article : Google Scholar
|
|
43
|
Panier S and Boulton SJ: Double-strand
break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol.
15:7–18. 2014. View Article : Google Scholar
|
|
44
|
Vandesompele J, De Preter K, Pattyn F,
Poppe B, Van Roy N, De Paepe A and Speleman F: Accurate
normalization of real-time quantitative RT-PCR data by geometric
averaging of multiple internal control genes. Genome Biol.
3:RESEARCH00342002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xia CQ and Smith PG: Drug efflux
transporters and multidrug resistance in acute leukemia:
Therapeutic impact and novel approaches to mediation. Mol
Pharmacol. 82:1008–1021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mirzaei S, Gholami MH, Hashemi F, Zabolian
A, Farahani MV, Hushmandi K, Zarrabi A, Goldman A, Ashrafizadeh M
and Orive G: Advances in understanding the role of P-gp in
doxorubicin resistance: Molecular pathways, therapeutic strategies,
and prospects. Drug Discov Today. 27:436–455. 2022. View Article : Google Scholar
|
|
47
|
Tewey KM, Rowe TC, Yang L, Halligan BD and
Liu LF: Adriamycin-induced DNA damage mediated by mammalian DNA
topoisomerase II. Science. 226:466–468. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Johnson M and Keyes D: Anthracycline
Toxicity. Disclosure: Daniel Keyes declares no relevant financial
relationships with ineligible companies. StatPearls; Treasure
Island, FL: 2024
|
|
49
|
Jin MH and Oh DY: ATM in DNA repair in
cancer. Pharmacol Ther. 203:1073912019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Garzon-Hernandez C, Ramirez-Merino N and
Soberon MCM: Molecular targeted therapy in oncology focusing on DNA
repair mechanisms. Arch Med Res. 53:807–817. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang R, Sun Y, Li C, Xue Y and Ba X:
Targeting the DNA damage response for cancer therapy. Int J Mol
Sci. 24:159072023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Basu B, Yap TA, Molife LR and de Bono JS:
Targeting the DNA damage response in oncology: Past, present and
future perspectives. Curr Opin Oncol. 24:316–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yusa K and Tsuruo T: Reversal mechanism of
multidrug resistance by verapamil: Direct binding of verapamil to
P-glycoprotein on specific sites and transport of verapamil outward
across the plasma membrane of K562/ADM cells. Cancer Res.
49:5002–5006. 1989.PubMed/NCBI
|
|
54
|
Yan T, Deng S, Metzger A, Godtel-Armbrust
U, Porter AC and Wojnowski L: Topoisomerase II{alpha}-dependent and
-independent apoptotic effects of dexrazoxane and doxorubicin. Mol
Cancer Ther. 8:1075–1085. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jirkovský E, Jirkovská A,
Bavlovič-Piskáčková H, Skalická V, Pokorná Z, Karabanovich G,
Kollárová-Brázdová P, Kubeš J, Lenčová-Popelová O, Mazurová Y, et
al: Clinically translatable prevention of anthracycline
cardiotoxicity by dexrazoxane is mediated by topoisomerase II beta
and not metal chelation. Circ Heart Fail. 14:e0082092021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jirkovska A, Karabanovich G, Kubes J,
Skalická V, Melnikova I, Korábečný J, Kučera T, Jirkovský E,
Nováková L, Bavlovič Piskáčková H, et al: Structure-activity
relationship study of dexrazoxane analogues reveals ICRF-193 as the
most potent bisdioxopiperazine against anthracycline toxicity to
cardiomyocytes due to its strong topoisomerase IIβ interactions. J
Med Chem. 64:3997–4019. 2021. View Article : Google Scholar
|
|
57
|
Huelsenbeck SC, Schorr A, Roos WP,
Huelsenbeck J, Henninger C, Kaina B and Fritz G: Rac1 protein
signaling is required for DNA damage response stimulated by
topoisomerase II poisons. J Biol Chem. 287:38590–38599. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wartlick F, Bopp A, Henninger C and Fritz
G: DNA damage response (DDR) induced by topoisomerase II poisons
requires nuclear function of the small GTPase Rac. Biochim Biophys
Acta. 1833:3093–3103. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Skvortsov S, Dudas J, Eichberger P,
Witsch-Baumgartner M, Loeffler-Ragg J, Pritz C, Schartinger VH,
Maier H, Hall J, Debbage P, et al: Rac1 as a potential therapeutic
target for chemo-radioresistant head and neck squamous cell
carcinomas (HNSCC). Br J Cancer. 110:2677–2687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li Q, Qin T, Bi Z, Hong H, Ding L, Chen J,
Wu W, Lin X, Fu W, Zheng F, et al: Rac1 activates non-oxidative
pentose phosphate pathway to induce chemoresistance of breast
cancer. Nat Commun. 11:14562020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kraft FB, Biermann L, Schäker-Hübner L,
Hanl M, Hamacher A, Kassack MU and Hansen FK: Hydrazide-based class
I selective HDAC inhibitors completely reverse chemoresistance
synergistically in platinum-resistant solid cancer cells. J Med
Chem. 67:17796–17819. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tang SW, Thomas A, Murai J, Trepel JB,
Bates SE, Rajapakse VN and Pommier Y: Overcoming resistance to
DNA-targeted agents by epigenetic activation of schlafen 11
(SLFN11) expression with class I histone deacetylase inhibitors.
Clin Cancer Res. 24:1944–1953. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vollmer J, Ecker J, Hielscher T,
Valinciute G, Ridinger J, Jamaladdin N, Peterziel H, van Tilburg
CM, Oehme I, Witt O, et al: Class I HDAC inhibition reduces DNA
damage repair capacity of MYC-amplified medulloblastoma cells. J
Neurooncol. 164:617–632. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bangert A, Hacker S, Cristofanon S,
Debatin KM and Fulda S: Chemosensitization of glioblastoma cells by
the histone deacetylase inhibitor MS275. Anticancer Drugs.
22:494–499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gianni L, Herman EH, Lipshultz SE, Minotti
G, Sarvazyan N and Sawyer DB: Anthracycline cardiotoxicity: From
bench to bedside. J Clin Oncol. 26:3777–3784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ferreira AL, Matsubara LS and Matsubara
BB: Anthracycline-induced cardiotoxicity. Cardiovasc Hematol Agents
Med Chem. 6:278–281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Henninger C and Fritz G: Statins in
anthracycline-induced cardiotoxicity: Rac and Rho, and the
heartbreakers. Cell Death Dis. 8:e25642017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kucuk P, Abbey L, Schmitt J, Henninger C
and Fritz G: Cardiomyocytes, cardiac endothelial cells and
fibroblasts contribute to anthracycline-induced cardiac injury
through RAS-homologous small GTPases RAC1 and CDC42. Pharmacol Res.
203:1071652024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yoshida M, Shiojima I, Ikeda H and Komuro
I: Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA
damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin
through the inhibition of Rac1 activity. J Mol Cell Cardiol.
47:698–705. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ma J, Wang Y, Zheng D, Wei M, Xu H and
Peng T: Rac1 signalling mediates doxorubicin-induced cardiotoxicity
through both reactive oxygen species-dependent and -independent
pathways. Cardiovasc Res. 97:77–87. 2013. View Article : Google Scholar
|
|
71
|
McGowan JV, Chung R, Maulik A, Piotrowska
I, Walker JM and Yellon DM: Anthracycline chemotherapy and
cardiotoxicity. Cardiovasc Drugs Ther. 31:63–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Gewirtz DA: A critical evaluation of the
mechanisms of action proposed for the antitumor effects of the
anthracycline antibiotics adriamycin and daunorubicin. Biochem
Pharmacol. 57:727–741. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fry AM, Chresta CM, Davies SM, Walker MC,
Harris AL, Hartley JA, Masters JR and Hickson ID: Relationship
between topoisomerase II level and chemosensitivity in human tumor
cell lines. Cancer Res. 51:6592–6595. 1991.PubMed/NCBI
|
|
74
|
Sun X and Kaufman PD: Ki-67: More than a
proliferation marker. Chromosoma. 127:175–186. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ledvin L, Gassaway BM, Tawil J, Urso O,
Pizzo D, Welsh KA, Bolhuis DL, Fisher D, Bonni A, Gygi SP, et al:
The anaphase-promoting complex controls a
ubiquitination-phosphoprotein axis in chromatin during
neurodevelopment. Dev Cell. 58:2666–2683.e9. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Uxa S, Castillo-Binder P, Kohler R,
Stangner K, Muller GA and Engeland K: Ki-67 gene expression. Cell
Death Differ. 28:3357–3370. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lamberti G, Andrini E, Sisi M, Federico AD
and Ricciuti B: Targeting DNA damage response and repair genes to
enhance anticancer immunotherapy: Rationale and clinical
implication. Future Oncol. 16:1751–1766. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Friedrich A, Assmann AS, Schumacher L,
Stuijvenberg JV, Kassack MU, Schulz WA, Roos WP, Hansen FK,
Pflieger M, Kurz T and Fritz G: In vitro assessment of the
genotoxic hazard of novel hydroxamic acid- and benzamide-type
histone deacetylase inhibitors (HDACi). Int J Mol Sci. 21:47472020.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sandrock K, Bielek H, Schradi K, Schmidt G
and Klugbauer N: The nuclear import of the small GTPase Rac1 is
mediated by the direct interaction with karyopherin alpha2.
Traffic. 11:198–209. 2010. View Article : Google Scholar
|
|
80
|
Kitzinger R, Fritz G and Henninger C:
Nuclear RAC1 is a modulator of the doxorubicin-induced DNA damage
response. Biochim Biophys Acta Mol Cell Res. 1869:1193202022.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fritz G and Henninger C: Rho GTPases:
Novel players in the regulation of the DNA damage response?
Biomolecules. 5:2417–2434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Thurn KT, Thomas S, Raha P, Qureshi I and
Munster PN: Histone deacetylase regulation of ATM-mediated DNA
damage signaling. Mol Cancer Ther. 12:2078–2087. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Roos WP and Krumm A: The multifaceted
influence of histone deacetylases on DNA damage signalling and DNA
repair. Nucleic Acids Res. 44:10017–10030. 2016.PubMed/NCBI
|
|
84
|
Kachhap SK, Rosmus N, Collis SJ,
Kortenhorst MS, Wissing MD, Hedayati M, Shabbeer S, Mendonca J,
Deangelis J, Marchionni L, et al: Downregulation of homologous
recombination DNA repair genes by HDAC inhibition in prostate
cancer is mediated through the E2F1 transcription factor. PLoS One.
5:e112082010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Karwatsky J, Lincoln MC and Georges E: A
mechanism for P-glycoprotein-mediated apoptosis as revealed by
verapamil hypersensitivity. Biochemistry. 42:12163–12173. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Bellamy WT, Dorr RT, Dalton WS and Alberts
DS: Direct relation of DNA lesions in multidrug-resistant human
myeloma cells to intracellular doxorubicin concentration. Cancer
Res. 48:6360–6364. 1988.PubMed/NCBI
|
|
87
|
Bellamy WT, Dalton WS, Kailey JM, Gleason
MC, McCloskey TM, Dorr RT and Alberts DS: Verapamil reversal of
doxorubicin resistance in multidrug-resistant human myeloma cells
and association with drug accumulation and DNA damage. Cancer Res.
48:6365–6370. 1988.PubMed/NCBI
|
|
88
|
Huelsenbeck J, Henninger C, Schad A,
Lackner KJ, Kaina B and Fritz G: Inhibition of Rac1 signaling by
lovastatin protects against anthracycline-induced cardiac toxicity.
Cell Death Dis. 2:e1902011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ohlig J, Henninger C, Zander S, Merx M,
Kelm M and Fritz G: Rac1-mediated cardiac damage causes diastolic
dysfunction in a mouse model of subacute doxorubicin-induced
cardiotoxicity. Arch Toxicol. 92:441–453. 2018. View Article : Google Scholar
|