Introduction
Globally, bladder cancer is ranked as the tenth most
frequently diagnosed cancer and is the thirteenth leading cause of
cancer-related mortalities (1,2).
According to 2024 global cancer statistics, the number of new
bladder cancer cases is estimated to have exceeded 600,000
worldwide. The age-standardized incidence rate is ~6.0 cases per
100,000 individuals, showing a slow upward trend. In the same year,
the number of bladder cancer-related deaths is projected to be
~220,000, with a mortality rate maintained at ~2.0 per 100,000
individuals. In recent years, with advancements in diagnostic
technologies and optimization of treatment regimens, the survival
rate of patients with bladder cancer has significantly improved in
some regions with high levels of development (1). The survival rate of bladder cancer
still requires improvement through early detection or targeted
anticancer therapy (3,4). Therefore, early diagnostic biomarkers
and the development of novel therapeutic targets for bladder cancer
remain a focus of our research.
Human plasma proteins exhibit great potential in
disease diagnosis and treatment monitoring, with several successes
achieved over the decades (5,6).
Previous studies have identified various proteins such as membrane
proteins, α-2-macroglobulin, and SPP1 protein, as being associated
with bladder cancer risk (7-9).
However, research on bladder cancer still faces limitations. In
particular, the investigation into proteins associated with the
development and progression of bladder cancer has not been
comprehensively carried out.
Large-scale proteomic investigations have led to the
identification of over 18,000 protein quantitative trait loci
(pQTL) that cover upwards of 4,800 proteins. Genetic variations
related to the levels of multiple plasma proteins have been
identified in previous pQTL studies (10-14).
These research efforts provide valuable data resources for
elucidating the causal role of plasma proteins in the risk of
bladder cancer through Mendelian randomization (MR). MR has been
widely used in studying the pathogenesis and developing preventive
strategies for various cancers, effectively distinguishing the
effects of genetic factors from environmental factors, and aiding
researchers identify potential disease biomarkers and therapeutic
targets (15,16).
In the present study, a proteome-wide MR analysis
was conducted by integrating human plasma proteomics and genomics
data to systematically identify biomarkers associated with bladder
cancer risk. Prognostic models were constructed based on the
identified genes and further validated. Single-cell expression
analysis was performed to detect their expression in bladder cancer
tissues. Finally, druggability evaluation was conducted to explore
their potential as therapeutic targets for bladder cancer.
Materials and methods
Data acquisition
The pQTL data were obtained from the UK Biobank
Pharma Proteomics Project (UKB-PPP) database (http://ukb-ppp.gwas.eu/), with baseline cohort data
selected for this analysis, describing pQTLs from 34,557
participants of European ancestry (17). Outcome data were primarily derived
from genome-wide association studies (GWAS) involving populations
of European ancestry. Summary statistics for the training set were
sourced from the FinnGen biobank database
(finngen_R10_C3_BLADDER_EXALLC), including 2,193 cases and 314,193
controls, while validation set data were retrieved from the EBI
database (ID no. GCST90041858), comprising 762 cases and 455,586
controls. Additionally, RNA-seq data from 431 specimens (19 normal
and 412 tumor samples) were collected from the TCGA database
(https://portal.gdc.cancer.gov/), and
bladder cancer-related single-cell data were downloaded from the
GEO database (https://www.ncbi.nlm.nih.gov/geo/) under accession no.
GSE129845(18), including 3
samples.
PQTL MR analysis
The UKB-PPP characterized the plasma proteomics
features of UKB participants. GWAS-based proteomics studies
(17) were used to extract
relevant causal relationships in pQTLs from the outcome summary
data, by selecting single nucleotide polymorphisms (SNPs)
associated with each gene at a genome-wide significance threshold
(P<10-6) as potential instrumental variables (IVs).
While the genome-wide significance threshold is typically set at
P<1x10-8, relaxing it to P<1x10-6 is
considered acceptable in specific scenarios, such as candidate gene
association analysis or fine-mapping analyses including sensitivity
analysis focused on specific loci. When the research objective is
exploratory analysis (rather than strict causal inference), this
threshold increases the number of IVs and avoids missing potential
associations. The LD between SNPs was calculated, retaining only
SNPs with R2<0.001 (clumping window size=10,000 kb), and weak
instruments with F>10 were excluded. Notably, four statistical
methods were used to assess the reliability of causal
relationships: Inverse Variance Weighted (IVW) (19), MR Egger (20), Weighted Median (21), and Weighted Mode (22). If only one SNP was present in the
causal relationship, the Wald ratio method was used. The
reliability of the identified causal relationships was verified
using leave-one-out analysis.
Colocalization analysis
The coloc method (23) was used with expression quantitative
trait locus (eQTL) summary data and bladder cancer GWAS data for
colocalization analysis. The posterior probability was calculated
for the 100-kilobase region around the index SNP. In coloc results,
H3 represents the posterior probability that two traits (gene
expression and bladder cancer) are associated but have different
causal variants; H4 represents the posterior probability that two
traits are associated and share a single causal variant. SNP.PP.H4
>0.95 was used as the colocalization threshold.
GSEA enrichment analysis
GSEA (24) is a
method that utilizes predefined gene sets. It ranks genes according
to the differential expression levels between two sample classes
and then determines whether these predefined gene sets are over,
represented at either the top or bottom of the ranking. In the
present study, GSEA was employed to contrast the signaling pathways
in the high-expression and low-expression groups. This was
performed to investigate the molecular mechanisms underlying key
genes in the 2 patient groups. The number of permutations was
configured as 1,000 and the permutation type was designated as
phenotype.
Tumor mutation burden (TMB) and
microsatellite instability (MSI) data analysis
TMB is characterized as the cumulative count of
somatic gene coding inaccuracies, including base substitutions,
insertions, or deletions, identified per million bases. In the
present research, the determination of TMB for each tumor sample
involved calculating the mutation frequency and the number of
mutations relative to the exome length. Specifically, TMB was used
by dividing the number of non-synonymous mutation sites by the
total length of the protein-coding region (25).
Nomogram model construction
A nomogram was developed through regression
analysis, incorporating gene expression levels and clinical
symptoms. This nomogram visually represents the relationships
between variables within the prediction model by means of scaled
lines plotted on a single plane. A multivariate regression model
was constructed and the contribution of each influencing factor to
the outcome variable (regression coefficient) was used to assign
scores to each level of each influencing factor. The scores were
summed to obtain a total score, from which the predicted value was
calculated.
Drug sensitivity analysis
Using the largest pharmacogenomics database (GDSC;
https://www.cancerrxgene.org/), the R
package ‘pRRophetic’ (26) was
used to predict chemotherapy sensitivity for each tumor sample.
Regression methods were used to obtain IC50 estimates
for each specific chemotherapy drug treatment, and 10-fold
cross-validation was performed on the GDSC training set to verify
regression and prediction accuracy. All parameters were set to
default values, including the removal of batch effects with
‘combat’ and averaging of replicated gene expression values.
Single-cell analysis
First, the Seurat package (27) was employed to read the expression
profiles. Genes with low expression were filtered out based on the
criteria: nFeature_RNA >50 and percent.mt <15. Subsequently,
the data underwent normalization and scaling, followed by
processing using principal component analysis (PCA). The ElbowPlot
function was used to ascertain the optimal number of principal
components. To visualize the positions of each cluster,
t-distributed Stochastic Neighbor Embedding (28) analysis was carried out. Annotation
of the clusters was performed using the celldex package (29), enabling the identification of cell
types that are significantly associated with the occurrence of the
disease. Each subunit of single cells was annotated using the R
package SingleR (30).
Disease gene co-expression
network
Disease-related genes were obtained from the
GeneCards database (https://www.genecards.org/). Genes with high relevance
scores were selected, and a correlation analysis was conducted to
explore the co-expression network between key genes and
disease-related genes. The Pearson correlation method was used for
the statistical analysis.
Cell culture
The immortalized cells of human ureteral epithelium
(SV-HUC-1), and human bladder cancer cell lines (T24, 5637, UMUC3,
J82) were obtained from the American Type Culture Collection
(ATCC). SV-HUC-1 cells were cultured in F-12K medium (Gibco; Thermo
Fisher Scientific, Inc.). T24, 5637, UMUC3, and J82 cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.). All media were supplemented with 10% fetal bovine serum
(FBS; Corning, Inc.). The cells were incubated in a humidified
atmosphere containing 5% CO2 at 37˚C. Regular
observation of the structural state of the cells during cell
culture and regular checks for mycoplasma contamination were
performed.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was utilized to extract RNA from cells and
tissues. Subsequently, complementary DNA (cDNA) synthesis was
carried out with a reverse transcription system (FastKing One Step
RT-PCR kit; Tiangen Biotech Co., Ltd.), following the
manufacturer's instructions precisely. RT-qPCR was performed in
triplicate using the Applied Biosystems 7500 Real-Time PCR System
(Thermo Fisher Scientific, Inc.) and SYBR Green PCR Master Mix
(TransStart® Top Green qPCR SuperMix; TransGen Biotech
Co., Ltd.) on 96-well optical plates according to the
manufacturer's instructions. The thermocycling conditions were as
follows: Pre-denaturation at 95˚C for 3 min, denaturation at 95˚C
for 30 sec, annealing extension at 60˚C for 30 sec, deformation and
annealing extension for 40 cycles. The dissolution curve was
increased by 0.5˚C every 2 cycles to 95˚C. The results were
normalized to the endogenous control, β-actin. The primers used are
listed in Table SI. The
2-ΔΔCq method was used for quantification (31).
Statistical analysis
Reliable MR analysis is based on three assumptions:
i) Relevance assumption (instrumental variables are closely related
to the exposure but not directly related to the outcome); ii)
independence assumption (instrumental variables are not related to
confounding factors); and iii) exclusion restriction assumption
(instrumental variables only affect the outcome through the
exposure; when IVs can affect the outcome through other pathways,
pleiotropy is present). All analyses were conducted using R
language (version 4.0). Continuous variables were compared using
paired and unpaired Student's t-test, or one-way ANOVA
(single-factor analysis) followed by Bonferroni's post hoc test for
multiple-group comparisons. Data are presented as the mean ±
standard deviation (SD) from three independent biological
replicates. All statistical tests were two-sided. P<0.05 was
considered to indicate a statistically significant difference.
Results
pQTL MR analysis in the training
set
The outcome ID finngen_R10_C3_BLADDER_EXALLC,
related to bladder cancer, was obtained from summary statistics of
316,386 samples (2,193 cases and 314,193 controls). Using the
extract_instruments and extract_outcome_data functions, 342 pairs
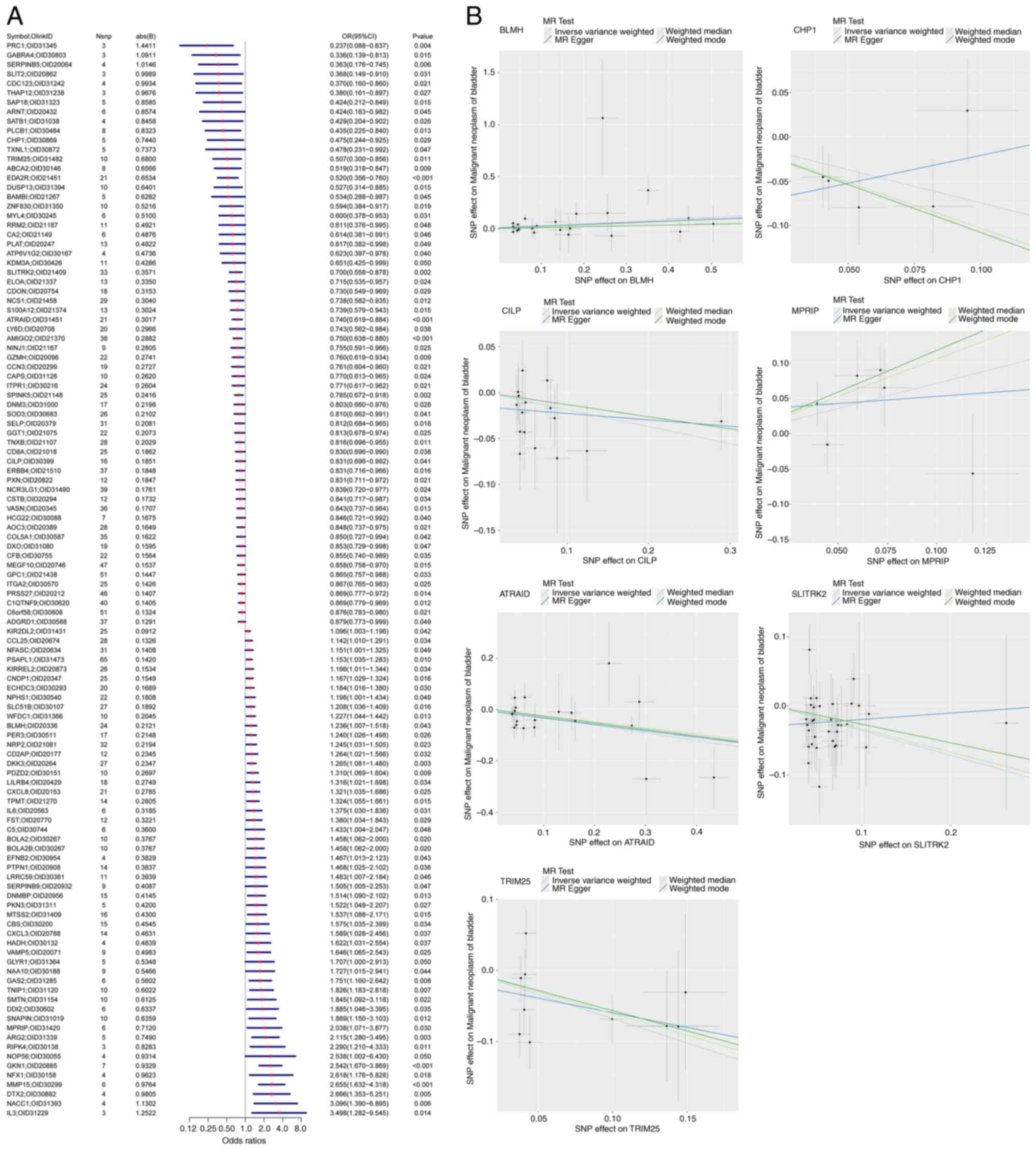
of gene-outcome causal relationships were extracted (Table SII). Further MR analysis
identified 114 gene-pQTL-positive causal relationships (Fig. 1A; IVW, P<0.05). Sensitivity
analysis was performed on the causal relationships of the 114 genes
to verify their reliability. The results indicated that the removal
of any SNP did not significantly affect the overall error lines,
suggesting that the 114 causal relationships were robust.
pQTL MR analysis
To further identify key genes among the 114 pQTL
genes associated with bladder cancer, the outcome ID GCST90041858
was obtained from the summary statistics of 456,348 samples related
to bladder cancer (762 cases and 455,586 controls). Using the
extract_instruments and extract_outcome_data functions, 22 pairs of
gene-outcome causal relationships were extracted (Table SIII). Further MR analysis
identified seven gene-pQTL-positive causal relationships (Fig. 1B; P<0.05), corresponding to the
genes all-trans retinoic acid-induced differentiation factor
(ATRAID), bleomycin hydrolase (BLMH), calcineurin B homologous
protein 1 (CHP1), cartilage intermediate layer protein (CILP),
myosin phosphatase Rho-interacting protein (MPRIP), SLIT and
NTRK-like family member 2 (SLITRK2), and tripartite motif
containing 25 (TRIM25). The genes ATRAID, BLMH, MPRIP, and TRIM25
were potentially associated with a low risk of bladder cancer. The
genes CHP1, CILP, and SLITRK2 were potentially associated with a
high risk of bladder cancer. Sensitivity analysis was performed on
the causal relationships of the seven genes to verify their
reliability. The results indicated that the removal of any SNP did
not significantly affect the overall error lines, suggesting that
the seven causal relationships were robust (Fig. 2A-G). Additionally, colocalization
analysis at the pQTL-GWAS level identified two genes with
colocalization SNP.PP.H4 >0.95 (Fig. 2H and I), specifically ATRAID and TRIM25.
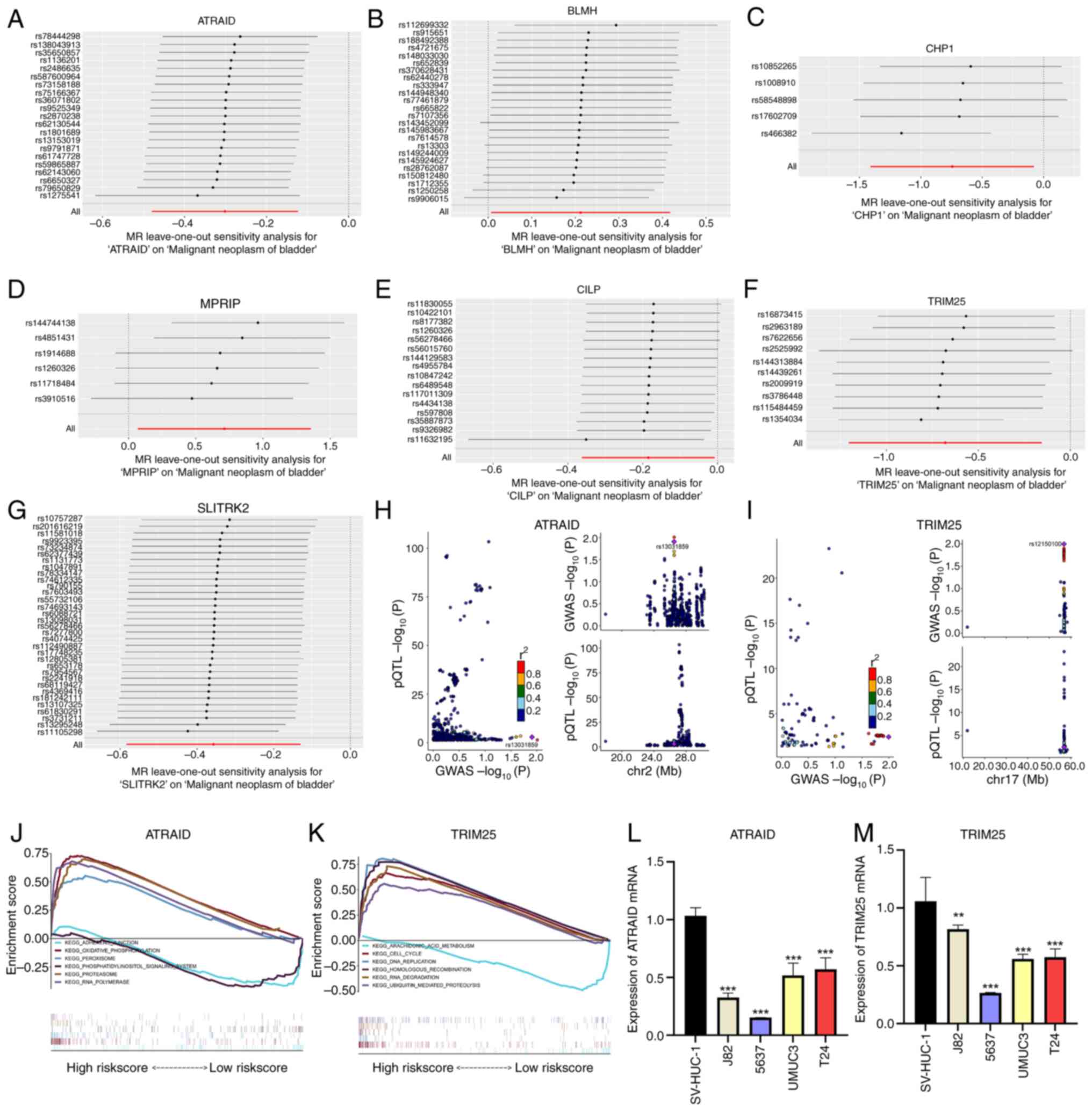 | Figure 2Key gene screening and reverse
transcription-quantitative PCR validation. (A-G) Forest plots of
leave-one-out analysis for SNPs corresponding to the seven genes.
(H and I) Associations between SNPs of ATRAID and TRIM25 with the
disease, where each point indicates a statistically significant
SNP-disease association. The x-axis represents the P-value of GWAS,
and the y-axis represents the P-value of the pQTL analysis. (J and
K) KEGG signaling pathways involving ATRAID and TRIM25, including
genes involved in pathway regulation. (L and M) mRNA expression
levels of ATRAID and TRIM25 in bladder cancer cell lines.
**P<0.01 and ***P<0.001. SNP, single
nucleotide polymorphism; ATRAID, all-trans retinoic acid-induced
differentiation factor; TRIM25, tripartite motif containing 25;
GWAS, genome-wide association studies; pQTL, protein quantitative
trait loci; KEGG, Kyoto, Encyclopedia of Genes and Genomes. |
Exploration of specific signaling
mechanisms related to key genes
The specific signaling pathways involved with these
two key genes and their impact on disease progression-related
signaling pathways were explored. GSEA results indicated that
ATRAID was enriched in ‘ADHERENS_JUNCTION’,
‘OXIDATIVE_PHOSPHORYLATION’, and ‘PEROXISOME’ signaling pathways
(Fig. 2J). TRIM25 was enriched in
‘ARACHIDONIC_ACID_METABOLISM’, ‘CELL_CYCLE’, and ‘DNA_REPLICATION’
signaling pathways (Fig. 2K). GSEA
was further used to analyze the impact of these key genes on
signaling pathways. RT-qPCR was performed to investigate the mRNA
expression of ATRAID and TRIM25 in bladder cancer cell lines. The
results of RT-qPCR, based on bladder cancer cell lines,
demonstrated that the mRNA expression levels of ATRAID and TRIM25
were lower in tumor cell lines than in SV-HUC-1 cells (Fig. 2L and M).
Clinical predictive value of key
genes
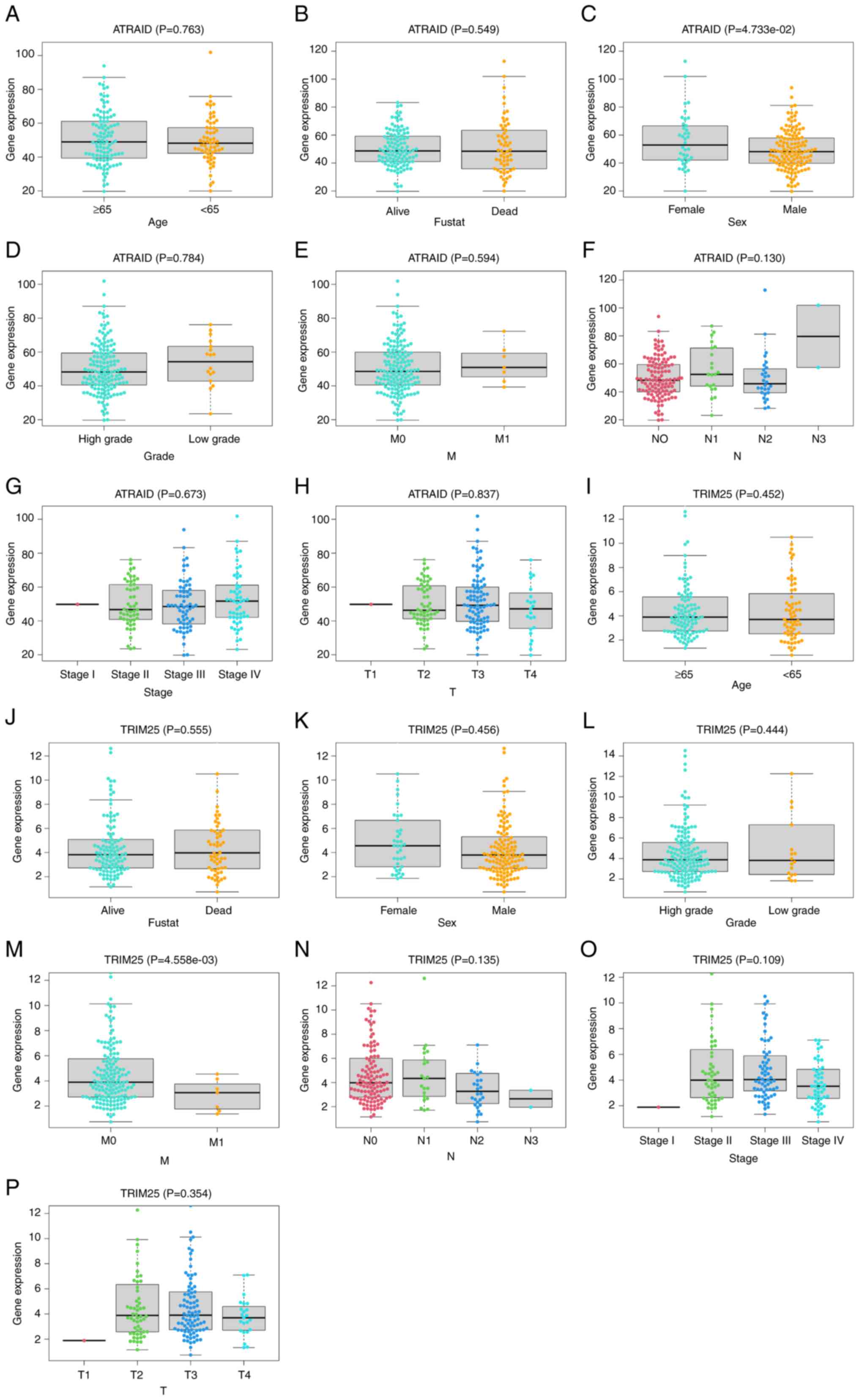
The relationship between key gene expression and
clinical symptoms was analyzed. The analysis revealed a significant
difference between ATRAID and the sex of patients (Fig. 3A-H), and a significant difference
between TRIM25 and the M stage of patients (Fig. 3I-P). These findings may provide
guidance and support for disease diagnosis and treatment, drug
development, and disease mechanism research.
Mutation profile and differences in
TMB/MSI of key genes
Processed SNP-related data for bladder cancer were
downloaded and the top 30 genes were selected with high mutation
frequency for display, comparing the differences in mutated genes
between the two groups of patients. Using the R package
ComplexHeatmap, a mutation landscape map was plotted. The results
revealed that there was a significant difference in the mutation
frequency of AT-rich interaction domain 1 (ARID1A) between patients
with high and low ATRAID expression (P=0.005), with a higher
mutation proportion of ARID1A in the high-expression group
(Fig. 4A). Similarly, significant
differences in mutation frequencies of ARID1A (P=0.036),
microtubule-actin crosslinking factor 1 (MACF1; P=0.010), spectrin
repeat containing nuclear envelope protein 2 (SYNE2; P=0.026), and
CREB binding lysine acetyltransferase (CREBBP; P<0.001) were
also observed between subgroups based on TRIM25 expression. The
high TRIM25-expression group had a higher mutation proportion of
ARID1A, MACF1, SYNE2, and CREBBP compared with the low
TRIM25-expression group (Fig. 4B).
The relationship between the two key genes and common
immunotherapy-related tumor markers were further explored,
revealing the relationship between TMB and MSI in the high- and
low-expression groups of key genes (Fig. 4C-F). The results indicated a
statistically significant relationship between ATRAID and MSI
(Fig. 4C).
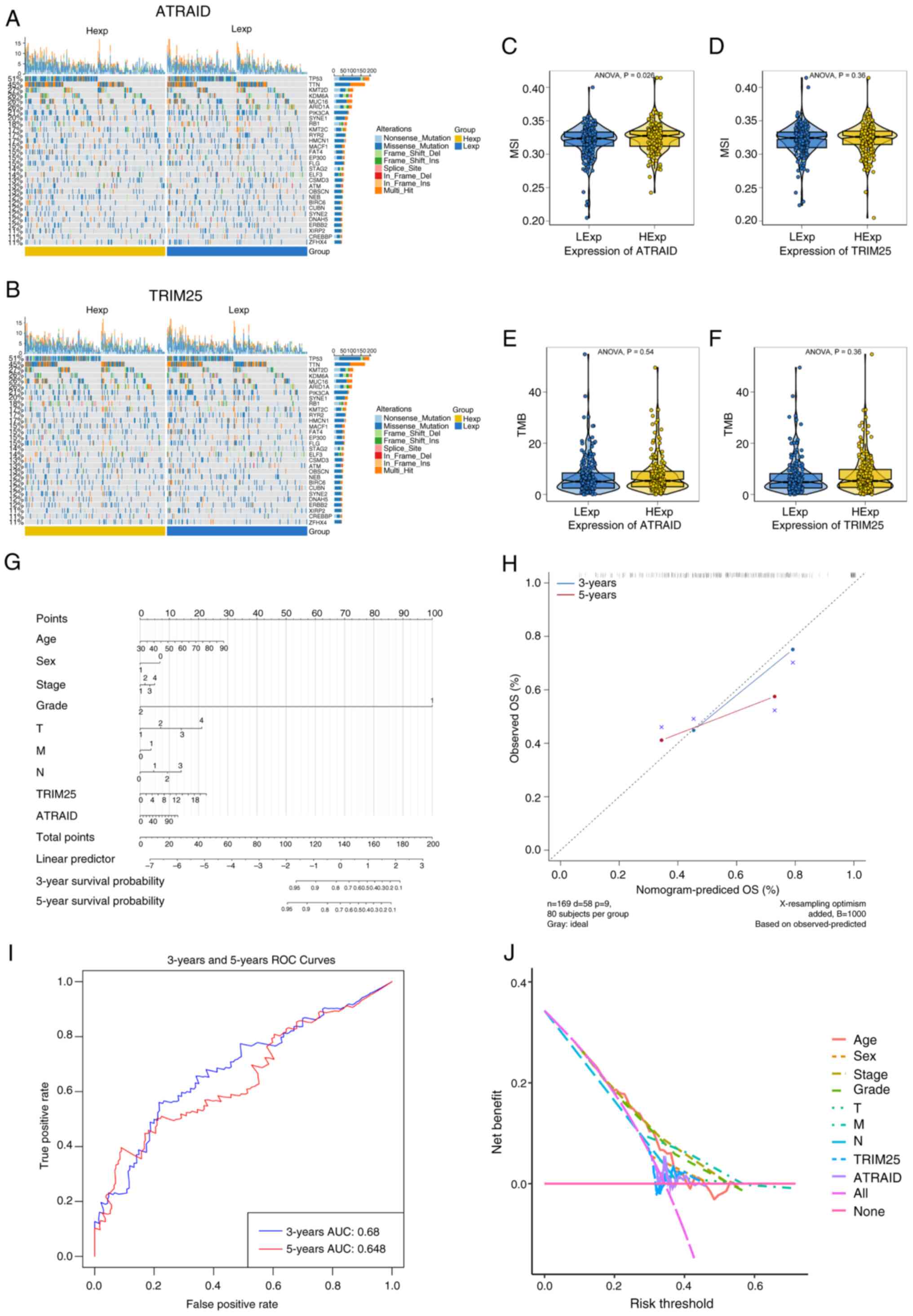 | Figure 4Mutational landscape of key genes,
differences in TMB/MSI, and construction of the Nomogram model. (A
and B) SNP-related data of bladder cancer, showing the top 30 genes
with high mutation frequencies in 2 patient groups. (C-F)
Differences in MSI and TMB between ATRAID and TRIM25. (G) Column
chart of the key gene model. (H) Prediction analysis of OS for 3
and 5 years. (I and J) ROC curve and decision curve analysis. TMB,
tumor mutation burden; MSI, microsatellite instability; SNP, single
nucleotide polymorphism; ATRAID, all-trans retinoic acid-induced
differentiation factor; TRIM25, tripartite motif containing 25; OS,
overall survival; ROC, receiver operating characteristic; LExp; low
expression; HExp, high expression; AUC, area under the curve. |
Nomogram model construction
The expression levels of key genes were used to
present the results of the regression analysis in the form of a
nomogram. The regression analysis results indicated that various
indicators of bladder cancer, along with the distribution of key
gene expression, contributed to different extents in the overall
scoring process (Fig. 4G). A
predictive analysis for the 3 and 5-year overall survival (OS)
periods was also conducted. The results indicated that the
predicted OS closely matched the observed OS, demonstrating the
good predictive performance of the nomogram model. In the
calibration curve, the data points in blue (3-year) and red
(5-year) are both closely distributed along the diagonal line,
indicating that the predicted OS aligns well with the observed OS
(Fig. 4H). In the receiver
operating characteristic (ROC) curve, the area under the curve
(AUC) for the 3-year OS prediction was 0.68, and the AUC for the
5-year OS prediction was 0.648. Although these AUC values were not
particularly high, they indicate that the model has a certain
ability to distinguish between the 3 and 5-year survival outcomes
of patients (Fig. 4I). In the
decision curve, the purple line representing the integration of key
genes and clinical indicators (‘All’) exhibited a higher net
benefit than lines incorporating only partial factors or no factors
(Fig. 4J). These findings suggest
that the nomogram model constructed by combining the clinical
indicators and key gene expression levels (corresponding to the
‘All’ curve) can provide greater net benefits to patients accross
appropriate risk thresholds in real-world clinical
decision-making.
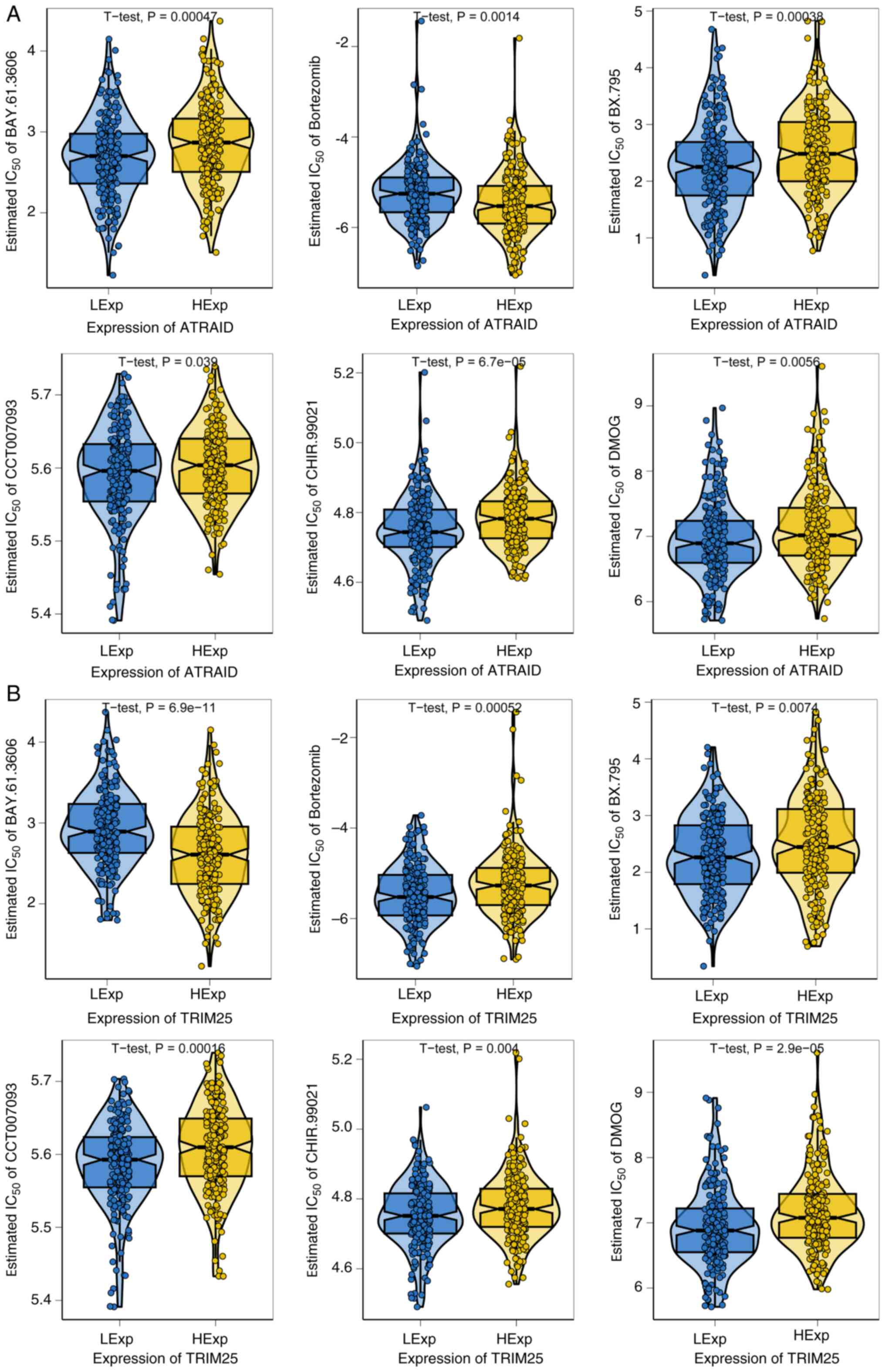
Drug sensitivity analysis
Early-stage bladder cancer is effectively treated
with surgery combined with chemotherapy (32). The present study used drug
sensitivity data from the GDSC database and the R package
‘pRRophetic’ to predict the chemotherapy sensitivity of each tumor
sample, and further explored the sensitivity to common chemotherapy
drugs. The results indicated that the two key genes, ATRAID and
TRIM25, were significantly associated with sensitivity to
BAY.61.3606, bortezomib, BX.795, CCT007093, CHIR.99021, and DMOG.
High ATRAID expression was associated with increased resistance of
bladder cancer cells to BAY.61.3606, BX.795, CCT007093, CHIR.99021,
and DMOG (higher IC50 values indicate a need for higher
drug concentrations to inhibit 50% of cells), while enhancing
sensitivity to bortezomib (Fig.
5A). Conversely, high TRIM25 expression led to increased
resistance to bortezomib, BX.795, CCT007093, CHIR.99021, and DMOG,
but enhanced sensitivity to BAY.61.3606 (Fig. 5B).
Expression of key genes at the
single-cell level
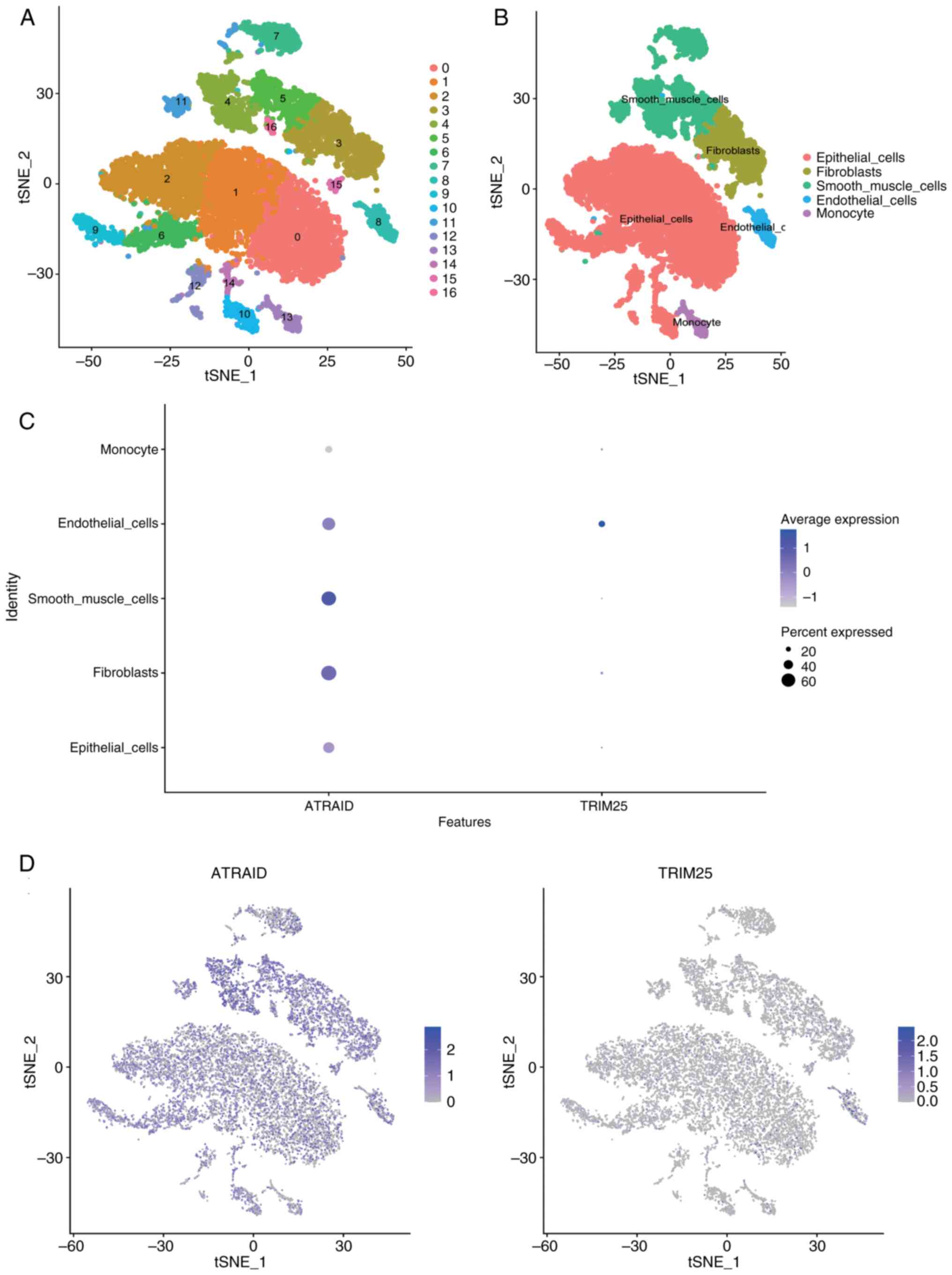
The analysis of the present study included
expression profiles from three bladder cancer-related tissue
samples. Using the FindClusters function from the Seurat package,
17 subtypes were identified (Fig.
6A). The R package SingleR was used to annotate each subtype,
identifying 17 clusters into five cell types: Epithelial_cells,
Fibroblasts, Smooth_muscle_cells, Endothelial_cells, and Monocytes
(Fig. 6B). The expression of the
two key genes in cell subtypes was analyzed and the results using
bubble plots and scatter plots are displayed (Fig. 6C and D). The findings revealed that ATRAID and
TRIM25 were expressed in multiple cell types, but their expression
was more widespread or higher in certain cells (such as epithelial
cells and fibroblasts). Multiple key genes were found to be
co-expressed with tumor genes. Co-expression relationships
(including both positive and negative co-expression) reflect the
correlation trends in expression levels between genes. When two
genes exhibit positive co-expression, their expression levels tend
to change in the same direction across samples.
Apoptosis-associated transcript in bladder cancer (AATBC) exhibited
a negative co-expression association with both the ATRAID and
TRIM25 genes (Fig. S1A and
B). Fibroblast growth factor
receptor 3 (FGFR3) was positively co-expressed with ATRAID and
negatively co-expressed with TRIM25 (Fig. S1C and D). Hras proto-oncogene, GTPase (HRAS)
was positively co-expressed with both ATRAID and TRIM25 (Figs. S1E and S2A). RB transcriptional corepressor 1
(RB1) was positively co-expressed with ATRAID and negatively
expressed with TRIM25 (Fig. S2B
and C). Tumor ptotein 53 (TP53)
was positively co-expressed with ATRAID and negatively expressed
with TRIM25 (Fig. S2D and
E). Positive co-expression
between two genes may indicate their involvement in the same
biological process or signaling pathway, achieving functional
coordination through synergistic expression. Alternatively, one
gene may act as an upstream regulatory factor for the other. By
contrast, negative co-expression of two genes, may reflect opposing
roles in biological processes, an antagonistic relationship within
a shared pathway, or mutual inhibitory regulation.
Discussion
In the present study, seven plasma proteins were
detected and the gene expression levels of these proteins were
found to be significantly associated with the risk of bladder
cancer. Among them, the genes ATRAID and TRIM25 had a probability
>0.95 of affecting the same phenotype in both pQTL and GWAS
data, suggesting a high likelihood that these two genes are
involved in the same biological pathway. The present study
identified novel biomarkers for bladder cancer, which may provide
new insights into the genetics and pathogenesis of bladder
cancer.
ATRAID, also known as APR3, is a gene associated
with apoptosis (33,34). Located on human chromosome 2p22.3,
it can affect the cell cycle by altering cyclin D1 expression,
thereby influencing malignant tumor occurrence and growth, and
promoting cell differentiation (35). In the present study, its potential
as a diagnostic marker for bladder cancer was identified,
suggesting a suppressive role in the development of bladder cancer.
However, previous studies have found increased expression of ATRAID
in ovarian cancer, cervical cancer, and lung adenocarcinoma,
suggesting an oncogenic role (35-37),
although no studies, to the best of the authors' knowledge, have
yet been conducted on ATRAID in bladder cancer. Pathway enrichment
analysis of ATRAID in the present study revealed significant
enrichment in adhesion function and oxidative stress. Li et
al (33) similarly found that
upregulation of APR3 expression in ARPE-19 cells significantly
increased intracellular reactive oxygen species (ROS) levels and
altered mitochondrial morphology, promoting cell oxidative stress
by affecting mitochondrial function. ATRAID may also exert its
effects in bladder cancer through similar oxidative stress
pathways.
In the analysis of the relationship between ATRAID
and clinical characteristics, it was determined that the expression
of ATRAID was lower in male patients than in female patients. It
was speculated that this difference may be related to sex
chromosome-associated genes regulating ATRAID, and it is also
possible that sex hormones such as estrogen, progesterone, and
testosterone may affect the expression of ATRAID. These findings
suggest a potential direction for future research. In immune
infiltration analysis, high expression of ATRAID was significantly
positively associated with MSI status, indicating its important
role in MSI tumors and possibly serving as a novel therapeutic
target.
TRIM25 is an E3 ubiquitin ligase confirmed to be an
RNA-binding protein (RBP) through its spry domain, and it plays a
crucial role in gene regulation, controlling cell proliferation,
and cancer cell migration (38-40).
The regulation of TRIM25 in tumors is largely through regulation of
the endoplasmic reticulum (ER), which can regulate apoptosis of
tumors through the IRE1-JNK signal, upregulating the unfolded
protein response and ER-associated degradation pathways (41,42).
TRIM25 has been shown to play a role in the development of colon
cancer, hepatocellular carcinoma, endometrial cancer, gastric
cancer, prostate cancer, lung cancer, breast cancer, ovarian
cancer, and renal cancer (43-53).
Although several studies have explored the role of TRIM25 in
bladder cancer, research on its role in this cancer type remains
limited and requires further investigation (54-56).
Pathway enrichment analysis of TRIM25 in the present study showed
significant enrichment in ‘ARACHIDONIC_ACID_METABOLISM’,
‘CELL_CYCLE’, and ‘DNA_REPLICATION’ pathways. The enrichment of
TRIM25 in the cell cycle pathway indicated its potential role in
regulating cell cycle progression.
In the analysis of the relationship between TRIM25
and clinical characteristics, it was found that the expression of
TRIM25 was lower in patients with distant metastases than in those
without, which may indicate that the expression of TRIM25 is
related to the occurrence of tumor metastasis, possibly exerting an
inhibitory effect in the invasion and metastasis process of bladder
cancer cells.
By combining ATRAID and TRIM25 with patient clinical
data, bladder cancer 3 and 5-year survival prediction models were
constructed, with model AUCs of 0.68 and 0.648, respectively. The 3
and 5-year prognostic models constructed in the present study
demonstrated relatively high predictive accuracy, which can assist
clinicians in predicting the prognosis of individual patients. For
patients predicted to have a shorter survival time, more aggressive
treatment options can be considered, while for those predicted with
a longer predicted survival, more conservative treatment strategies
can be selected.
Drug sensitivity analysis of ATRAID and TRIM25
revealed that high expression of ATRAID was associated with
increased resistance of bladder cancer cells to BAY.61.3606,
BX.795, CCT007093, CHIR.99021, and DMOG (higher IC50
values indicate higher drug concentrations required to inhibit 50%
of cells), while enhancing sensitivity to bortezomib. Conversely,
high expression of TRIM25 led to increased resistance to
bortezomib, BX.795, CCT007093, CHIR.99021, and DMOG, but enhanced
sensitivity to BAY.61.3606. From the perspective of drug
sensitivity, these findings suggest that the expression levels of
ATRAID and TRIM25 may serve as drug response prediction indicators
(patients with high expression may exhibit poorer efficacy to these
drugs). This implies that the expression levels of these two genes
may influence the response of tumor cells to these drugs, and the
drugs may regulate bladder cancer by affecting signaling pathways
associated with ATRAID and TRIM25. Further investigation of these
associations could improve our understanding of tumor drug response
mechanisms and provide novel insights and strategies for
personalized therapy.
ATRAID was mainly expressed in ‘Fibroblasts’ and
‘Smooth_muscle_cells’, indicating that ATRAID may play an important
role in tumor stromal remodeling or microenvironment regulation.
TRIM25 was mainly expressed in ‘Endothelial_cells’, indicating that
TRIM25 may play an important role in encoding vascular growth
factors and regulating vascular maturation and stability.
In the present study, the association between plasma
protein biomarkers and bladder cancer risk was comprehensively
explored. A two-stage screening MR design was employed, which is
characterized by a large sample size and a minimal likelihood of
reverse causation and confounding bias. The consistency of multiple
rigorous analysis results confirms the robustness of the findings
of the present study. By screening ATRAID and TRIM25, a predictive
model was constructed to effectively predict the 3 and 5-year
survival rates. The small sample size in the single-cell analysis
of bladder cancer may fail to accurately reflect cellular
heterogeneity, which is also one of the limitations of the present
study. In addition, only qPCR was used to verify the RNA expression
levels of the two genes, ATRAID and TRIM25. Verification at the
protein level using western blotting and immunohistochemistry has
yet to be carried out. Similarly, the functional verification of
the ATRAID and TRIM25 genes in bladder cancer and the exploration
of the mechanisms by which these two genes participate in the
occurrence and development of bladder cancer have not been
performed. Subsequently, bladder cancer cell lines with knockdown
and overexpression of the ATRAID and TRIM25 genes will be
constructed for functional exploration. Furthermore, the specific
mechanisms affected by these two genes will be investigated,
particularly in the direction of bladder cancer drug resistance. In
addition, an in-depth research on their mechanisms will be
conducted, so as to provide valuable insights for future studies on
drug resistance in bladder cancer. In the present study, ATRAID and
TRIM25 were identified as biomarkers associated with bladder cancer
risk. These findings not only provide novel perspectives on the
causes of bladder cancer, but also offer potential targets for the
development of innovative biomarkers and therapeutic drugs against
bladder cancer.
Supplementary Material
Co-expression with disease genes.
(A-E) Co-expression analysis of disease-related genes with ATRAID
and TRIM25 in single-cell data, including the correlation of
co-expressed genes. ATRAID, all-trans retinoic acid-induced
differentiation factor; TRIM25, tripartite motif containing 25;
AATBC, apoptosis-associated transcript in bladder cancer; FGFR3,
fibroblast growth factor receptor 3; HRAS, Hras proto-oncogene,
GTPase; t-SNE, t-distributed Stochastic Neighbor Embedding.
Co-expression with disease genes.
(A-E) Co-expression analysis of disease-related genes with ATRAID
and TRIM25 in single-cell data, including the correlation of
co-expressed genes. ATRAID, all-trans retinoic acid-induced
differentiation factor; TRIM25, tripartite motif containing 25;
HRAS, Hras proto-oncogene, GTPase; RB1, RB transcriptional
corepressor 1; TP53, tumor protein 53; t-SNE, t-distributed
Stochastic Neighbor Embedding.
Primer sequences.
Mendelian randomization analysis of
342 gene-outcome causal relationships in the training set of
pQTL.
Mendelian randomization analysis of 22
gene-outcome causal relationships in the validation set of
pQTL.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Central
Government Guiding Local Funds for Science and Technology
Development (grant no. 236Z7710G), and the Peking University First
Hospital-Miyun Hospital Joint Program (grant no.
2023-study029-001).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
WG, YL, ZF, ZG, YaG, XJ, YJ and YuG conceptualized
and designed the present study. WG, YL, ZF, ZG, YaG, XJ, YJ and YuG
provided administrative support. WG, YL and ZF conducted the
collation of public data. WG, YL and ZF collected and assembled the
data of the present study. WG, YL and ZF analyzed and interpretated
the data. WG and YuG confirm the authenticity of all the raw data.
All authors assisted with the writing of the present study, and
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was carried out in compliance with
the Declaration of Helsinki (revised in 2013). Additionally, the
present study was approved (approval no. 2023-029-001) by the
Ethics Committee of Peking University First Hospital-Miyun Hospital
(Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang Y, Rumgay H, Li M, Yu H, Pan H and
Ni J: The global landscape of bladder cancer incidence and
mortality in 2020 and projections to 2040. J Glob Health.
13(04109)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Peng M, Chu X, Peng Y, Li D, Zhang Z, Wang
W, Zhou X, Xiao D and Yang X: Targeted therapies in bladder cancer:
Signaling pathways, applications, and challenges. MedComm (2020).
4(e455)2023.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Thomas J and Sonpavde G: Molecularly
targeted therapy towards genetic alterations in advanced bladder
cancer. Cancers (Basel). 14(1795)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Anderson NL and Anderson NG: The human
plasma proteome: History, character, and diagnostic prospects. Mol
Cell Proteomics. 1:845–867. 2002.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Suhre K, McCarthy MI and Schwenk JM:
Genetics meets proteomics: Perspectives for large population-based
studies. Nat Rev Genet. 22:19–37. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Roslan A, Sulaiman N, Mohd Ghani KA and
Nurdin A: Cancer-associated membrane protein as targeted therapy
for bladder cancer. Pharmaceutics. 14(2218)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sarafidis M, Lambrou GI, Zoumpourlis V and
Koutsouris D: An integrated bioinformatics analysis towards the
identification of diagnostic, prognostic, and predictive key
biomarkers for urinary bladder cancer. Cancers (Basel).
14(3358)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kojima T, Kawai K, Miyazaki J and
Nishiyama H: Biomarkers for precision medicine in bladder cancer.
Int J Clin Oncol. 22:207–213. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pietzner M, Wheeler E, Carrasco-Zanini J,
Cortes A, Koprulu M, Wörheide MA, Oerton E, Cook J, Stewart ID,
Kerrison ND, et al: Mapping the proteo-genomic convergence of human
diseases. Science. 374(eabj1541)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ferkingstad E, Sulem P, Atlason BA,
Sveinbjornsson G, Magnusson MI, Styrmisdottir EL, Gunnarsdottir K,
Helgason A, Oddsson A, Halldorsson BV, et al: Large-scale
integration of the plasma proteome with genetics and disease. Nat
Genet. 53:1712–1721. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang J, Dutta D, Köttgen A, Tin A,
Schlosser P, Grams ME and Harvey B: CKDGen Consortium. Yu B,
Boerwinkle E, et al: Plasma proteome analyses in individuals of
European and African ancestry identify cis-pQTLs and models for
proteome-wide association studies. Nat Genet. 54:593–602.
2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sun BB, Maranville JC, Peters JE, Stacey
D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran
P, et al: Genomic atlas of the human plasma proteome. Nature.
558:73–79. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Suhre K, Arnold M, Bhagwat AM, Cotton RJ,
Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, DeLisle RK, et
al: Connecting genetic risk to disease end points through the human
blood plasma proteome. Nat Commun. 8(14357)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Davies NM, Holmes MV and Davey Smith G:
Reading mendelian randomisation studies: A guide, glossary, and
checklist for clinicians. BMJ. 362(k601)2018.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Xin J, Gu D, Chen S, Ben S, Li H, Zhang Z,
Du M and Wang M: SUMMER: A Mendelian randomization interactive
server to systematically evaluate the causal effects of risk
factors and circulating biomarkers on pan-cancer survival. Nucleic
Acids Res. 51 (D1):D1160–D1167. 2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sun BB, Chiou J, Traylor M, Benner C, Hsu
YH, Richardson TG, Surendran P, Mahajan A, Robins C,
Vasquez-Grinnell SG, et al: Plasma proteomic associations with
genetics and health in the UK Biobank. Nature. 622:329–338.
2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yu Z, Liao J, Chen Y, Zou C, Zhang H,
Cheng J, Liu D, Li T, Zhang Q, Li J, et al: Single-cell
transcriptomic map of the human and mouse bladders. J Am Soc
Nephrol. 30:2159–2176. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Burgess S, Butterworth A and Thompson SG:
Mendelian randomization analysis with multiple genetic variants
using summarized data. Genet Epidemiol. 37:658–665. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bowden J, Davey Smith G and Burgess S:
Mendelian randomization with invalid instruments: Effect estimation
and bias detection through Egger regression. Int J Epidemiol.
44:512–525. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bowden J, Davey Smith G, Haycock PC and
Burgess S: Consistent estimation in mendelian randomization with
some invalid instruments using a weighted median estimator. Genet
Epidemiol. 40:304–314. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hartwig FP, Davey Smith G and Bowden J:
Robust inference in summary data Mendelian randomization via the
zero modal pleiotropy assumption. Int J Epidemiol. 46:1985–1998.
2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Giambartolomei C, Vukcevic D, Schadt EE,
Franke L, Hingorani AD, Wallace C and Plagnol V: Bayesian test for
colocalisation between pairs of genetic association studies using
summary statistics. PLoS Genet. 10(e1004383)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bonneville R, Krook MA, Kautto EA, Miya J,
Wing MR, Chen HZ, Reeser JW, Yu L and Roychowdhury S: Landscape of
microsatellite instability across 39 cancer types. JCO Precis
Oncol. 2017(PO.17.00073)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Geeleher P, Cox N and Huang RS:
pRRophetic: An R package for prediction of clinical
chemotherapeutic response from tumor gene expression levels. PLoS
One. 9(e107468)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hao Y, Hao S, Andersen-Nissen E, Mauck WM
III, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al:
Integrated analysis of multimodal single-cell data. Cell.
184:3573–3587.e29. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cieslak MC, Castelfranco AM, Roncalli V,
Lenz PH and Hartline DK: t-Distributed stochastic neighbor
embedding (t-SNE): A tool for eco-physiological transcriptomic
analysis. Mar Genomics. 51(100723)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang Q, Yu B, Zhang Y, Tian Y, Yang S,
Chen Y and Wu H: Combination of single-cell and bulk RNA seq
reveals the immune infiltration landscape and targeted therapeutic
drugs in spinal cord injury. Front Immunol.
14(1068359)2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Aran D, Looney AP, Liu L, Wu E, Fong V,
Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al:
Reference-based analysis of lung single-cell sequencing reveals a
transitional profibrotic macrophage. Nat. Immunol. 20:163–172.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang W, Lu Z, Wang M, Liu Z, Wu B, Yang C,
Huan H and Gong P: The cuproptosis-related signature associated
with the tumor environment and prognosis of patients with glioma.
Front Immunol. 13(998236)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li Y, Zou X, Gao J, Cao K, Feng Z and Liu
J: APR3 modulates oxidative stress and mitochondrial function in
ARPE-19 cells. FASEB J: fj201800001RR, 2018 (Epub ahead of
print).
|
|
34
|
Zhu F, Yan W, Zhao ZL, Chai YB, Lu F, Wang
Q, Peng WD, Yang AG and Wang CJ: Improved PCR-based subtractive
hybridization strategy for cloning differentially expressed genes.
Biotechniques. 29:310–313. 2000.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang Y, Li Q, Huang W, Zhang J, Han Z,
Wei H, Cui J, Wang Y and Yan W: Increased expression of
apoptosis-related protein 3 is highly associated with tumorigenesis
and progression of cervical squamous cell carcinoma. Hum Pathol.
44:388–393. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang P, Zhou C, Jing Q, Gao Y, Yang L, Li
Y, Du J, Tong X and Wang Y: Role of APR3 in cancer: Apoptosis,
autophagy, oxidative stress, and cancer therapy. Apoptosis.
28:1520–1533. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cao HL, Gu MQ, Sun Z and Chen ZJ:
miR-144-3p contributes to the development of thyroid tumors through
the PTEN/PI3K/AKT pathway. Cancer Manag Res. 12:9845–9855.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kwon SC, Yi H, Eichelbaum K, Föhr S,
Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW and Kim VN:
The RNA-binding protein repertoire of embryonic stem cells. Nat
Struct Mol Biol. 20:1122–1130. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Choudhury NR, Heikel G, Trubitsyna M,
Kubik P, Nowak JS, Webb S, Granneman S, Spanos C, Rappsilber J,
Castello A and Michlewski G: RNA-binding activity of TRIM25 is
mediated by its PRY/SPRY domain and is required for ubiquitination.
BMC Biol. 15(105)2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Heikel G, Choudhury NR and Michlewski G:
The role of Trim25 in development, disease and RNA metabolism.
Biochem Soc Trans. 44:1045–1050. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Liu Y, Tao S, Liao L, Li Y, Li H, Li Z,
Lin L, Wan X, Yang X and Chen L: TRIM25 promotes the cell survival
and growth of hepatocellular carcinoma through targeting Keap1-Nrf2
pathway. Nat Commun. 11(348)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Rahimi-Tesiye M, Zaersabet M, Salehiyeh S
and Jafari SZ: The role of TRIM25 in the occurrence and development
of cancers and inflammatory diseases. Biochim Biophys Acta Rev
Cancer. 1878(188954)2023.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Shu XS, Zhao Y, Sun Y, Zhong L, Cheng Y,
Zhang Y, Ning K, Tao Q, Wang Y and Ying Y: The epigenetic modifier
PBRM1 restricts the basal activity of the innate immune system by
repressing retinoic acid-inducible gene-I-like receptor signalling
and is a potential prognostic biomarker for colon cancer. J Pathol.
244:36–48. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Li YH, Zhong M, Zang HL and Tian XF: The
E3 ligase for metastasis associated 1 protein, TRIM25, is targeted
by microRNA-873 in hepatocellular carcinoma. Exp Cell Res.
368:37–41. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wang J, Yin G, Bian H, Yang J, Zhou P, Yan
K, Liu C, Chen P, Zhu J, Li Z and Xue T: LncRNA XIST upregulates
TRIM25 via negatively regulating miR-192 in hepatitis B
virus-related hepatocellular carcinoma. Mol Med.
27(41)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Dai H, Zhao S, Xu L, Chen A and Dai S:
Expression of Efp, VEGF and bFGF in normal, hyperplastic and
malignant endometrial tissue. Oncol Rep. 23:795–799.
2010.PubMed/NCBI
|
|
47
|
Dai H, Zhang P, Zhao S, Zhang J and Wang
B: Regulation of the vascular endothelial growth factor and growth
by estrogen and antiestrogens through Efp in Ishikawa endometrial
carcinoma cells. Oncol Rep. 21:395–401. 2009.PubMed/NCBI
|
|
48
|
Yang KS, Xu CQ and Lv J: Identification
and validation of the prognostic value of cyclic GMP-AMP
synthase-stimulator of interferon (cGAS-STING) related genes in
gastric cancer. Bioengineered. 12:1238–1250. 2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wang S, Kollipara RK, Humphries CG, Ma SH,
Hutchinson R, Li R, Siddiqui J, Tomlins SA, Raj GV and Kittler R:
The ubiquitin ligase TRIM25 targets ERG for degradation in prostate
cancer. Oncotarget. 7:64921–64931. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Qin X, Chen S, Qiu Z, Zhang Y and Qiu F:
Proteomic analysis of ubiquitination-associated proteins in a
cisplatin-resistant human lung adenocarcinoma cell line. Int J Mol
Med. 29:791–800. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Walsh LA, Alvarez MJ, Sabio EY, Reyngold
M, Makarov V, Mukherjee S, Lee KW, Desrichard A, Turcan Ş, Dalin
MG, et al: An integrated systems biology approach identifies TRIM25
as a key determinant of breast cancer metastasis. Cell Rep.
20:1623–1640. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li F, Sun Q, Liu K, Zhang L, Lin N, You K,
Liu M, Kon N, Tian F, Mao Z, et al: OTUD5 cooperates with TRIM25 in
transcriptional regulation and tumor progression via
deubiquitination activity. Nat Commun. 11(4184)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhenilo S, Deyev I, Litvinova E, Zhigalova
N, Kaplun D, Sokolov A, Mazur A and Prokhortchouk E: DeSUMOylation
switches Kaiso from activator to repressor upon hyperosmotic
stress. Cell Death Differ. 25:1938–1951. 2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Tang H, Li X, Jiang L and Liu Z, Chen L,
Chen J, Deng M, Zhou F, Zheng X and Liu Z: RITA1 drives the growth
of bladder cancer cells by recruiting TRIM25 to facilitate the
proteasomal degradation of RBPJ. Cancer Sci. 113:3071–3084.
2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Li W, Shen Y, Yang C, Ye F, Liang Y, Cheng
Z, Ou Y, Chen W, Chen Z, Zou L, et al: Identification of a novel
ferroptosis-inducing micropeptide in bladder cancer. Cancer Lett.
582(216515)2024.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Eberhardt W, Nasrullah U and Pfeilschifter
J: TRIM25: A global player of cell death pathways and promising
target of tumor-sensitizing therapies. Cells. 14(65)2025.PubMed/NCBI View Article : Google Scholar
|