Introduction
Hepatocellular carcinoma (HCC), one of the highest
prevalence of liver cancer, caused by hepatitis B virus (HBV), and
more than 350 million people were affected by HBV worldwide
(1,2). All over the world, majority of
HBV-related deaths were closely associated with HCC for each year,
which indicates the third leading cause of cancer deaths (2,3). As we
known, the HBV was a major etiologic agent for HCC, and prevalent
in China, Southeast Asia and sub-Saharan Africa (4). Notably, an increasing number of
studies indicated that HBV was implicated in tumorigenesis, of
which the main mechanism as follows (3-7):
i) Expression of viral proteins, in particular, from HBV gene X
(HBx), to modulate cell proliferation and viability, ii)
accumulation of genetic damage due to hepatic inflammation mediated
by virus-specific T cells, and iii) it is intriguing that
integration of HBV DNA into the host genome to alter the function
of endogenous genes or induce chromosomal instability.
There are increasing evidences that the events of
the HBV integration occurred in the HCC, and affected function of
HCC genome. Some researchers found that HBV DNA integration
distributed different chromosomal sites in the host genome
(8), and they could trigger
chromosomal changes, genomic instability, or changes in the
expression of human genes (9). It
was reported that HBV integration events are involved in chromosome
fragile sites or repetitive sequences and are usually followed by
local rearrangement, all of which relate to higher genomic
instability (10). HBV insertion
was found to target the retinoic acid receptor-β (RARB) gene or the
human cyclin A2 (CCNA2) and to generate chimeric oncogenic proteins
(11). Until now, all reports
involving HBV DNA integration implied that integration plays a role
in the transformation (12-14).
HBV integration into the host cell genome has also been reported,
which resulted in gene mutations, insertions, deletions or
rearrangements of the host genome (15-17).
Recently, a great number of insertion sites were identified by
next-generation sequencing (4,18,19).
Interestingly, telomerase reverse transcriptase (TERT) and
mixed-lineage leukemia 4 (MLL4) gene were frequently targeted by
HBV in HCC tissue, and the latter may play a major role in HCC
carcinogenesis (20). Since firstly
discovered at the TERT gene, HBV integration breakpoints have been
widely reported in some gene targets, such as the fatty acyl CoA
reductase 2 (FAR2), inositol triphosphate receptor type 1
(ITPR1,IP3R1), Interleukin-1-receptor-associated kinase 2 (IRAK2),
mitogen associated protein kinase 1 (MAPK1), mixed-lineage leukemia
2 (MLL2) and MLL4 genes (2,21,22).
Although HBV integration event has been reported in the HCC, and
its mechanism is not clear. Therefore, systematic analysis of HBV
integration targets could elucidate HCC physiologies and diseases
development processes, and predict novel therapeutic targets. Here,
to directly detect HBV integration breakpoints at whole genome
level, we constructed four small sequencing libraries and
characterized the HBV integration profiles from four patients with
HCC. Therefore, our research revealed HBV integration events in the
HCC, and acquired insight into the pathogenesis of HCC.
Materials and methods
Human samples and DNA extractions
Four tumor samples were collected from the patients
who underwent curative primary hepatectomy or liver transplantation
in the Guilin No. 924 Hospital. They were have precisely diagnosed
with HCC and associated with HBV infection in the department of
pathology, and the hepatitis B surface antigen (HBsAg) was
positively expressed and HBV-DNA quantification was greater than
103 copies/ml (Fig. 1A
and B and Table I). Subsequently, total DNA was
extracted from the tumor samples using QIAamp DNA Micro kit (Qiagen
Ltd.) according to the manufacturer's methods. Informed consent was
obtained from the participant donors, and the protocol for the
research project has been approved by a Ethics Committee of the
Guilin No. 924 Hospital accord with China's Guidelines.
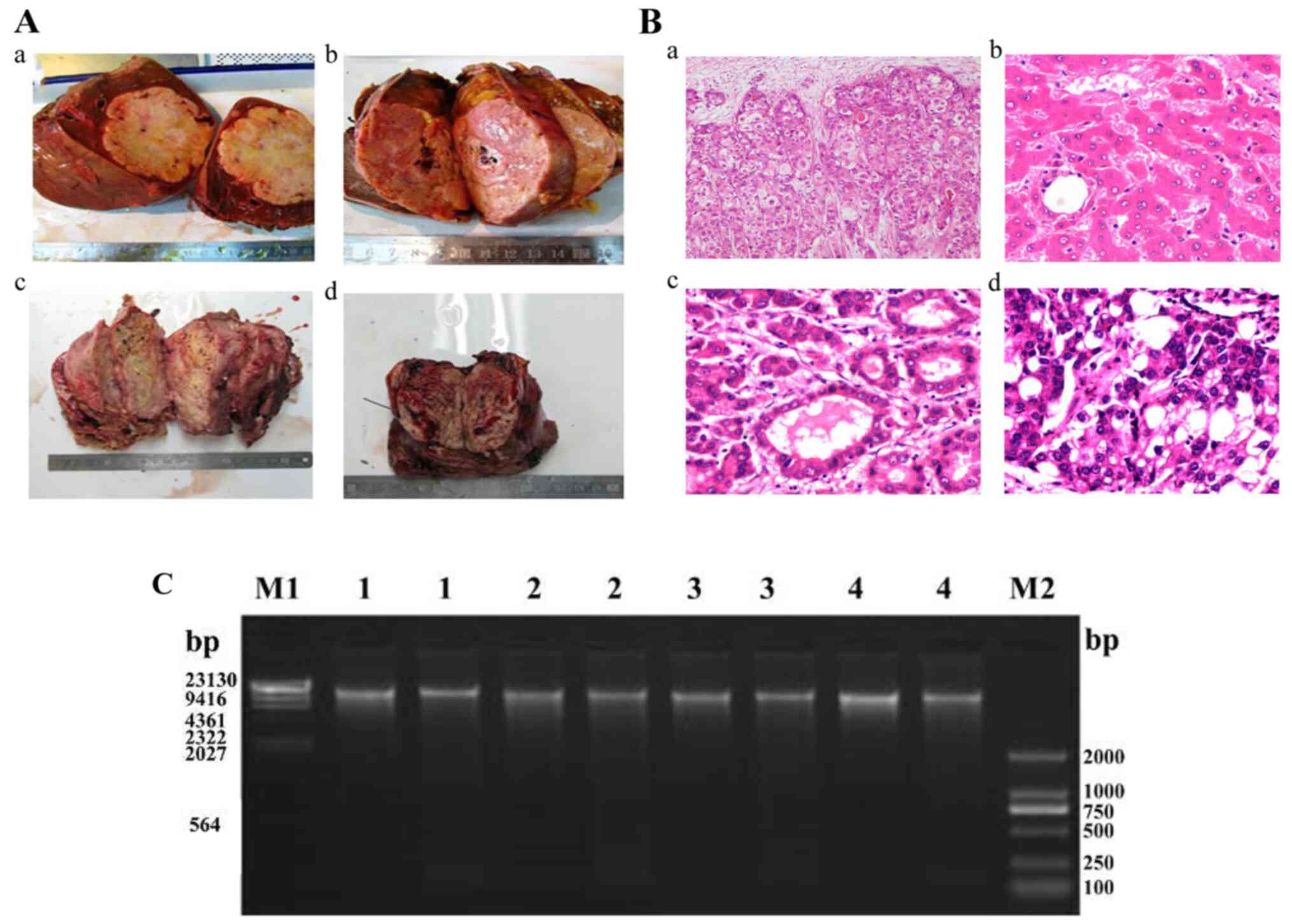 | Figure 1Clinical characteristics of the four
patients enrolled for deep sequencing. (A) Excised tumors of the
four patients with HCC included in the present study (Aa, patient
1; Ab, patient 2; Ac, patient 3; Ad, patient 4). (B) Pathological
images of tumor tissues from the four patients with HCC
(magnification, x20; Ba, patient 1; Bb, patient 2; Bc, patient 3;
Bd, patient 4). (C) Integrity analysis of total DNA using
preparative agarose gel electrophoresis. HCC, hepatocellular
carcinoma. |
 | Table IClinical and biochemical
characteristics of the four patients prior to whole-genome
sequencing. |
Table I
Clinical and biochemical
characteristics of the four patients prior to whole-genome
sequencing.
| ID | Age (years) | Sex | HBsAg | Anti-HBs | Anti-HBc | AFP | HBV (copies/ml) |
|---|
| 1 | 38 | Male | + | - | + | + | 9,337 |
| 2 | 52 | Male | + | - | + | + | 6,060 |
| 3 | 42 | Male | + | - | + | + | 3,504 |
| 4 | 50 | Male | + | - | + | + | 2,139 |
Hybridized Libraries construction and
sequencing
DNA purification and library preparation were
conducted as previously reported methods (2). Brief, integrity and quality of total
DNA were evaluated by the Qubit Fluorometer and agarose gel
electrophoresis (Fig. 1). Next, the
genomic DNA were fragmented randomly by a Bioruptor Pico
(Diagenode, B01060001) into the target DNA fragments (170 bp) from
3 µg of total DNA. And then, the target fragments were subjected to
perform DNA end repair, and added a single ‘A’ nucleotide in the 3'
ends of the target DNA fragments. Subsequently, PCR were performed
after adapters ligating, size-selection and tailed random primers
addition to obtain sufficient amplification products for libraries
construction. For these constructed libraries, we used the virus
probe to hybridize, and enriched the target DNA fragments. The
hybridized target DNA fragments were eluted using AW2 Buffer in
Elution column (QIAamp minElute Column) And then, PCR were
performed to obtain sufficient amplification products for the
hybridized libraries construction. PCR products were collected and
preceded to 101 cycle's paired-end index sequencing in the Illumina
HiSeq 2,000 sequencer according to manufacturer's instructions
(Illumina Inc.).
Hybrid reference genome construction
and alignment analysis
The advanced analysis begins with raw data generated
from the Illumina platform. The sequence tags with adapter ligation
or low quality or Ns and low base quality were filtered out to
obtain clean tags, which were subjected to further analyze. Next,
we combined the human reference genome (hg19) and the HBV genome
(NC_003977.1) together to build a hybrid reference genome for
alignment analysis. The clean tags were aligned to the hybrid
reference genome by Burrows-Wheeler Aligner (BWA) (23), and the alignment results were saved
in BAM format files. These files were further preprocessed to be
the final BAM files for the HBV integration detection, such as
sorting according to the alignment position, marking duplicate
reads caused by PCR.
SNPs and InDels detection and
annotation
Further bioinformatic analysis for the final BAM
files, single Nucleotide Polymorphisms (SNPs) are detected by GATK
(24). The unique genotypes were
identified for each individual, which has the highest probability
at a given locus, the consensus sequence tags were collected and
saved as CNS format. And then, the high confidence SNP datasets
were acquired by filtering the consensus sequence tags. In
addition, we detect the small Insertion and Deletion (InDels) using
pair-end reads for gap alignment.
HBV integration detection
We detected the HBV integrations by seeksv, and the
seeksv was an in-house tool. The HBV integration positions were
filtered, because of the sequences with a sum of junction read
number and abnormal read pair number smaller than 2. After
integration positions identification, the distribution of the HBV
integrations was analyzed, which could evaluate the numbers of
integrations.
Results
Analysis of sequencing data from four
tumor samples
Integrated genomic DNA was captured from tumor
tissues of four patients with HCC and sequenced by Illumina HiSeq
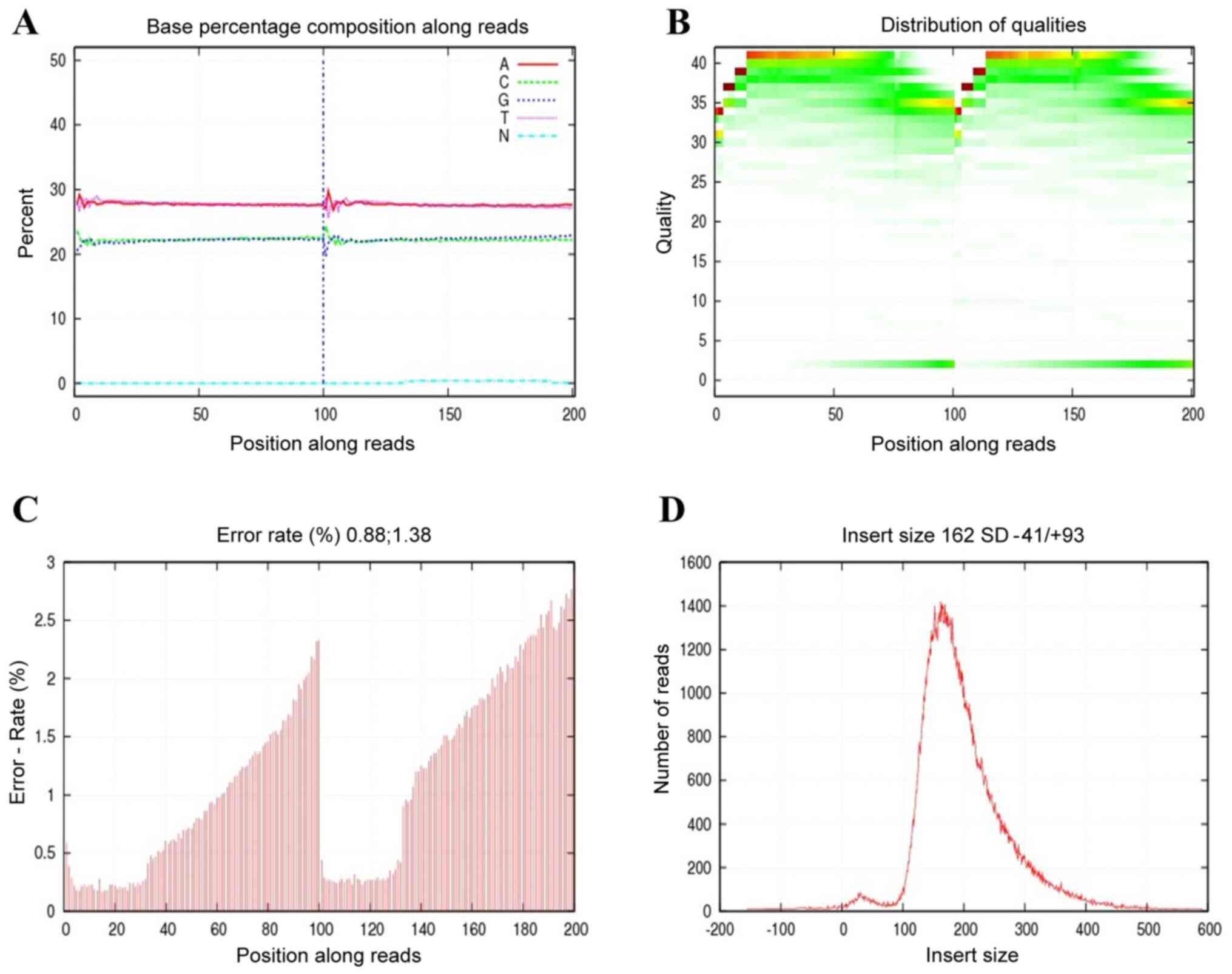
2,000. Before doing any further analysis, quality control is
essential to detect whether the data is qualified. As shown in
Fig. 2, the sequencing data was
good, which satisfied the subsequent advanced analysis. These raw
reads with adapters, low quality and unknown bases were removed.
The remaining data were called as clean read data, which were
submitted for further bioinformatic analysis. In total, an average
of 11,413,090 raw reads were obtained for each sample. A Phred
quality score is a measure that was used to evaluate the quality
for sequencing data. Phred quality scores sQ were defined as the
sequencing data quality from tumor tissues of four patients, and
the E indicated the sequencing error rate. They had a
relationship as follows: sQ=-10log10E. It's worth
noting that the GC content accounted for 43.68-44.64% when the
Phred score was >30.
Further statistical analysis for clean reads, we
identified 11,800,974, 11,216,998, 11,026,546 and 11,607,842 clean
reads for Patient 1-3 and 4, of which 92.82, 95.95, 97.21 and
97.29%, respectively, were properly aligned to the hybrid reference
genome. The average sequencing depths were 0.07, 0.08, 0.05 and
0.07-fold, and 5.47, 6.17, 3.63 and 4.93 of the hybrid reference
genome were covered by the clean reads, respectively. Coverage at
least 20-fold were 0.01% for four tumor tissues (Table II).
| Table IIQuality control results of alignment
data. |
Table II
Quality control results of alignment
data.
| Categories | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|
| Clean reads | 11,800,974 | 11,216,998 | 11,026,546 | 11,607,842 |
| Clean bases (bp) | 1,180,097,400 | 1,121,699,800 | 1,102,654,600 | 1,160,784,200 |
| Mapped reads | 10,953,113 | 10,763,243 | 10,718,360 | 11,293,616 |
| Mapped bases
(bp) | 1,075,438,128 | 1,058,305,701 | 1,049,564,676 | 1,108,297,233 |
| Mapping rate
(%) | 92.82 | 95.95 | 97.21 | 97.29 |
| Uniq reads | 10,295,380 | 10,118,513 | 10,040,852 | 10,627,022 |
| Uniq bases
(bp) | 1,012,882,982 | 996,468,729 | 985,514,390 | 1,045,019,717 |
| Unique rate
(%) | 94.00 | 94.01 | 93.68 | 94.10 |
| Duplicate
reads | 8,692,617 | 8,282,575 | 9,152,300 | 9,265,976 |
| Duplicate rate
(%) | 79.36 | 76.95 | 85.39 | 82.05 |
| Mismatch bases
(bp) | 5,924,651 | 4,942,501 | 5,471,027 | 4,874,040 |
| Mismatch rate
(%) | 0.55 | 0.47 | 0.52 | 0.44 |
| Average sequencing
depth | 0.07 | 0.08 | 0.05 | 0.07 |
| Coverage (%) | 5.47 | 6.17 | 3.63 | 4.93 |
| Coverage at least
4X (%) | 0.05 | 0.05 | 0.05 | 0.05 |
| Coverage at least
10X (%) | 0.02 | 0.02 | 0.02 | 0.02 |
| Coverage at least
20X (%) | 0.01 | 0.01 | 0.01 | 0.01 |
HBV integration analysis for four
tumor tissues
In order to explore the HBV integration events in
the HCC patients, we used the seeksv to detect the HBV integration
sites, and conducted the annotation and classification by ANNOVAR.
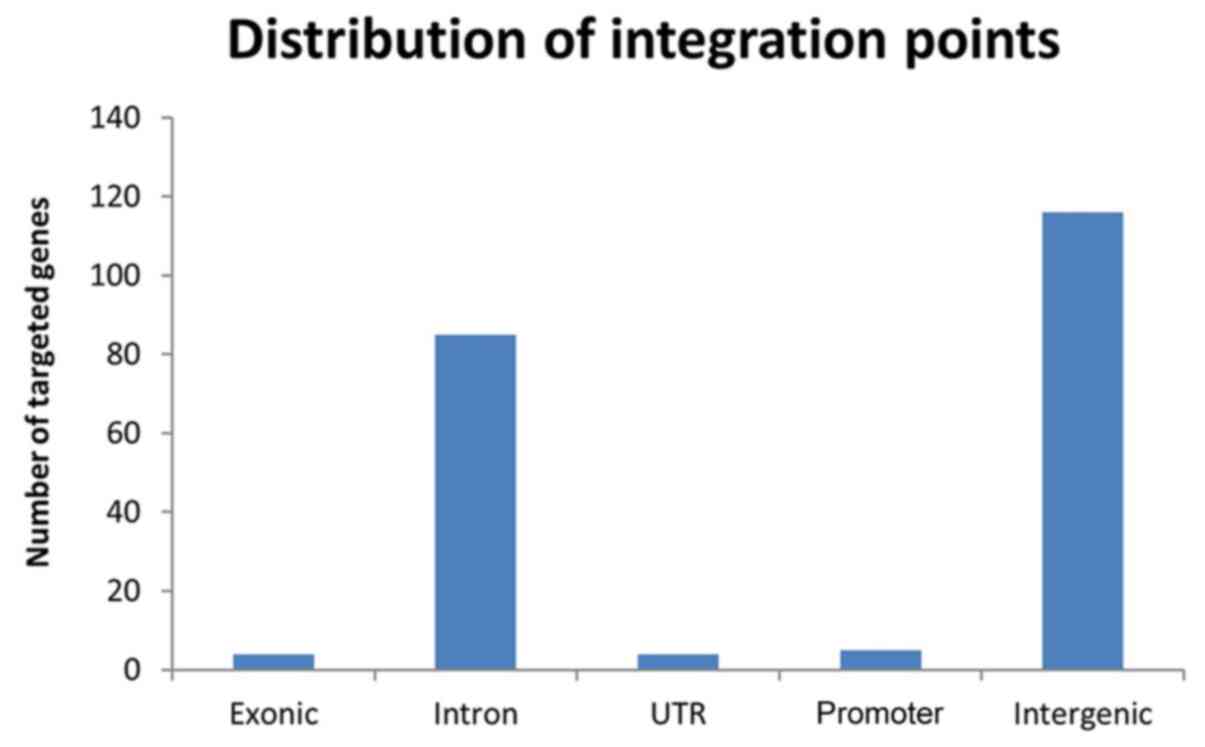
In total, the 220 HBV integration events were detected from the
tumor tissues of four HCC patients, and an average of 55
breakpoints for each sample. The integration breakpoints
distributions for four samples were 143, 40, 25 and 12,
respectively (Fig. 3), which
evaluated the numbers of integrations located in different gene
regions. Our results indicated that the HBV integration breakpoints
had a preference for chromosomes 2, 3 and 5 (Table III). Majority of HBV breakpoints
in HCC were found near coding genes (116 of 220 breakpoints). Eight
of the 220 HBV breakpoints were located in known coding genes, such
as ITGA9, FAM19A4, TTBK1, POT1, FLNC, OR51V1, HSPA4, ITGA4, and
these breakpoints were significantly over-represented in exon and
promoter (defined as 0 to -2 kb relative to the transcriptional
start site) regions. In contrast, 85 of 220 HBV breakpoints were
mainly located in introns. In addition, we identified 688 potential
mutations of SNPs four samples by aligning to the hybrid reference
genome. Majority of SNPs (666/688) were somatic single-base
mutations, while the minority (22/688) occured small insertions and
deletions (InDels), which were detected by pair-end reads for gap
alignment (Table SI).
| Table IIIDistribution of the targets in
chromosomes. |
Table III
Distribution of the targets in
chromosomes.
| chr | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|
| chr1 | 9 | 3 | 1 | 0 |
| chr2 | 10 | 4 | 6 | 8 |
| chr3 | 11 | 3 | 6 | 0 |
| chr4 | 4 | 0 | 2 | 0 |
| chr5 | 10 | 1 | 8 | 0 |
| chr6 | 5 | 1 | 0 | 2 |
| chr7 | 6 | 0 | 3 | 0 |
| chr8 | 3 | 1 | 0 | 1 |
| chr9 | 6 | 2 | 0 | 0 |
| chr10 | 6 | 3 | 0 | 0 |
| chr11 | 7 | 0 | 1 | 0 |
| chr12 | 3 | 0 | 2 | 0 |
| chr13 | 10 | 0 | 1 | 0 |
| chr14 | 8 | 0 | 0 | 0 |
| chr15 | 8 | 2 | 1 | 0 |
| chr16 | 4 | 2 | 0 | 0 |
| chr17 | 6 | 0 | 3 | 0 |
| chr18 | 3 | 2 | 0 | 0 |
| chr19 | 3 | 0 | 0 | 1 |
| chr20 | 8 | 0 | 2 | 0 |
| chr21 | 0 | 0 | 0 | 0 |
| chr22 | 5 | 0 | 0 | 0 |
| chrX | 2 | 0 | 0 | 0 |
Discussion
Hepatocellular carcinoma (HCC) is one of the most
lethal malignancies, and it's development was a multifactorial
process because of several direct and indirect mechanisms (25). HBV DNA integration events were
frequently detected in the HCC patients, which was observed more
usually in the tumors than in adjacent liver tissues, and was
associated with patient's survival (4,21).
Exactly as our clinical examination results for four HCC patients,
the hepatitis B surface antigen (HBsAg) was positively expressed,
and HBV DNA integration was frequently detected in the most HCC
patients (26). Linghao Zhao and
his colleagues identified 4,225 HBV integration events in tumor and
adjacent non-tumor samples from 426 patients with HCC, and found
that they preferred rare fragile sites and functional genomic
regions, such as CpG islands (27).
In addition, some researchers observed massive genomic
perturbations near viral integration sites, such as direct gene
disruption, viral promoter-driven human transcription, viral-human
transcript fusion and DNA copy number alteration (28).
Traditionally, majority of the HBV integration
events were detected by PCR-based methods, such as Alu-HBV PCR, and
preferred the Alu regions (2). As a
result, the HBV integration that located in the Alu regions can be
frequently detected (2). Several
studies have shown that the whole-genome sequencing combined with
HBV DNA capture for HBV DNA integration events detection was
feasible and effective, with considerable savings in time and cost,
and could help improve the early diagnosis of hepatocellular
carcinomas, and thus the survival rate. (4,21,27).
In our study, we selected the whole-genome sequencing combined with
HBV DNA capture to identify the HBV integration breakpoints from
four tumor samples, which learns about the biological
characteristics of HBV integrated into the human genome and
provides some references for targeted therapy of HCC patients in
the future.
Notably, we successfully identified the HBV
integration breakpoints at the single base level, which can
effectively validate confidence of HBV capture sequencing (2). Our data indicate that a large
proportion of SNPs (666/688) were identified, which suggested the
main tendency of HBV integration, and the InDels occasionally
occurred in the HBV integration events. Subsequently, we analyzed
the HBV integration breakpoints that distributed in distinct
genomic elements, and found that majority of HBV integration
breakpoints in the four tumor samples located in the coding region
(116 of 220 HBV integration breakpoints). Of the 220 HBV
integration breakpoints, 9 were located in known coding genes that
significantly over-represented in exon and promoter regions. In
contrast, 85 of 220 HBV integration breakpoints were mainly located
in the introns, which suggested the preference among the HBV
integration breakpoints. It is intriguing that virus genes inserted
into host genomic DNA in totally random ways in the previously
studies (29). However, some recent
studies indicated that virus genes integration events had
preference among the different regions (4,9,19). Our
HBV integration profiles also demonstrated that HBV integration
breakpoints preferentially landed in transcription units and
specific chromosomes. Ding et al (9) found the HBV integration preference
located in chromosomes 11 and 17. However, a preference for
chromosome 3 has been reported in chronic hepatitis tissues without
HCC by Alu-PCR (21). Our results
indicated that the HBV integration breakpoints had a preference for
chromosomes 2, 3 and 5. There are some potential reasons for the
preferences, including the great HCC heterogeneity, different
subclasses of HCC, sub-genotypes with different integration
capabilities, etc. Therefore, we need a large number of samples to
verify the results of this study, further explore the function of
these target genes in the pathological mechanism of HCC and the
characteristics they are in the TCGA database, which will provide
more references for our future to discussion of ‘therapeutics in
HCC patients. Additionally, future analysis for the potential
relationship between HBV subtypes and their integration frequencies
was needed in the future.
In summary, our results strengthened understanding
that the HBV DNA integration events are implicated in HCC
physiologies and diseases, and further demonstrated that the HBV
insertional sequence capturing may be a useful tool to study the
related human diseases.
Supplementary Material
Summary of single nucleotide
polymorphisms and InDels from thefour tumor samples.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Scientific
Research and Technology Development Planning Project of Guilin
(grant no. 2016012702-1), the Science and Technology Planning
Project of Guangdong Province, China (grant no. 2017B020209001) and
the Science and Technology Planning Project of Guangdong Province,
China (grant no. 2016A020215027).
Availability of data and materials
The datasets used and/or analyzed are available from
the corresponding author on reasonable request.
Authors' contributions
WS, MY and YD conceived and designed the
experiments. GY, FL, MO, CL, JC, HL, YZ, WX, YW and YX performed
the experiments. GY, FL, MO, CL and WX analyzed the data. GY, WS
and MY drafted the manuscript. GY and YD revised the manuscript
critically for important intellectual content. MY and YD obtained
the funding. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was performed in accordance with
the Helsinki Declaration and approved by the Ethics Committee of
the Guilin no. 924 Hospital. Informed consent was obtained from the
participant donors.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ringelhan M, Pfister D, O'Connor T,
Pikarsky E and Heikenwalder M: The immunology of hepatocellular
carcinoma. Nat Immunol. 19:222–232. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li W, Zeng X, Lee NP, Liu X, Chen S, Guo
B, Yi S, Zhuang X, Chen F, Wang G, et al: HIVID: An efficient
method to detect HBV integration using low coverage sequencing.
Genomics. 102:338–344. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wu G, Ding H and Zeng C: Overview of HBV
whole genome data in public repositories and the Chinese HBV
reference sequences. Prog Nat Sci. 18:13–20. 2008.
|
|
4
|
Sung WK, Zheng H, Li S, Chen R, Liu X, Li
Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al: Genome-wide
survey of recurrent HBV integration in hepatocellular carcinoma.
Nat Genet. 44:765–769. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Gehring AJ, Ho ZZ, Tan AT, Aung MO, Lee
KH, Tan KC, Lim SG and Bertoletti A: Profile of tumor
antigen-specific CD8 T cells in patients with hepatitis B
virus-related hepatocellular carcinoma. Gastroenterology.
137:682–690. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lau CC, Sun T, Ching AK, He M, Li JW, Wong
AM, Co NN, Chan AW, Li PS, Lung RW, et al: Viral-human chimeric
transcript predisposes risk to liver cancer development and
progression. Cancer Cell. 25:335–349. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Neuveut C, Wei Y and Buendia MA:
Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol.
52:594–604. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tokino T and Matsubara K: Chromosomal
sites for hepatitis B virus integration in human hepatocellular
carcinoma. J Virol. 65:6761–6764. 1991.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ding D, Lou X, Hua D, Yu W, Li L, Wang J,
Gao F, Zhao N, Ren G, Li L and Lin B: Recurrent targeted genes of
hepatitis B virus in the liver cancer genomes identified by a
next-generation sequencing-based approach. PLoS Genet.
8(e1003065)2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Feitelson MA and Lee J: Hepatitis B virus
integration, fragile sites, and hepatocarcinogenesis. Cancer Lett.
252:157–170. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Taha SE, El-Hady SA, Ahmed TM and Ahmed
IZ: Detection of occult HBV infection by nested PCR assay among
chronic hepatitis C patients with and without hepatocellular
carcinoma. Egypt J Med Hum Genet. 14:353–360. 2013.
|
|
12
|
Li X, Zhang J, Yang Z, Kang J, Jiang S,
Zhang T, Chen T, Li M, Lv Q, Chen X, et al: The function of
targeted host genes determines the oncogenicity of HBV integration
in hepatocellular carcinoma. J Hepatol. 60:975–984. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Guerrieri F, Belloni L, Pediconi N and
Levrero M: Molecular mechanisms of HBV-associated
hepatocarcinogenesis. Semin Liver Dis. 33:147–156. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rey-Cuille MA, Njouom R, Bekondi C, Seck
A, Gody C, Bata P, Garin B, Maylin S, Chartier L, Simon F and Vray
M: Hepatitis B virus exposure during childhood in Cameroon, Central
African Republic and Senegal after the integration of HBV vaccine
in the expanded program on immunization. Pediatr Infect Dis J.
32:1110–1115. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bonilla Guerrero R and Roberts LR: The
role of hepatitis B virus integrations in the pathogenesis of human
hepatocellular carcinoma. J Hepatol. 42:760–777. 2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bok J, Kim KJ, Park MH, Cho SH, Lee HJ,
Lee EJ, Park C and Lee JY: Identification and extensive analysis of
inverted-duplicated HBV integration in a human hepatocellular
carcinoma cell line. BMB Rep. 45:365–370. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Arzumanyan A, Reis HM and Feitelson MA:
Pathogenic mechanisms in HBV- and HCV-associated hepatocellular
carcinoma. Nat Rev Cancer. 13:123–135. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Jiang Z, Jhunjhunwala S, Liu J, Haverty
PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S,
et al: The effects of hepatitis B virus integration into the
genomes of hepatocellular carcinoma patients. Genome Res.
22:593–601. 2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S,
Nault JC, Jaoui D, Gaston Mathe Y, Laurent C, Laurent A,
Bioulac-Sage P, et al: Integration of tumour and viral genomic
characterisations in HBV-related hepatocellular carcinomas. Gut.
64:820–829. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Murakami Y, Saigo K, Takashima H, Minami
M, Okanoue T, Bréchot C and Paterlini-Bréchot P: Large scaled
analysis of hepatitis B virus (HBV) DNA integration in HBV related
hepatocellular carcinomas. Gut. 54:1162–1168. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Saigo K, Yoshida K, Ikeda R, Sakamoto Y,
Murakami Y, Urashima T, Asano T, Kenmochi T and Inoue I:
Integration of hepatitis B virus DNA into the myeloid/lymphoid or
mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in
human hepatocellular carcinoma. Hum Mutat. 29:703–708.
2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bauer DC: Variant calling comparison
CASAVA1.8 and GATK. Nat Prec, 2011.
|
|
25
|
Lim L, Tran BM, Vincan E, Locarnini S and
Warner N: HBV-related hepatocellular carcinoma: The role of
integration, viral proteins and miRNA. Future Virol. 7:1237–1249.
2012.
|
|
26
|
Tamori A, Nishiguchi S, Kubo S, Narimatsu
T, Habu D, Takeda T, Hirohashi K and Shiomi S: HBV DNA integration
and HBV-transcript expression in non-B, non-C hepatocellular
carcinoma in Japan. J Med Virol. 71:492–498. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhao LH, Liu X, Yan HX, Li WY, Zeng X,
Yang Y, Zhao J, Liu SP, Zhuang XH, Lin C, et al: Genomic and
oncogenic preference of HBV integration in hepatocellular
carcinoma. Nat Commun. 7(12992)2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang Z: Abstract LB-400: The effects of
hepatitis B virus integration into the genomes of hepatocellular
carcinoma patients. Cancer Res. 72 (Suppl 8):LB–400.
2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bréchot C, Gozuacik D, Murakami Y and
Paterlini-Bréchot P: Molecular bases for the development of
hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC).
Semin Cancer Biol. 10:211–231. 2000.PubMed/NCBI View Article : Google Scholar
|