Introduction
The rising incidence of early-onset cancer has
become a critical global health concern, with the age at diagnosis
continuously decreasing. Gastric cancer (GC), ranked as the fifth
most common malignancy globally, presents substantial clinical
challenges due to its frequently hidden early symptoms and the
absence of effective diagnostic tools (1,2).
Consequently, numerous patients are diagnosed at an advanced stage,
resulting in a poor five-year survival rate of ~20-30% (3,4).
Despite therapeutic progress, including surgery,
chemotherapy, radiotherapy and immunotherapy, the management of GC
remains challenging. Tumor heterogeneity, a complex tumor
microenvironment (TME), and frequent therapeutic resistance often
result in recurrence and unfavorable outcomes (5-9).
Therefore, identifying biomarkers that accurately predict
therapeutic efficacy in GC is imperative.
Recent studies emphasize the critical role of
ribosome biogenesis (RB) in cancer development. This complex
process involves the synthesis, modification, and assembly of
ribosomal RNA (rRNA). RB is fundamental to cellular function and is
tightly regulated. UTP4, also known as CIRH1A, functions as a
ribosomal RNA processing factor involved in the biosynthesis of the
small ribosomal subunit (10).
Elevated UTP4 expression levels have been closely associated with
tumor growth and disease progression across multiple cancer
types.
For instance, reduced UTP4 expression has been shown
to significantly inhibit ribosome synthesis (11). Additionally, mutations in the UTP4
gene are linked to North American Indian Childhood Cirrhosis
(NAIC), further highlighting its essential role in RB (12,13).
In colorectal cancer (CRC) cells, RNA interference (RNAi)-mediated
suppression of UTP4 notably decreased cell proliferation and
promoted apoptosis (14).
Similarly, in gliomas, high levels of UTP4 have been strongly
correlated with rapid tumor progression and poor clinical outcomes
(15). These collective findings
suggest that UTP4 serves as a pivotal regulator in RB and
potentially contributes to cancer progression.
The present study aims to evaluate UTP4 as a
pan-cancer prognostic biomarker and to validate its biological
functions in GC. By utilizing multi-omics data from TCGA and other
databases, the expression patterns of UTP4 were comprehensively
analyzed across multiple cancer types and its associations with
patient prognosis were examined. Additionally, through in vitro
experiments, the influence of UTP4 on GC cell proliferation,
migration and invasion was explored. The present findings offer
novel insights into UTP4's role in cancer biology and underscore
its potential as a prognostic biomarker and therapeutic target for
GC.
Materials and methods
Data collection
RNA sequencing data and clinical details regarding
UTP4 were retrieved from The Cancer Genome Atlas (TCGA) database
(http://cancergenome.nih.gov/). Protein
expression data were obtained from the UALCAN database (http://ualcan.path.uab.edu/index.html).
Additionally, whole-genome CRISPR-Cas9 screening data were
downloaded from the DepMap portal (https://depmap.org/portal/download/).
Genomic and protein alterations of
UTP4 in pan-cancer
RNA sequencing and clinical data covering 33 tumor
types were carefully analyzed after download from the TCGA
database. Expression levels in transcripts per million (TPM) were
converted using log2 transformation for accuracy. Differences in
UTP4 expression across tumor types were further analyzed by
constructing boxplots and paired plots in R (version 4.2.1) using
packages such as ggplot2 (version 3.3.6), stats (version 4.2.1),
and car (version 3.1-0). The Tumor Immune Estimation Resource
version 2 (TIMER2.0; http://timer.cistrome.org/) was also used to compare
UTP4 expression between tumor and normal tissues.
The UALCAN platform (http://ualcan.path.uab.edu/index.html) was utilized
for additional analyses. The proteomics module within UALCAN
allowed comparisons of UTP4 protein expression between tumor and
normal tissues, as well as analyses of its association with
clinical characteristics, including cancer stage.
UTP4 expression at single-cell spatial
transcriptomics level
To further validate differences in UTP4 expression
in cancer and examine its gene expression profiles in specific cell
types, the Sparkle database (https://grswsci.top/) was employed. The Spatial
Transcriptomics module was selected, and five datasets were
analyzed, namely ‘CESC: Human_Cervical_Cancer (10x database)’,
‘GIST2: GSE203612-GSM6177609’, ‘HNSC2:
GSE181300-GSM5494476_HNSCC201125T05’, ‘KIRC2:
GSE175540-GSM5924030_ffpe_c_2’, and ‘CRC2: IntestineCancer_10x_FFPE
(10x database)’, to explore differences in UTP4 expression at
single-cell resolution within spatial transcriptomic contexts.
Prognostic and diagnostic value
analysis
RNA sequencing data and clinical information for the
33 tumor types were standardized and analyzed for prognostic
significance. Using the R ‘survival’ package, Kaplan-Meier survival
curves were constructed, proportional hazards assumption tests were
performed, and survival regression models were fitted.
Receiver operating characteristic (ROC) curve
analysis was conducted using the timeROC package (version 0.4) to
evaluate diagnostic performance. The area under the ROC curve (AUC)
values, ranging from 0.5 to 1, were assessed to determine
diagnostic capability, with higher AUC values indicating superior
diagnostic accuracy (16).
UTP4 gene alteration analysis
To investigate UTP4 gene alterations across
different cancers, the cBioPortal platform (https://www.cbioportal.org/) was utilized, an
interactive tool designed for analyzing multidimensional cancer
genomic datasets. The study cohort was selected from the ‘TCGA
PanCancer Atlas Studies’, and the UTP4 gene was queried using
cBioPortal's module to obtain relevant mutation information. The
‘Cancer Types Summary’ module provided an overview of mutation
frequencies and types associated with UTP4 across various cancers.
Additionally, the ‘Mutations’ module offered detailed insights into
mutation sites, counts, and types within the UTP4 gene. Mutation
data were subsequently visualized using cBioPortal's visualization
tools. To further analyze the mutational landscape, the ‘Mutation
Effect-Expression’ module of the Sparkle database (https://grswsci.top/) was employed. This database
enabled analysis of UTP4 mutation types and single nucleotide
variations (SNVs), revealing potential roles for UTP4 gene
alterations in cancer progression.
Immune-related analysis
Multiple databases were used to investigate the
relationship between UTP4 and immune processes. Initially, immune
infiltration associated with UTP4 expression across various tumors
was analyzed using the Xiantao Academy (www.xiantaozi.com). Subsequently, the Sparkle database
(https://grswsci.top/) was utilized to examine
associations between UTP4 expression and the tumor immune
microenvironment, including immune cells specifically in GC.
Finally, correlations between pan-cancer UTP4 expression and tumor
stemness, as well as immune checkpoints, were explored using the
SangerBox tool (http://sangerbox.com/home.html).
Protein-protein interaction network
and enrichment analysis
The protein interaction network involving UTP4 was
obtained from the STRING database (https://string-db.org/) (17). Next, the top 100 genes correlated
with UTP4 expression were retrieved from GEPIA2.0 (http://timer.cistrome.org/) (18). Utilizing the ‘clusterProfiler
(version 4.4.4)’ package in R, enrichment analysis was performed on
the top 10 correlated genes. This allowed the identification of
crucial phenotypes and signaling pathways involving UTP4 across
pan-cancer contexts. Additionally, Pearson correlations between
UTP4 expression z-scores and GSVA scores for 14 tumor states were
analyzed using the ‘Pathway Analysis-Activity Score’ template
provided by the Sparkle database (https://grswsci.top/) (19).
Univariate Cox regression analysis in
pan-cancer and multivariate cox regression analysis in GC
The proportional hazards assumption was verified
using the ‘survival’ package in R, followed by univariate Cox
regression analysis conducted across various tumors. Specifically
focusing on GC, multivariate Cox proportional hazards regression
analysis was performed to assess the independent prognostic
significance of UTP4, adjusting for clinical variables including
age, sex and tumor stage. The results were illustrated through
forest plots displaying hazard ratios (HRs), 95% confidence
intervals (CIs), and P-values for UTP4 and other clinical
factors.
Bayesian colocalization analysis
Mendelian colocalization analysis, a statistical
method used to investigate genetic associations with diseases
across multiple phenotypes, was employed to identify shared genetic
factors. To explore genetic associations of UTP4 in pan-cancer and
GC contexts, datasets for pan-cancer (ieu-b-4966) and GC
(ebi-a-GCST90018629) were downloaded from the OpenGWAS database.
Colocalization analysis was conducted using the ‘coloc’ package.
Results were visualized through the ‘locuscompare’ function within
the ‘locuscompare’ package and the ‘stack_assoc_plot’ function from
the ‘gassocplot2’ package. A posterior probability (PP.H4.abf)
greater than 80% was set as the threshold indicating robust
evidence of colocalization (20).
Dependency score of UTP4 in various
tumor cell lines
Genome-wide CRISPR-Cas9 screening data were obtained
from the DepMap portal (https://depmap.org/portal/download/). This dataset
includes dependency scores for candidate genes across numerous
cancer cell lines, calculated using the CERES algorithm. The CERES
dependency scores for UTP4, which were scaled such that 0 indicates
a non-essential gene and -1 denotes the median dependency of core
essential genes, were specifically analyzed. The 200 cell lines
exhibiting the most negative UTP4 dependency scores were selected
and visualized using bar plots.
Cell culture and transfection
The human normal gastric epithelial cell line GES-1
and GC cell lines AGS and MKN-45 were purchased from Sai Bai Kang
(Shanghai) Biotechnology Co., Ltd. Cells were cultured in medium
containing 5% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C with 5% CO2. Cells in optimal growth
conditions were transfected with CRISPR-Cas9 small guide RNA
(sgRNA) lentiviral vectors. A total of three distinct sgRNAs were
designed against the human UTP4 gene. Their RNA guide sequences
(5'-3') are as follows: sgRNA#1: AUGCCUUUGGAGGACCUAUU; sgRNA#2:
CGUCAGCAUUAGCGAUGAGA; and sgRNA#3: GUAGCCCGAGACUCAUGACC.
The knockdown efficiency of all three sgRNAs was
initially evaluated by reverse transcription-quantitative PCR
(RT-qPCR). sgRNA#2 showed the strongest and most consistent
reduction in UTP4 mRNA levels and was therefore chosen to generate
stable knockdown cell pools for all subsequent functional
experiments. A custom non-targeting control (NTC) sgRNA was
included in parallel to account for potential non-specific effects
associated with lentiviral transduction and Cas9 activity. Its RNA
guide sequence is: GGCAGCGAGAUAACUUGACUC. All sgRNA lentiviral
vectors were supplied by Shenggong Bioengineering (Shanghai) Co.,
Ltd. The initial qPCR screening data assessing the knockdown
efficiency of the three sgRNAs are provided in Fig. S1 and Table SIII.
Cell Counting Kit-8 (CCK-8) assay
Cell viability following sgRNA transfection was
measured using the CCK-8 from Shanghai Taosu Biotechnology Co.,
Ltd. Transfected cells were seeded into 96-well plates at a density
of 1x103 cells per well and incubated overnight at 37˚C.
At 0, 1, 2, and 3 days, 10 µl of CCK-8 solution was added to each
well, and absorbance was measured at a wavelength of 450 nm.
RT-qPCR
Total RNA was extracted from cells using TRIzol
Universal RNA extraction reagent according to the manufacturer's
protocol. RT-qPCR were performed using the Vazyme genomic RT-qPCR
kit and an ABI 7900HT Fast Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions for PCR amplification were as follows: Initial
denaturation at 95˚C for 30 sec; 40 cycles of denaturation at 95˚C
for 10 sec, annealing at 60˚C for 30 sec, and extension at 72˚C for
30 sec. Quantification was performed using the
2-ΔΔCq method (21). Each experiment was repeated three
times. The primer sequences used were as follows: UTP4 forward,
5'-AGGTCCATCGAGTACGTTTCT-3' and reverse,
5'-CACAGTGCCATCTGTTCGTG-3'. GAPDH was used as an endogenous control
for normalization. The primer sequences for GAPDH were as follows:
forward, 5'-GAAGGTGAAGGTCGGAGTC-3' and reverse,
5'-GAAGATGGTGATGGGATTTC-3'.
Wound healing assay
Prior to the scratch assay, cells were serum-starved
in medium containing 0.5% FBS for 12 h to suppress cell
proliferation. A baseline was drawn horizontally at the bottom of
each well in a six-well plate for reference. Once cells reached
near-confluence, a sterile pipette tip was gently used to scratch a
straight line along the baseline, creating a wound. Images of the
initial wound state were immediately captured under a light
microscope, and its width was recorded. After 48 h, images of the
wound area were captured again. The wound area was precisely
measured using ImageJ software (version 1.53t; National Institutes
of Health) to assess cell migration rates and wound closure
efficiency.
Apoptosis assay
Cells were cultured in six-well plates to an
appropriate density. Cell apoptosis was assessed following the
manufacturer's protocol using the Annexin V-AF488/PI Apoptosis
Detection Kit (BioLegend-Shanghai Yaji Biotechnology Co., Ltd.).
The number of apoptotic cells was quantitatively determined using a
BD LSRFortessa flow cytometer (BD Biosciences) and analyzed with BD
FACSDiva software (version 9.0; BD Biosciences).
Statistical analysis
Statistical analyses were performed using R (version
4.3.0) and GraphPad Prism (version 10.3.0), with visualization and
specific tests conducted via R packages (ggplot2, survminer,
pheatmap, timeROC). A two-sided P<0.05 was considered to
indicate a statistically significant difference.
For public omics and clinical data, continuous
variables are presented as the mean ± SD or median (IQR). Group
comparisons used paired/independent t-tests, one-way ANOVA (with
Tukey's post hoc test), or Kaplan-Meier analysis (log-rank test).
The independent prognostic value of UTP4 was assessed by Cox
proportional hazards regression (assumption verified via Schoenfeld
residuals). Diagnostic performance was evaluated with
time-dependent ROC curves. Correlations were calculated using
Pearson's coefficient. Functional enrichment (GO/KEGG) was analyzed
with the clusterProfiler package (FDR-adjusted).
For in vitro data, results from ≥3
independent replicates are shown as mean ± SD. Comparisons used the
independent t-test (two groups) or two-way ANOVA with Tukey's post
hoc tests, as indicated.
Results
Abnormal expression of UTP4 in various
tumors and normal tissues
To examine differential UTP4 expression across
cancer types, RNA sequencing data from the TCGA database were
analyzed. UTP4 was significantly upregulated in several cancers,
including cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD),
esophageal cancer (ESCA), head and neck cancer (HNSC),
hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung
squamous cell carcinoma (LUSC), rectal adenocarcinoma (READ),
stomach adenocarcinoma (STAD), bladder cancer (BLCA) and
glioblastoma (GBM) (P<0.05) (Fig.
1A). These findings suggest that UTP4 may play an important
role in the initiation and progression of multiple malignancies.
Conversely, UTP4 expression was significantly decreased in breast
cancer (BRCA), kidney chromophobe (KICH) and prostate cancer (PRAD)
(P<0.05; Fig. 1A). A follow-up
analysis using cancer-paired samples further supported these
results. Pronounced differences in UTP4 expression were observed
across the corresponding tumor types, consistent with the initial
findings (Fig. 1B). Additional
validation using the TIMER2.0 database confirmed similar expression
patterns. Although the difference in UTP4 expression in GBM did not
reach statistical significance, other cancer types exhibited
expression trends consistent with the TCGA analysis. Notably,
cervical cancer (CESC) showed significant upregulation of UTP4
(P<0.05; Fig. 1C).
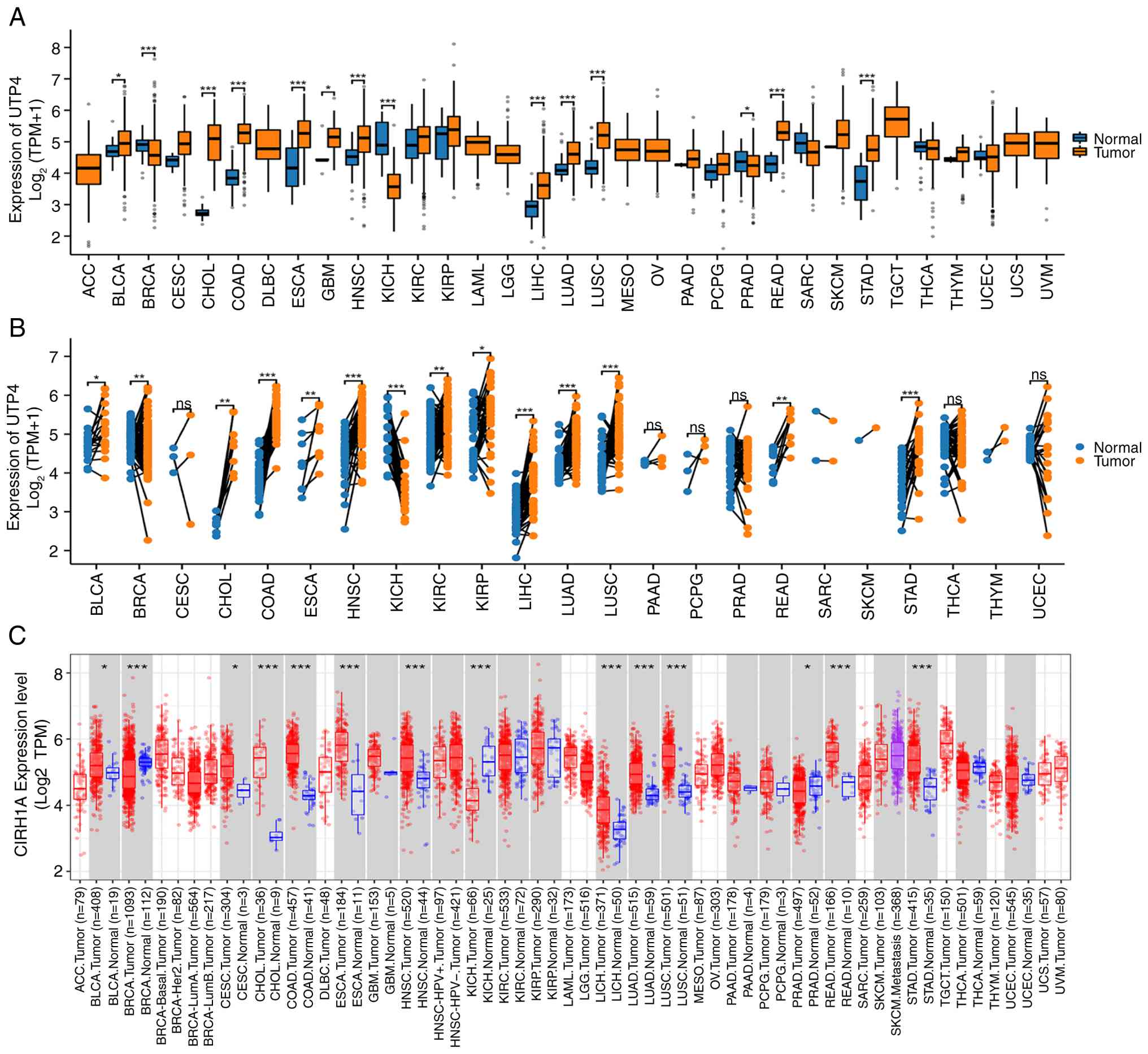 | Figure 1(A) UTP4 expression across pan-cancer
types based on the TCGA dataset. (B) Comparison of UTP4 expression
between paired tumor and normal samples from TCGA. (C) UTP4
expression across pan-cancer types using the TIMER2.0 database.
*P<0.05, **P<0.01 and
***P<0.001. TCGA, The Cancer Genome Atlas; ns, not
significant. ACC, adenoid cystic carcinoma; BLCA, bladder
urothelial carcinoma; BRCA, breast cancer; CESC, cervical squamous
cell carcinoma; CHOL, cholangiocarcinoma; COAD, colon
adenocarcinoma; DLBC, diffuse large B-cell lymphoma; ESCA,
esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and
neck cancer; KICH, kidney chromophobe; KIRC, kidney renal clear
cell carcinoma; KIRP, kidney renal papillary carcinoma; LAML, acute
myeloid leukemia; LGG, low-grade glioma; LIHC, liver hepatocellular
carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell
carcinoma; MESO, mesothelioma; OV, ovarian cancer; PAAD, pancreatic
adenocarcinoma; PCPG, pheochromocytoma; PRAD, prostate
adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM,
skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT,
tenosynovial giant cell tumor; THCA, thyroid carcinoma; THYM,
thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine
carcinosarcoma; UVM, uveal melanoma. |
Differential expression of UTP4 at the
protein level
To further characterize UTP4 expression at the
protein level, proteomic data from the UALCAN database were
analyzed. UTP4 protein levels were significantly elevated in
multiple cancer types, including COAD, HNSC, kidney renal clear
cell carcinoma (KIRC), pancreatic adenocarcinoma (PAAD),
endometrial carcinoma (UCEC), LUAD, LUSC, GBM and LIHC (P<0.05;
Fig. 2A). These findings reinforce
the hypothesis that UTP4 may contribute to tumor progression.
Moreover, UTP4 protein expression varied significantly across
pathological stages in COAD, HNSC, KIRC, PAAD, UCEC and lung cancer
(P<0.05; Fig. 2B). This
stage-dependent variation suggests that UTP4 overexpression is
closely linked to tumor development rather than being an isolated
molecular event, supporting its potential role in cancer
progression.
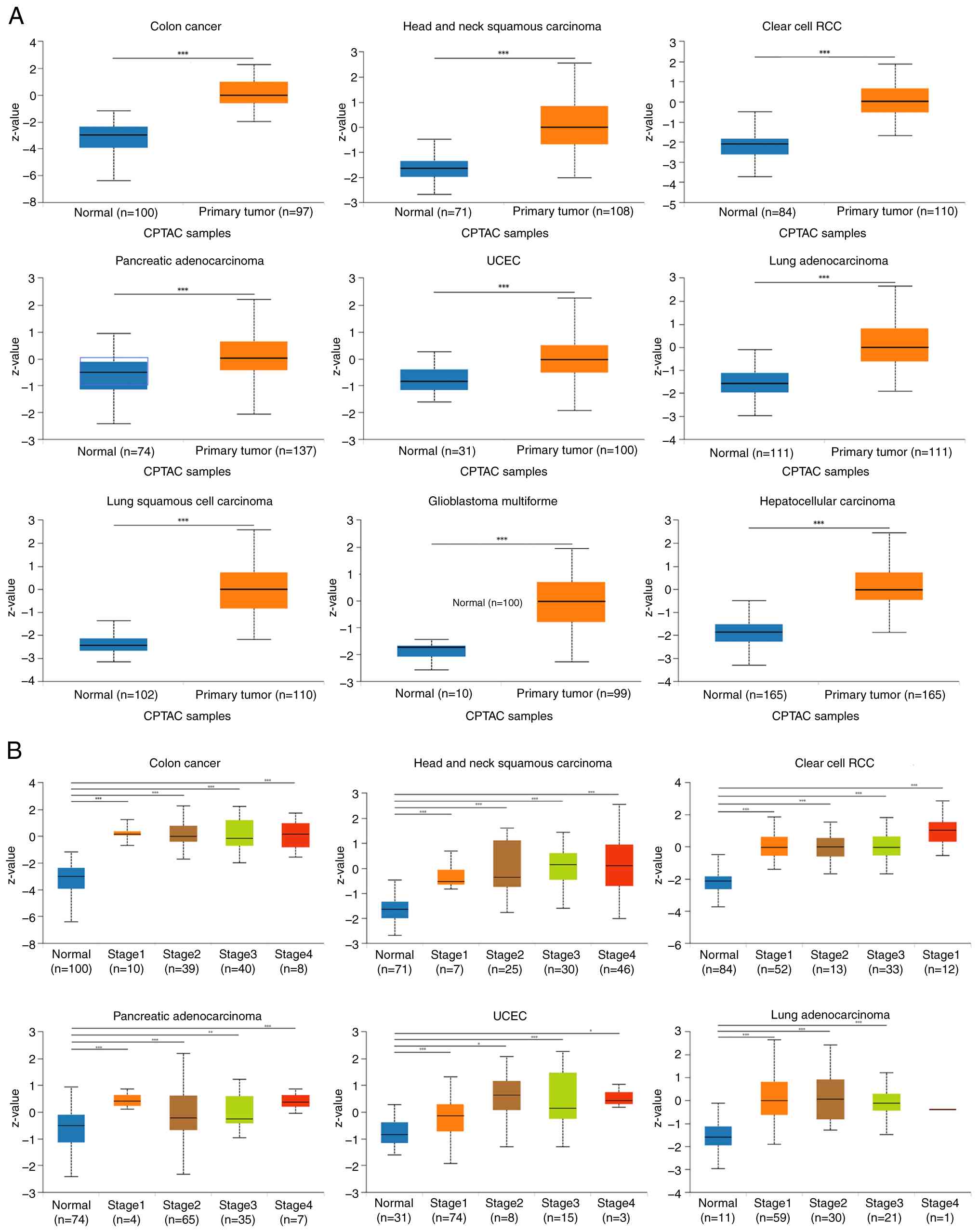 | Figure 2(A) UTP4 protein expression in COAD,
HNSC, KIRC, PAAD, UCEC, LUAD, LUSC, glioblastoma multiforme and
liver hepatocellular carcinoma from the UALCAN database. (B)
Differential UTP4 expression across pathological stages in COAD,
HNSC, KIRC, PAAD, UCEC and LUAD. *P<0.05,
**P<0.01 and ***P<0.001. COAD, colon
adenocarcinoma; HNSC, head and neck cancer; KIRC, kidney renal
clear cell carcinoma; PAAD, pancreatic adenocarcinoma; UCEC,
uterine corpus endometrial carcinoma; LUAD, lung adenocarcinoma;
RCC, renal cell carcinoma; ns, not significant. |
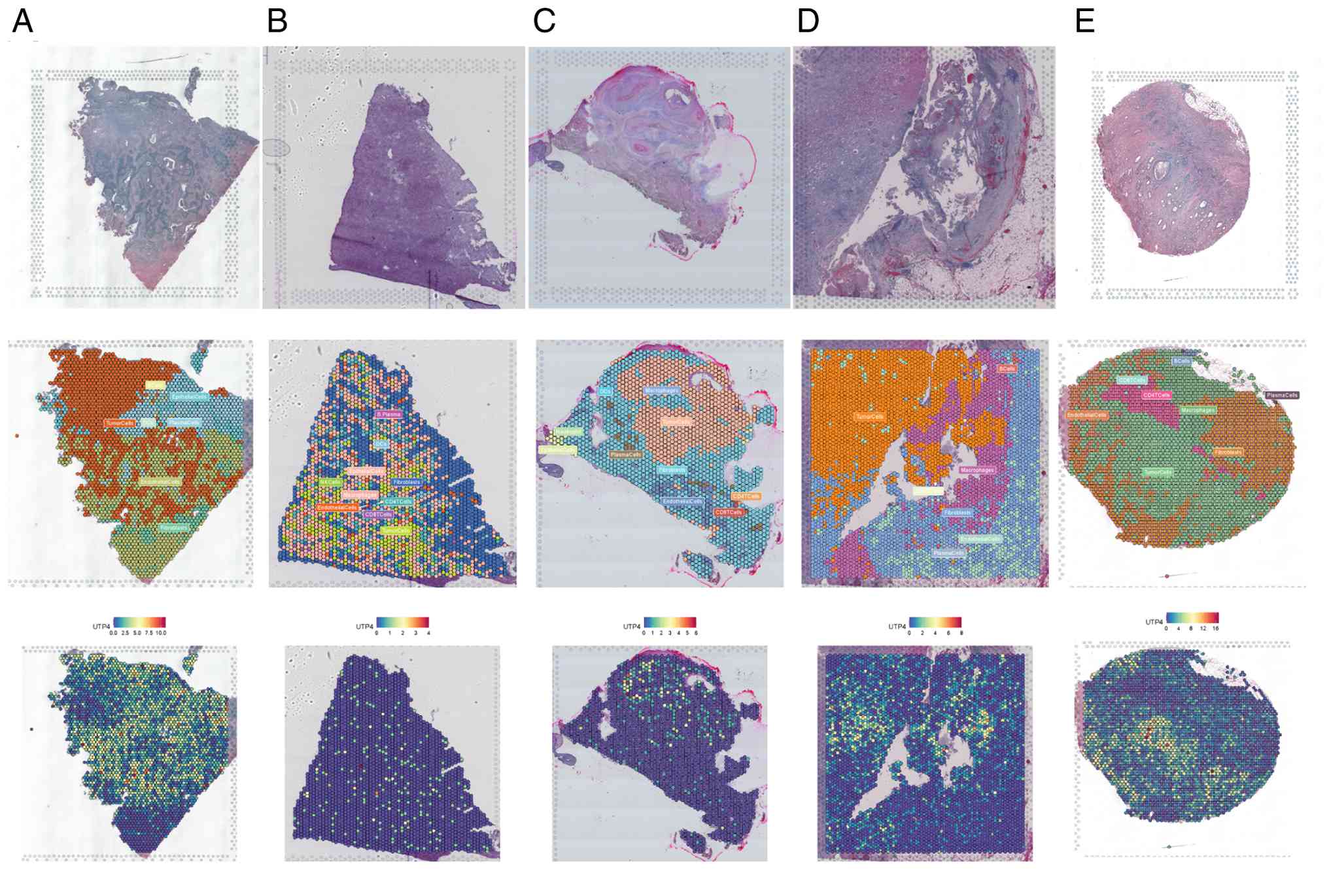
Differential expression of UTP4 in
single cells
Single-cell spatial transcriptomic analysis was
conducted to precisely evaluate UTP4 expression differences among
various cancers and cell types (22). This approach allowed detailed
exploration of UTP4 expression in specific cellular populations
(23,24). The results demonstrated that in
human cervical squamous cell carcinoma samples
(Human_Cervical_Cancer), UTP4 expression was mainly detected in
tumor and epithelial cells (Fig.
3A). Similarly, gastric stromal tumor (GSE203612-GSM6177609)
samples showed predominant UTP4 expression in tumor and epithelial
cells (Fig. 3B). In head and neck
squamous cell carcinoma (GSE181300-GSM5494476_HNSCC201125T05), UTP4
expression was concentrated predominantly in tumor cells (Fig. 3C). In clear cell renal cell
carcinoma (GSE175540-GSM5924030_ffpe_c_2) samples, UTP4 was
expressed not only in tumor cells but also in macrophages (Fig. 3D). In CRC
(IntestineCancer_10x_FFPE) samples, UTP4 expression was primarily
localized to tumor and epithelial cells (Fig. 3E). These single-cell spatial
transcriptomics findings highlight that UTP4 is predominantly
expressed in tumor and epithelial cells in various tumor tissues,
suggesting its critical involvement across multiple cancer
types.
Prognostic and diagnostic value of
UTP4 in pan-cancer
A comprehensive survival analysis was conducted to
evaluate the prognostic significance of UTP4. Elevated UTP4
expression significantly correlated with poor prognosis in multiple
cancers, including ACC (P=0.021), HNSC (P=0.008), GBMLGG
(P<0.001), LUAD (P=0.010), LGG (P=0.015), LIHC (P=0.036), PAAD
(P=0.025), SARC (P=0.048), STAD (P=0.023) and OSCC (P=0.038)
(Fig. 4A). These results imply
that UTP4 may be a robust indicator of adverse outcomes,
underscoring its importance in cancer progression and patient
survival. By contrast, high UTP4 expression correlated with
favorable prognosis in KIRC (P<0.001) (Fig. 4A). Furthermore, ROC curve analysis
evaluated the diagnostic accuracy of UTP4. UTP4 demonstrated strong
diagnostic performance in KICH (AUC=0.959), STAD (AUC=0.894), LIHC
(AUC=0.823) and OSCC (AUC=0.810) (Fig.
4B), and moderate diagnostic ability in HNSC (AUC=0.787),
GBMLGG (AUC=0.630), LUAD (AUC=0.795), and PAAD (AUC=0.682)
(Fig. 4B). Analysis of survival
rates by sex showed that the association between UTP4 expression
levels and prognosis was more significant in females (Fig. 4C). Notably, although UTP4
expression was significantly associated with overall survival (OS),
it showed no statistically significant correlation with the
disease-free interval (DFI) (P=0.39; Fig. S2). This discrepancy suggests that
high expression of UTP4 may not directly drive early tumor
recurrence but instead predominantly influences disease progression
after recurrence. These findings emphasize the significant
prognostic and diagnostic relevance of UTP4 across various cancer
types. Collectively, the results support the utility of UTP4 as a
potential biomarker and provide a basis for developing cancer
diagnostic and prognostic assessment tools.
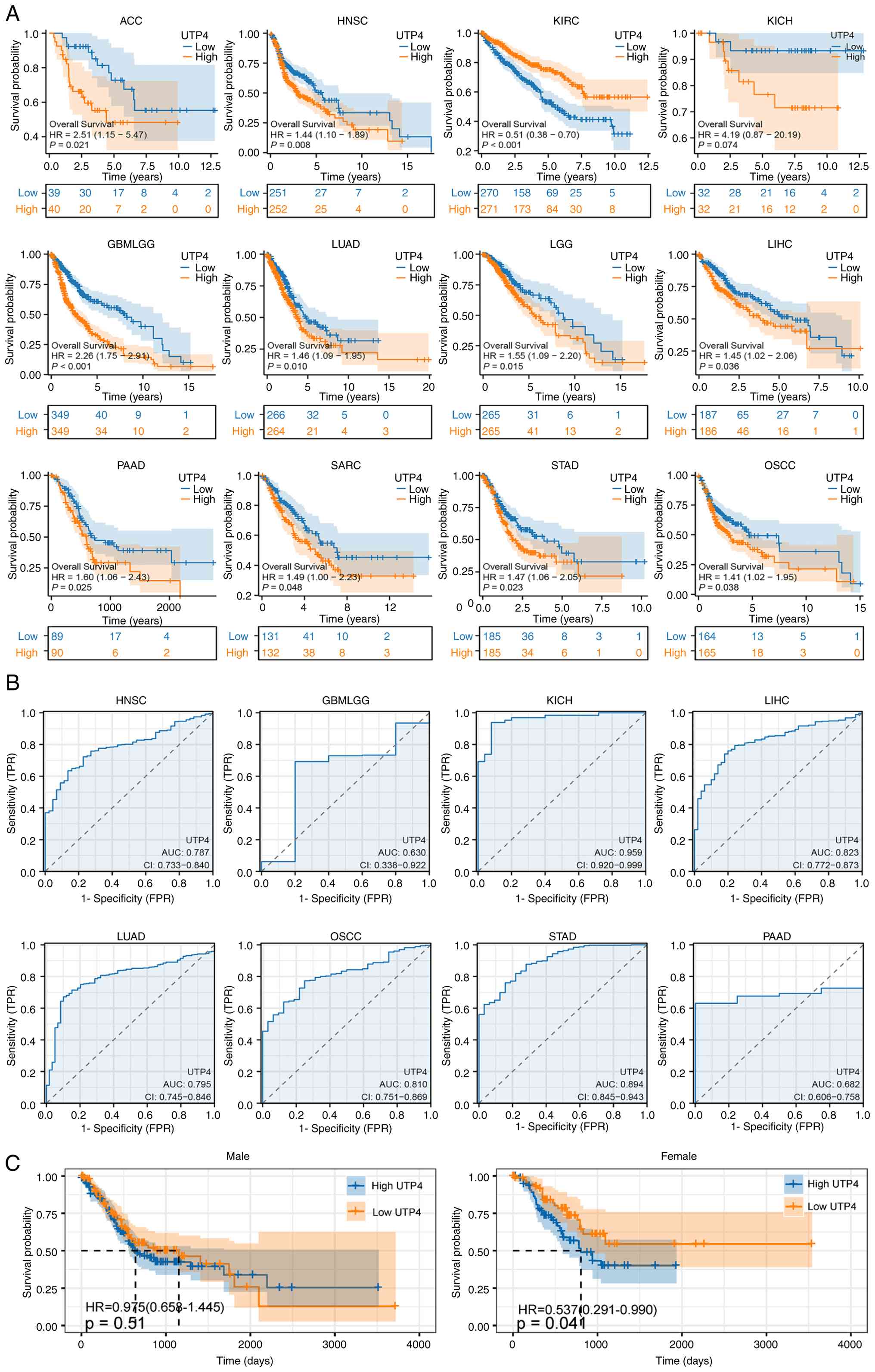 | Figure 4(A) Overall survival analysis of UTP4
using TCGA data. (B) ROC curves for UTP4 based on TCGA data. (C)
Comparison of overall survival by sex regarding UTP4 expression
based on TCGA data. TCGA, The Cancer Genome Atlas; ACC, adenoid
cystic carcinoma; HNSC, head and neck cancer; KIRC, kidney renal
clear cell carcinoma; KICH, kidney chromophobe; GBMLGG,
glioblastoma and lower-grade glioma; LUAD, lung adenocarcinoma;
LGG, low-grade glioma; LIHC, liver hepatocellular carcinoma; PAAD,
pancreatic adenocarcinoma; SARC, sarcoma; STAD, stomach
adenocarcinoma; OSCC, oral squamous cell carcinoma. |
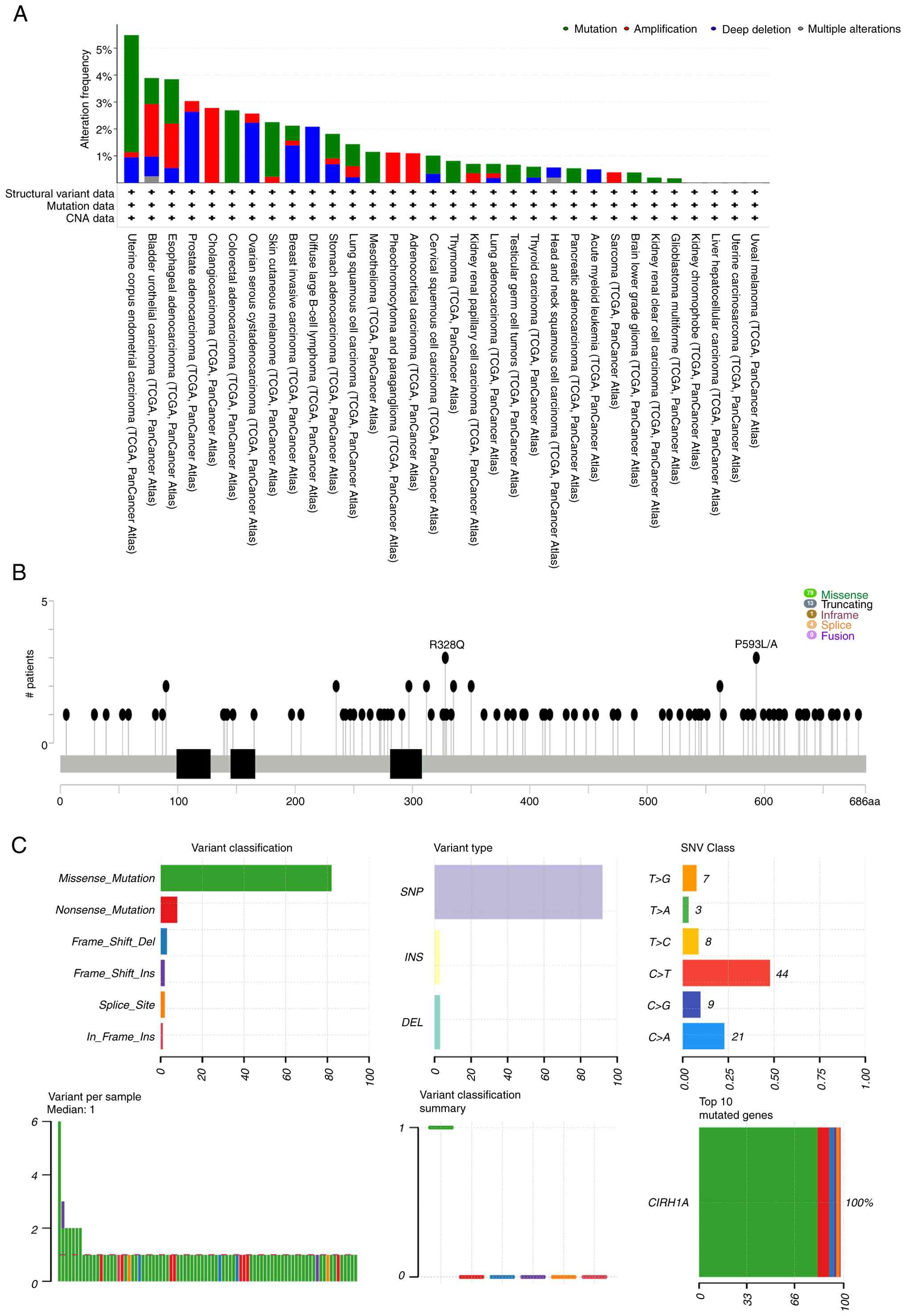
Mutation landscape of UTP4
To improve understanding of the mutational
characteristics of UTP4 across different cancers, detailed analyses
were conducted. Significant variations in UTP4 mutation frequencies
and types were observed among cancer types. High mutation
frequencies occurred in UCEC (mutation 4.35%, amplification 0.19%,
deep deletion 0.95%), BLCA (mutation 0.97%, amplification 1.95%,
deep deletion 0.73%, multiple alterations 0.24%), ESAD (mutation
1.65%, amplification 1.65%, deep deletion 0.55%), PRAD
(amplification 0.4%, deep deletion 2.63%), CHOL (amplification
2.78%), COADREAD (mutation 2.69%), OV (amplification 0.34%, deep
deletion 2.23%), SKCM (mutation 2.03%, amplification 0.23%), BRCA
(mutation 0.55%, amplification 0.18%, deep deletion 1.38%), DLBC
(deep deletion 2.08%), and STAD (mutation 0.91%, amplification
0.23%, deep deletion 0.68%) (Table
SI, Fig. 5A). Further analysis
using cBioPortal identified R328Q and P593L/A as the most frequent
mutation sites in UTP4. These mutations appeared in multiple
cancers and were each supported by at least three samples (Fig. 5B). Statistical evaluation revealed
that missense mutations predominated in UTP4, followed by nonsense
mutations, while SNVs were mainly single nucleotide polymorphisms
(SNPs) (25,26) (Fig.
5C). These findings indicate that UTP4 mutations vary
significantly across cancers, suggesting diverse functional roles.
Different mutation types might influence UTP4 functionality,
potentially affecting cancer initiation and progression. For
instance, missense mutations may alter UTP4 protein structure and
function, disrupting RB and cancer cell proliferation; nonsense
mutations may result in premature termination of UTP4 proteins and
loss of function; and SNV-type SNPs may affect the regulation of
UTP4 gene expression.
Relationship between UTP4 and immune
cells and microorganisms in the TME
Associations between UTP4 expression, immune cells
and microorganisms within the TME were further explored. UTP4
expression negatively correlated with most immune cells across
pan-cancer analyses, indicating its potential role in inhibiting
immune cell infiltration or function. However, positive
correlations were noted with specific immune cell subsets, such as
T helper cells, central memory T cells and Th2 cells (Fig. 6A). Moreover, UTP4 expression
exhibited significant correlations with various microorganisms in
the TME. Specifically, in STAD and COAD, UTP4 positively correlated
with most microorganisms; in ESCA, it showed negative correlations;
in READ and HNSC, both positive and negative correlations were
observed (Fig. 6B). These results
reflect the complex functional roles of UTP4 across cancers. In GC
specifically, elevated UTP4 expression negatively correlated with
infiltration of most immune cells but positively correlated with
particular immune subsets, including Common lymphoid
progenitor_XCELL, T cells CD4+ Th2_XCELL, and T cells CD4+
Th1_XCELL (Fig. 6C). Thus, UTP4
may regulate immune responses and microbial composition in the TME,
influencing cancer progression and patient prognosis.
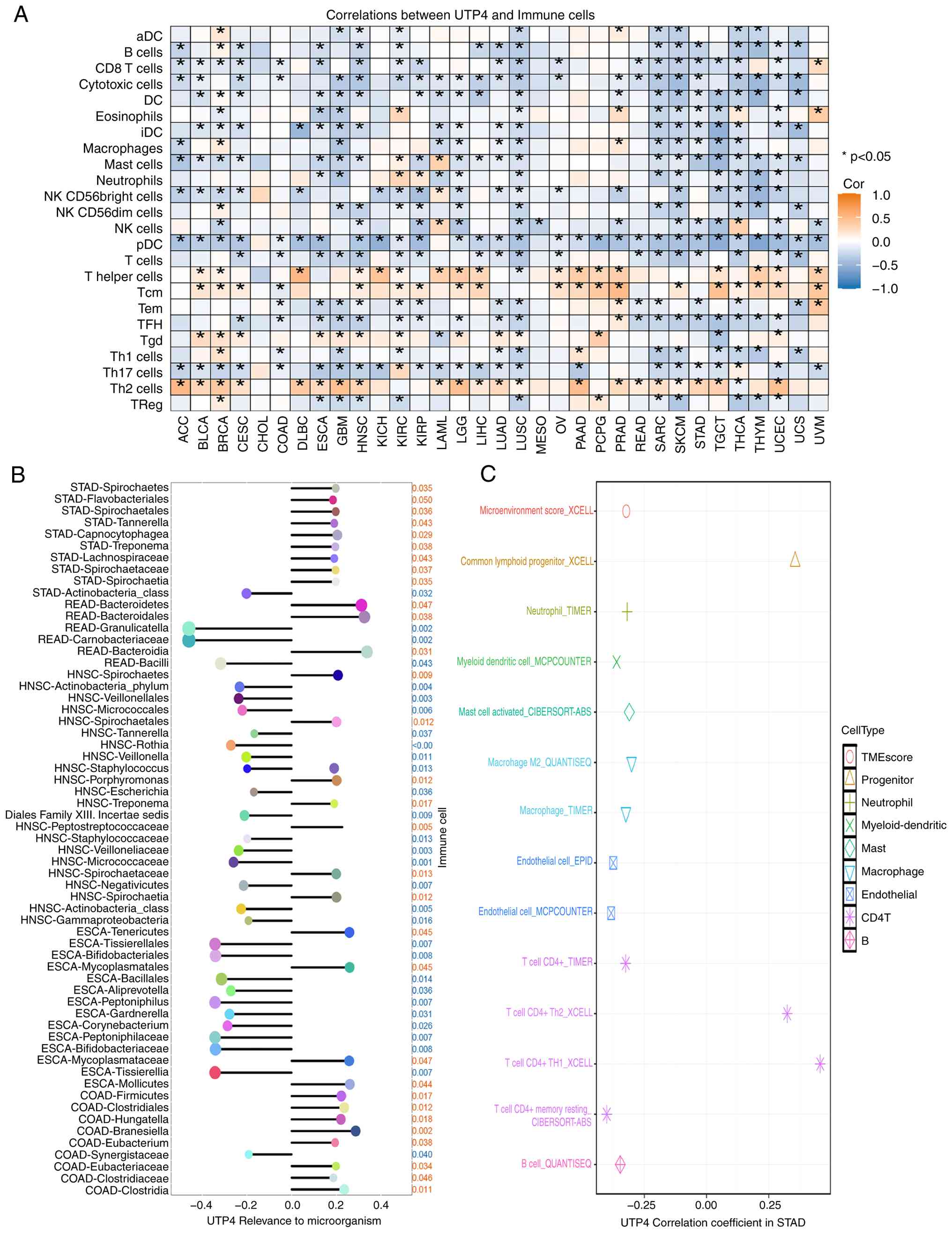 | Figure 6(A) Correlations between UTP4
expression and 24 immune cell types across pan-cancer.
*P<0.05. (B) Correlations between UTP4 expression and
intratumoral microbes in pan-cancer. (C) Correlations between UTP4
and immune cells specifically in gastric cancer. ACC, adenoid
cystic carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast
cancer; CESC, cervical squamous cell carcinoma; CHOL,
cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, diffuse large
B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma
multiforme; HNSC, head and neck cancer; KICH, kidney chromophobe;
KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal
papillary carcinoma; LAML, acute myeloid leukemia; LGG, low-grade
glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung
adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO,
mesothelioma; OV, ovarian cancer; PAAD, pancreatic adenocarcinoma;
PCPG, pheochromocytoma; PRAD, prostate adenocarcinoma; READ, rectum
adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD,
stomach adenocarcinoma; TGCT, tenosynovial giant cell tumor; THCA,
thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial
carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma. |
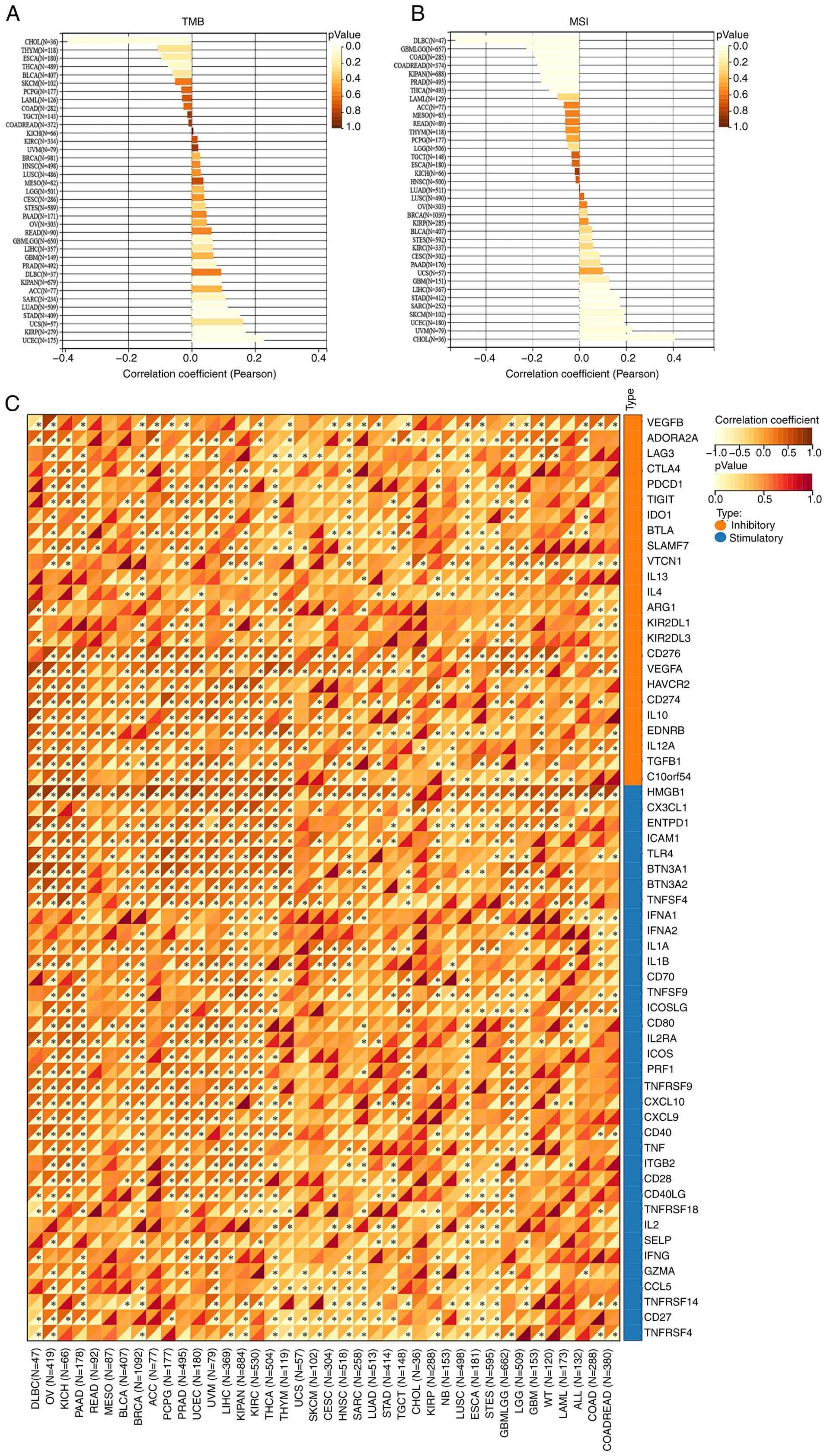
Correlation analysis of UTP4 with
tumor stemness and immune checkpoints
Tumor stemness refers to the stem cell-like capacity
of tumor cells for self-renewal and differentiation, enabling
aggressive proliferation and metastasis (27,28).
Tumor stem cells significantly contribute to tumor recurrence and
therapy resistance (29). To
investigate the role of UTP4 in tumor stemness, correlations
between UTP4 expression and tumor mutational burden (TMB) and
microsatellite instability (MSI) were analyzed. UTP4 expression
positively correlated with TMB in KIRC, UVM, BRCA, HNSC, LUSC,
MESO, LGG, CESC, STES, PAAD, OV, READ, GBMLGG, LIHC, GBM, PRAD,
DLBC, KIPAN, ACC, SARC, LUAD, STAD, UCS, KIRP and UCEC (Fig. 7A). Similarly, UTP4 expression
positively correlated with MSI in LUSC, OV, BRCA, KIRP, BLCA, STES,
KIRC, CESC, PAAD, UCS, GBM, LIHC, STAD, SARC, SKCM, UCEC, UVM and
CHOL (Fig. 7B). Immune checkpoint
molecules are crucial for tumor immune evasion. High UTP4
expression was significantly correlated with immune checkpoint
molecules in multiple cancers, including DLBC, OV, KICH, PAAD,
BLCA, BRCA, PCPG, PRAD, UCEC, UVM, LIHC, KIPAN, KIRC, THCA, THYM,
HNSC, KIRP, LUSC and GBMLGG (Fig.
7C). These results indicate that UTP4 may influence cancer
progression and metastasis by regulating tumor stemness, mutation
burden and immune checkpoint expression.
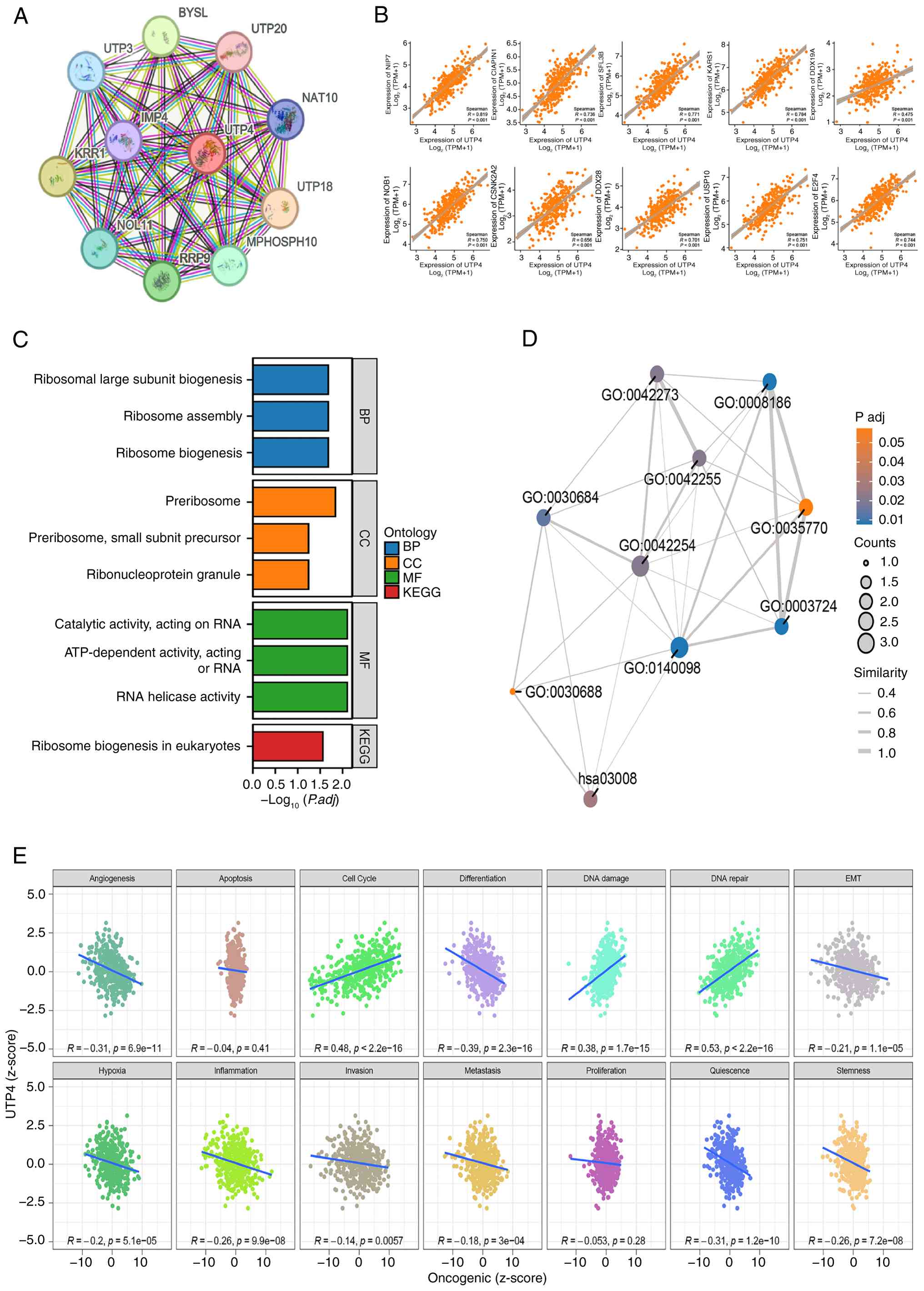
Enrichment analysis of UTP4-related
genes and correlation analysis of 14 tumor states in GC
To clarify the molecular mechanisms of UTP4 in
cancer, enrichment analysis of UTP4-associated genes was performed.
Firstly, a protein-protein interaction network involving 11
proteins related to UTP4 was obtained from the STRING database
(Fig. 8A). Subsequently, using
GEPIA2.0, the top 100 genes co-expressed with UTP4 were identified
(Table SII). Among these, the top
10 genes, including NIP7, CIAPIN1 and SF3B3, exhibited strong
pan-cancer correlations with UTP4, illustrated by scatter plots
(Fig. 8B). GO and KEGG enrichment
analyses (Fig. 8C and D) revealed significant involvement of
UTP4 in pathways such as RB, RNA processing, and ribonucleoprotein
particle assembly, highlighting its essential role in cellular
functions. RB is critical for cellular protein synthesis and normal
cellular operations (30,31). UTP4's participation suggests roles
in various stages of ribosome assembly, potentially supporting
rapid cancer cell proliferation. GO analysis results included
biological processes (BP), cellular components (CC), and molecular
functions (MF). The BP and CC analyses indicated that UTP4 is
involved in pre-ribosome assembly and biogenesis, processes
essential for efficient protein synthesis in cancer cells.
Additionally, CC analysis suggested that UTP4 might participate in
the formation and regulation of ribonucleoprotein granules, which
influence mRNA storage and translation. Dysregulated
ribonucleoprotein granules could disrupt gene expression control,
thereby promoting cancer development (32). BP results also indicated that UTP4
might exhibit catalytic functions, particularly ATP-dependent
RNA-related activities. Abnormal RNA processing and modification
due to altered UTP4 activity may disrupt RNA metabolism,
consequently supporting cancer cell proliferation and survival.
In GC, Pearson correlation analysis between UTP4
expression z-scores and GSVA scores of 14 tumor states showed
significant positive correlations with the cell cycle, DNA damage
and DNA repair (P<0.05; Fig.
8E). This suggests that UTP4 may facilitate cell proliferation
and tumor progression, and its elevated expression might indicate
increased DNA damage coupled with active repair mechanisms in tumor
cells. Conversely, UTP4 expression negatively correlated with
angiogenesis, differentiation, epithelial-mesenchymal transition,
hypoxia, inflammation, quiescence and stemness. However, the
precise roles of UTP4 in these states require further
investigation.
Prognostic univariate and multivariate
Cox regression
To assess the prognostic significance of UTP4,
pan-cancer univariate and GC multivariate Cox regression analyses
were performed using RNA-seq data and clinical information from 33
cancer types (TCGA database). Univariate analysis revealed that
UTP4 significantly influenced patient prognosis in ACC (P=0.0211),
HNSC (P=0.0082), KIRC (P=2.17x10-5), LGG (P=0.0149), LIHC
(P=0.0359), LUAD (P=0.0101), PAAD (P=0.0253), SARC (P=0.0485) and
STAD (P=0.0226) (Fig. 9A). These
findings indicate the potential utility of UTP4 as a prognostic
biomarker across multiple cancers.
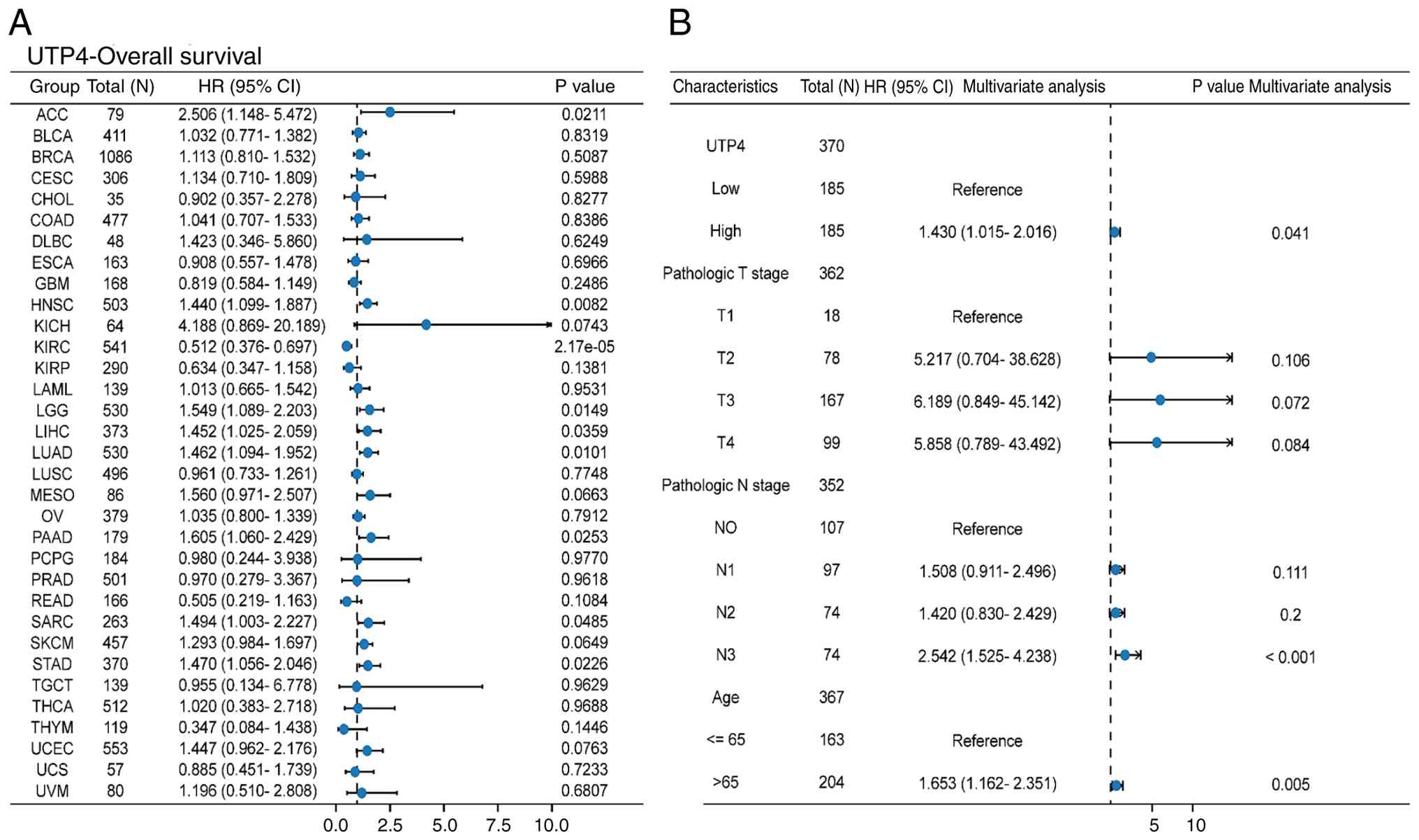 | Figure 9(A) Forest plot of univariate Cox
regression analysis results for UTP4 across pan-cancer. (B) Forest
plot of multivariate Cox regression analysis results for GC (STAD),
including variables pathological T stage, pathological N stage, age
and ‘UTP4 expression level’. ACC, adenoid cystic carcinoma; BLCA,
bladder urothelial carcinoma; BRCA, breast cancer; CESC, cervical
squamous cell carcinoma; CHOL, cholangiocarcinoma; COAD, colon
adenocarcinoma; DLBC, diffuse large B-cell lymphoma; ESCA,
esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and
neck cancer; KICH, kidney chromophobe; KIRC, kidney renal clear
cell carcinoma; KIRP, kidney renal papillary carcinoma; LAML, acute
myeloid leukemia; LGG, low-grade glioma; LIHC, liver hepatocellular
carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell
carcinoma; MESO, mesothelioma; OV, ovarian cancer; PAAD, pancreatic
adenocarcinoma; PCPG, pheochromocytoma; PRAD, prostate
adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM,
skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT,
tenosynovial giant cell tumor; THCA, thyroid carcinoma; THYM,
thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine
carcinosarcoma; UVM, uveal melanoma. |
Subsequently, multivariate Cox regression was
performed for GC (STAD), including clinical variables (pathological
T stage, pathological N stage, age) with P-values <0.1 from
univariate analyses along with UTP4 expression. The results
demonstrated that UTP4 expression remained statistically
significant (P=0.041) after adjusting for clinical factors
(Fig. 9B), further validating UTP4
as an independent prognostic factor in GC.
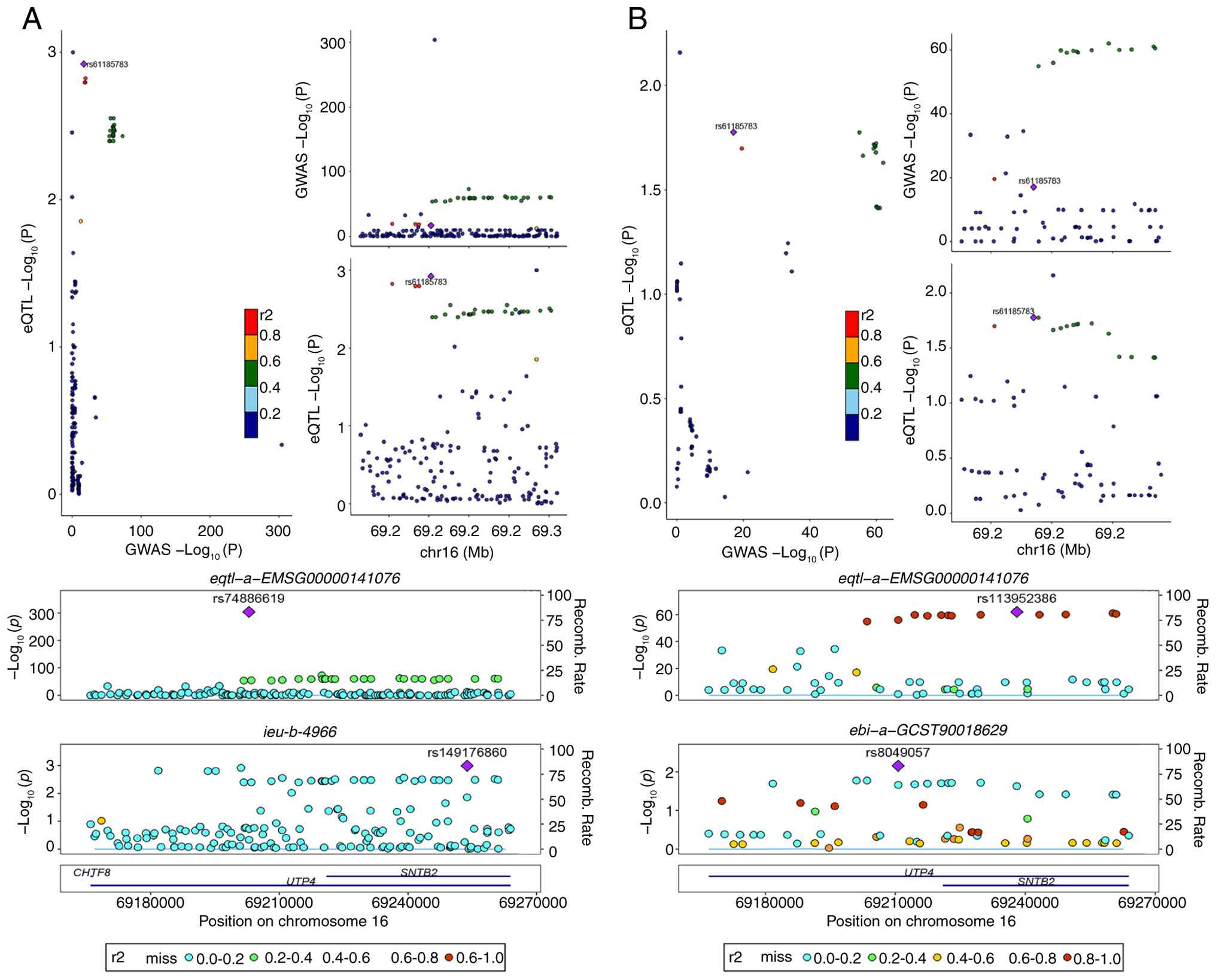
Colocalization analysis of UTP4 in
pan-cancer and GC
Colocalization analysis evaluates genetic
associations between genes and disease-related SNPs to elucidate
potential disease mechanisms (33). Colocalization of UTP4 with
pan-cancer GWAS data identified the locus rs74886619, displaying a
posterior probability of 1 (Fig.
10A). This result indicates potential shared genetic mechanisms
between UTP4 and pan-cancer, emphasizing its importance. Similarly,
in GC (STAD), colocalization analysis identified rs113952386 with a
posterior probability of 0.87 (Fig.
10B), suggesting a significant genetic role for UTP4 in GC
development.
Gene editing-CRISPR-Cas9 CERES growth
essentiality scores predict the effect of UTP4 knockout
To predict the effect of UTP4 knockout on cancer
cell proliferation, CRISPR-Cas9 screening data from the DepMap
database were analyzed to measure gene essentiality (34). Data scaling ensured accurate
scoring, with 0 representing non-essential genes and -1 indicating
median effects of core essential genes. UTP4 dependency scores were
frequently below -1.5, approaching -2 in various cancer cell lines,
such as BLCA, BRCA and CESC (Fig.
11). These scores indicate that UTP4 is essential in these cell
lines, suggesting that its knockout might significantly inhibit
cancer cell proliferation or induce apoptosis, thereby confirming
its role in cancer cell proliferation and survival.
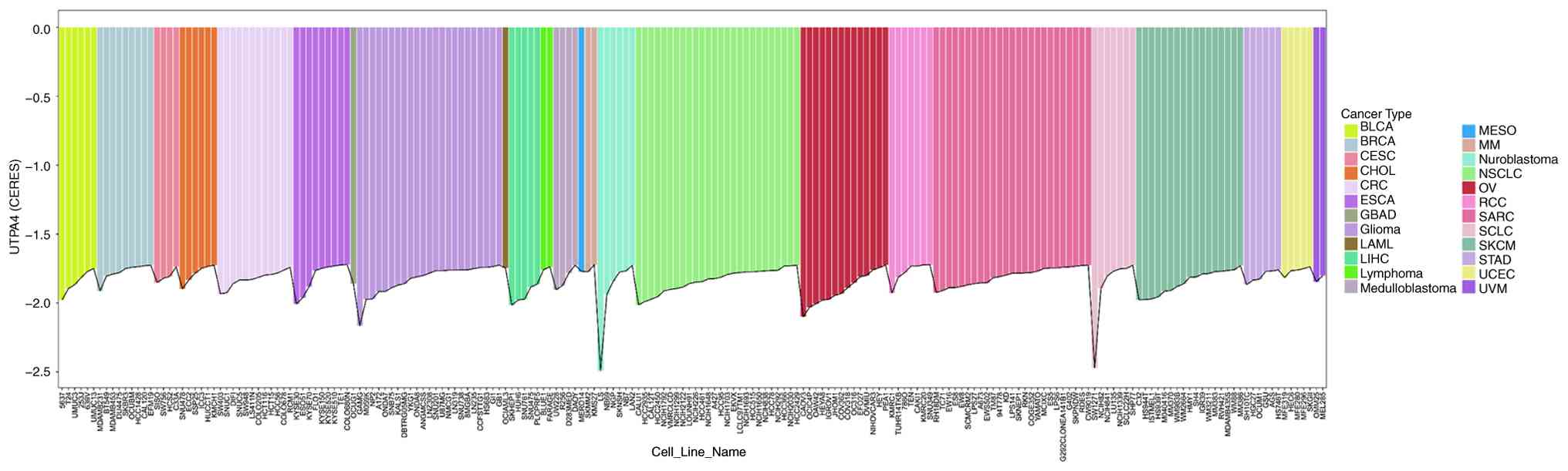 | Figure 11Visualization of CERES growth
essentiality scores for UTP4 across the top 200 pan-cancer cell
lines (y-axis represents CERES scores; x-axis indicates different
cell lines; colors represent different tumor types). BLCA, bladder
urothelial carcinoma; BRCA, breast cancer; CESC, cervical squamous
cell carcinoma; CHOL, cholangiocarcinoma; CRC, colorectal cancer;
ESCA, esophageal carcinoma; GBAD, gallbladder adenocarcinoma; LAML,
acute myeloid leukemia; LIHC, liver hepatocellular carcinoma; MESO,
mesothelioma; MM, multiple myeloma; NSCLC, non-small cell lung
cancer; OV, ovarian cancer; RCC, renal cell carcinoma; SARC,
sarcoma; SCLC, small cell lung cancer; SKCM, skin cutaneous
melanoma; STAD, stomach adenocarcinoma; UCEC, uterine corpus
endometrial carcinoma; UVM, uveal melanoma. |
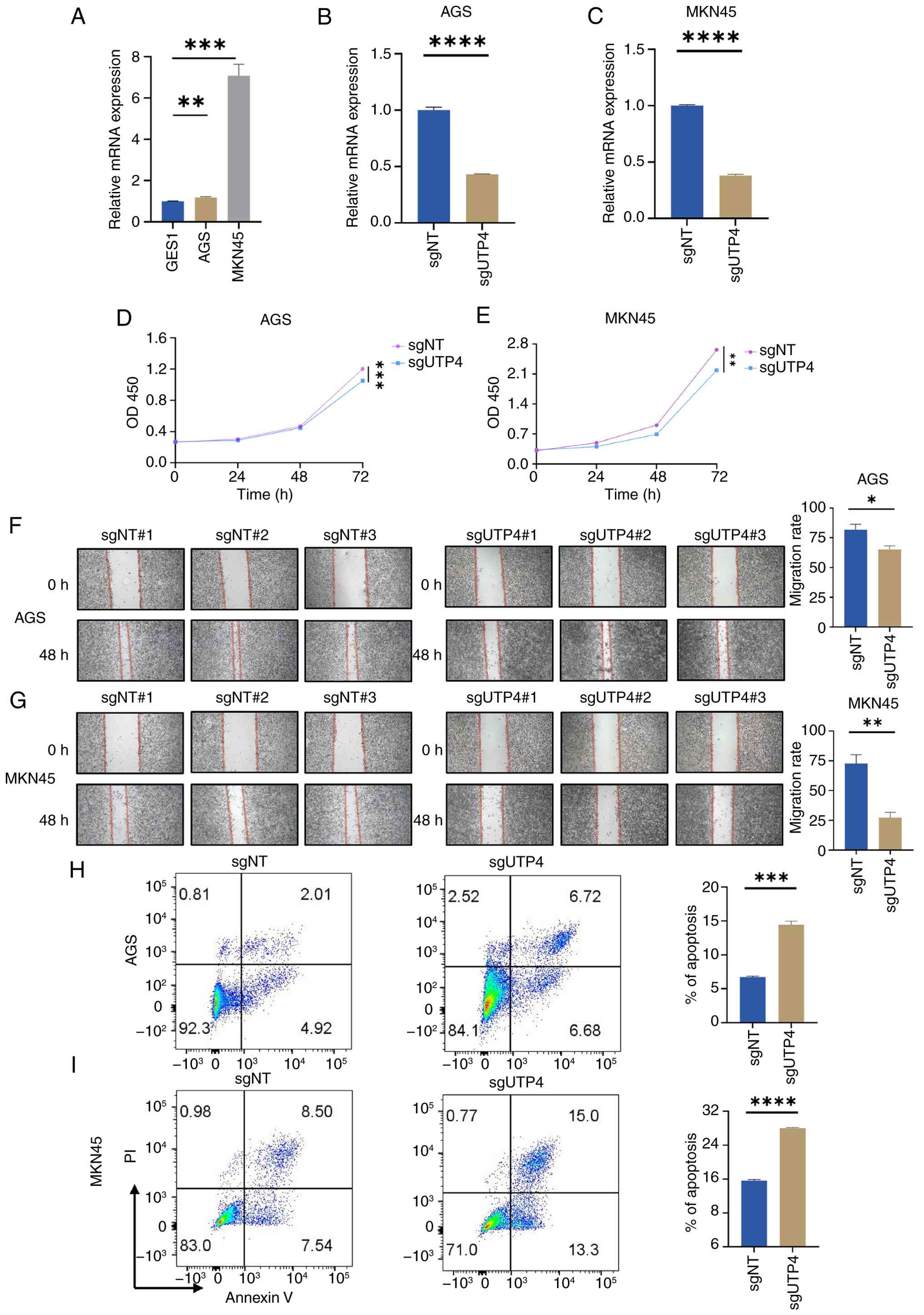
UTP4 knockout inhibits proliferation
and migration, promotes apoptosis in STAD
Based on genetic associations from colocalization
analysis and CRISPR-Cas9 screening predictions, the function of
UTP4 in GC cells was further investigated. First, qPCR was employed
to evaluate UTP4 mRNA expression in normal gastric epithelial cells
(GES-1) and GC cell lines (AGS and MKN-45). UTP4 expression was
significantly higher in GC cell lines compared with normal
epithelial cells (Fig. 12A).
Specific sgRNAs were designed to silence UTP4 expression
effectively in both GC cell lines, and the efficacy of the selected
sgRNA (sgUTP4#2) was confirmed via qPCR (Fig. 12B and C). Subsequent CCK-8, wound healing and
apoptosis assays showed a marked decrease in proliferation of AGS
and MKN-45 cells within 72 hours following UTP4 knockout (Fig. 12D and E). Furthermore, cell migration was
significantly reduced after 48 h (Fig. 12F and G). These findings substantiate the
critical role of UTP4 in GC cell migration, suggesting its
involvement in invasive processes related to cancer progression.
Additionally, apoptosis assays revealed a significant increase in
apoptosis rates following UTP4 depletion. The apoptosis rate in AGS
cells increased notably from 6.93 to 13.4%, and in MKN-45 cells
from 16.04 to 28.3% (Fig. 12H and
I). These results indicated that
UTP4 knockout significantly inhibits GC cell proliferation and
migration and promotes apoptosis, validating its critical role in
GC biology.
Discussion
Ribosomes are increasingly recognized as critical
drug targets and prognostic markers in cancer treatment. Their
central roles in protein synthesis, biomolecule assembly and
regulation of cell proliferation are closely associated with tumor
initiation and progression (35,36).
In particular, ribosomes regulate translation initiation, RB, and
cellular signaling, making them attractive targets for anticancer
therapy (37,38). Ribosome-targeted therapies have
effectively suppressed cancer cell proliferation, migration and
survival, particularly in drug-resistant tumor cells and cancer
stem cells (39,40). For example, specific drugs such as
CX-5461 (an RNA polymerase I inhibitor) and GCN2 inhibitors (genome
integrity protein kinase 2 inhibitors) block ribosomal RNA
synthesis, inhibit protein synthesis, and induce apoptosis in
cancer cells (40,41).
UTP4 is essential for pre-ribosomal RNA processing,
particularly in the nucleolar maturation of 18S rRNA, and is
crucial in RB. Elevated UTP4 expression potentially supports rapid
cancer cell proliferation by promoting RB. Moreover, studies
indicate that UTP4 may also function as a transcriptional regulator
(42). In the present study, the
expression patterns and prognostic implications of UTP4 were
systematically analyzed across multiple cancers and experimentally
confirmed its biological significance in GC.
In the present study, UTP4 expression was initially
assessed and validated in GC using RNA and protein data from the
TCGA cohort, confirming that UTP4 is significantly upregulated in
most cancer types. Single-cell spatial transcriptomic analysis
further verified high UTP4 expression in various tumor cell
populations. Given the strong prognostic and diagnostic
significance of UTP4 identified in the TCGA cohort, its functional
mechanisms were explored through multi-omics approaches. Functional
analyses indicated that UTP4 gene mutations, mainly missense and
nonsense mutations, commonly occur in multiple cancers, potentially
influencing UTP4 function and promoting tumorigenesis.
Additionally, UTP4 expression negatively correlated with the
infiltration of several immune cells but positively correlated with
specific populations such as T helper cells, Tcm cells and Th2
cells. This suggests UTP4 may contribute to an immunosuppressive
TME, enabling immune evasion. Moreover, high UTP4 expression
correlated positively with microbial presence in TMEs, especially
in GC and CRC, potentially further accelerating tumor progression.
The present findings also indicated that UTP4 may influence cancer
progression and metastasis by regulating tumor stemness, mutational
burden and immune checkpoint molecules. Univariate and multivariate
regression analyses confirmed that UTP4 serves as an independent
prognostic indicator in GC. Bayesian colocalization genetic
association analysis and CRISPR-Cas9 predictions further validated
UTP4's functional role. The present study further demonstrated that
high UTP4 expression was significantly correlated with poor OS but
not DFI. This discrepancy suggests UTP4 does not primarily drive
initial tumor recurrence; instead, it plays a key role in
accelerating disease progression and lethality post-recurrence.
Thus, UTP4 mainly acts in advanced disease stages, driving
progression and poor outcomes, supporting its potential as a
therapeutic target for advanced tumors. Specifically, UTP4
knockdown significantly reduced proliferation and migration while
promoting apoptosis in GC cell lines (AGS and MKN-45). These
results confirm the pivotal role of UTP4 in GC development,
reinforcing its potential as a therapeutic target and prognostic
biomarker.
Notably, UTP4 functions not only at the mRNA level
but also as a circular RNA (circRNA-CIRH1A), exhibiting significant
expression in various cancers associated with cell proliferation,
migration and invasion (43,44).
CircRNAs commonly act as ‘sponges’ for specific miRNAs, thus
regulating downstream signaling pathways (44,45).
For instance, circRNA-CIRH1A influences the PI3K/AKT and JAK2/STAT3
signaling pathways by sponging miR-1276, an interaction validated
in osteosarcoma cells (46). These
findings suggest that elevated UTP4 expression may affect cancer
progression via both protein-coding and circular RNA mechanisms.
Therefore, distinguishing whether the cancer-promoting effects of
UTP4 are mediated primarily by its mRNA or circRNA form is
critical. Although systematic pan-cancer analyses demonstrated that
UTP4 mRNA promotes tumorigenesis, the precise roles of its circRNA
form remain unclear. A recent study indicated that circular RNAs
function as inhibitors or activators influencing GC initiation and
progression, emphasizing their potential utility as diagnostic
biomarkers and therapeutic targets (47). The experimental findings of the
present study clearly demonstrated UTP4's impact on GC cells;
however, as the entire UTP4 gene was targeted for knockout,
potential effects of its circRNA form cannot be excluded.
Clarifying the distinct roles of UTP4 in different RNA forms will
provide deeper insights into its diverse functions in cancer
biology, opening new avenues for targeted therapeutic
development.
Currently, studies linking UTP4 abnormalities to
disease are limited, with most studies focused on North American
Indian childhood cirrhosis. The role of UTP4 in other diseases
remains largely unexplored. Research on UTP4's relationship with
cancer is predominantly restricted to CRC, leaving its involvement
in other cancers unclear. Studies indicate that cancer cells
produce neuroendocrine mediators, activating central neuroendocrine
axes and establishing bidirectional communication with autonomic
and sensory nerves. This process disrupts metabolic, immune, and
circadian homeostasis. UTP4 might facilitate the production of
neuroendocrine mediators by enhancing RB, thereby activating
central neuroendocrine pathways, regulating immune-metabolic
homeostasis, and promoting cancer-related homeostatic disruption
(48). Therefore, the present
study addresses an existing research gap, has innovative
significance, and provides a foundation for future research.
However, the current findings primarily depend on
publicly available databases. The inherent batch effect, variations
in data quality and sample processing heterogeneity in these
databases may introduce biases, impacting the accuracy of UTP4
expression measurements and cross-cancer comparisons. These factors
should be taken into consideration when interpreting the findings
of the present study and their generalizability. Additionally,
non-random sample selection could limit the generalizability of our
findings across different populations and cancer subtypes.
Therefore, additional clinical data are necessary to confirm the
potential of UTP4 as a reliable biomarker. Though the current
pan-cancer analysis points to a wide-ranging involvement of UTP4 in
tumorigenesis, functional validation has thus far been confined
exclusively to GC. Thus, subsequent studies are warranted to verify
its functional relevance in other malignancies. The present study
is predominantly reliant on analyses of public databases and lacks
validation using prospective clinical cohorts or fresh tissue
samples, which represents a major barrier to its clinical
translation. Furthermore, the current functional evidence remains
insufficient: The absence of in vivo data, particularly from murine
models, greatly impairs the rigorous validation of UTP4's role in
tumorigenesis and therapeutic response. Therefore, future studies
should integrate animal model experiments and clinical sample
validation to fully confirm its translational potential.
In conclusion, UTP4 exhibits high expression across
various cancers and correlates significantly with prognosis in
multiple cancer types, including GC. Its expression shows notable
associations with TMB, MSI, immune checkpoints, immune cells and
microorganisms in a pan-cancer context. In vitro experiments
validated the oncogenic function of UTP4 in GC cells, further
supporting its value as a prognostic biomarker.
Supplementary Material
Screening of UTP4-targeting sgRNAs by
reverse transcription-quantitative PCR in gastric cancer cells.
Relative UTP4 mRNA expression levels (normalized to GAPDH) in AGS
and MKN-45 cells after transfection with three different sgRNAs
targeting UTP4 (sgUTP4#1, #2, #3) or a NTC sgRNA. Data are
presented as the mean ± SD from three independent experiments.
Statistical significance was determined by unpaired Student's
t-test comparing each sgUTP4 group to the NTC group within the same
cell line. ***P<0.001. sgRNA#2 showed the most potent
knockdown efficiency and was selected for subsequent functional
studies. sgRNA, small guide RNA; NTC, non-targeting control.
Association between UTP4 expression
and DFI in patients with gastric cancer. Kaplan-Meier curve of
patients with STAD stratified by UTP4 expression (High/Low).
HR=0.884 (95% CI: 0.422-1.855), log-rank test, P=0.39. UTP4
expression was not significantly correlated with DFI, suggesting no
association with early tumor recurrence. DFI, disease-free
interval; STAD, stomach adenocarcinoma; HR, hazard ratio; CI,
confidence interval.
Mutation frequency and mutation types
of UTP4 across various cancers.
The top 100 genes co-expressed with
UTP4 in pan-cancer.
UTP4 Knockout Detection: qPCR Raw
Data.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Special Project
of Science and Technology Cooperation between Hubei and Chinese
Academy of Sciences (grant no. 42000021817T300000050), the Teaching
research project of Medical Faculty of Wuhan University in 2021
(grant no. 2021026) and Wuhan Medical Scientific Research Project
(grant no. WX23Q12).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
DW and QH designed the research. DW, TL and YC
analyzed the data and wrote the manuscript. All authors read and
approved the final version of the manuscript. DW and QH confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vishwanath A, Krishna S, Manudhane AP,
Hart PA and Krishna SG: Early-onset gastrointestinal malignancies:
An investigation into a rising concern. Cancers (Basel).
16(1553)2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
López MJ, Carbajal J, Alfaro AL, Saravia
LG, Zanabria D, Araujo JM, Quispe L, Zevallos A, Buleje JL, Cho CE,
et al: Characteristics of gastric cancer around the world. Crit Rev
Oncol Hematol. 181(103841)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Smyth EC, Nilsson M, Grabsch HI, van
Grieken NC and Lordick F: Gastric cancer. Lancet. 396:635–648.
2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Boilève J, Touchefeu Y and Matysiak-Budnik
T: Clinical management of gastric cancer treatment regimens. Curr
Top Microbiol Immunol. 444:279–304. 2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li S, Gao J, Xu Q, Zhang X, Huang M, Dai
X, Huang K and Liu L: A signature-based classification of gastric
cancer that stratifies tumor immunity and predicts responses to
PD-1 inhibitors. Front Immunol. 12(693314)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lordick F, Carneiro F, Cascinu S, Fleitas
T, Haustermans K, Piessen G, Vogel A and Smyth EC: ESMO Guidelines
Committee. Electronic address: clinicalguidelines@esmo.org. Gastric
cancer: ESMO clinical practice guideline for diagnosis, treatment
and follow-up. Ann Oncol. 33:1005–1020. 2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y,
Kato K, Chung HC, Chen JS, Muro K, Kang WK, et al: Nivolumab in
patients with advanced gastric or gastro-oesophageal junction
cancer refractory to, or intolerant of, at least two previous
chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised,
double-blind, placebo-controlled, phase 3 trial. Lancet.
390:2461–2471. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yasuda T and Wang YA: Gastric cancer
immunosuppressive microenvironment heterogeneity: Implications for
therapy development. Trends Cancer. 10:627–642. 2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kim R, An M, Lee H, Mehta A, Heo YJ, Kim
KM, Lee SY, Moon J, Kim ST, Min BH, et al: Early tumor-immune
microenvironmental remodeling and response to first-line
fluoropyrimidine and platinum chemotherapy in advanced gastric
cancer. Cancer Discov. 12:984–1001. 2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Calviño FR, Kornprobst M, Schermann G,
Birkle F, Wild K, Fischer T, Hurt E, Ahmed YL and Sinning I:
Structural basis for 5'-ETS recognition by Utp4 at the early stages
of ribosome biogenesis. PLoS One. 12(e0178752)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang C, Ma H, Baserga SJ, Pederson T and
Huang S: Nucleolar structure connects with global nuclear
organization. Mol Biol Cell. 34(ar114)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Freed EF and Baserga SJ: The C-terminus of
Utp4, mutated in childhood cirrhosis, is essential for ribosome
biogenesis. Nucleic Acids Res. 38:4798–4806. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wilkins BJ, Lorent K, Matthews RP and Pack
M: p53-mediated biliary defects caused by knockdown of cirh1a, the
zebrafish homolog of the gene responsible for North American Indian
childhood cirrhosis. PLoS One. 8(e77670)2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Guo F, Chen JJ and Tang WJ: CIRH1A
augments the proliferation of RKO colorectal cancer cells. Oncol
Rep. 37:2375–2381. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gahoi N, Syed P, Choudhary S, Epari S,
Moiyadi A, Varma SG, Gandhi MN and Srivastava S: A protein
microarray-based investigation of cerebrospinal fluid reveals
distinct autoantibody signature in low and high-grade gliomas.
Front Oncol. 10(543947)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hanley JA and McNeil BJ: The meaning and
use of the area under a receiver operating characteristic (ROC)
curve. Radiology. 143:29–36. 1982.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hänzelmann S, Castelo R and Guinney J:
GSVA: Gene set variation analysis for microarray and RNA-seq data.
BMC Bioinformatics. 14(7)2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Giambartolomei C, Vukcevic D, Schadt EE,
Franke L, Hingorani AD, Wallace C and Plagnol V: Bayesian test for
colocalisation between pairs of genetic association studies using
summary statistics. PLoS Genet. 10(e1004383)2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Longo SK, Guo MG, Ji AL and Khavari PA:
Integrating single-cell and spatial transcriptomics to elucidate
intercellular tissue dynamics. Nat Rev Genet. 22:627–644.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Rodriques SG, Stickels RR, Goeva A, Martin
CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F and Macosko
EZ: Slide-seq: A scalable technology for measuring genome-wide
expression at high spatial resolution. Science. 363:1463–1467.
2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ståhl PL, Salmén F, Vickovic S, Lundmark
A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss
M, et al: Visualization and analysis of gene expression in tissue
sections by spatial transcriptomics. Science. 353:78–82.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Brookes AJ: The essence of SNPs. Gene.
234:177–186. 1999.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vignal A, Milan D, SanCristobal M and
Eggen A: A review on SNP and other types of molecular markers and
their use in animal genetics. Genet Sel Evol. 34:275–305.
2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Clevers H: The cancer stem cell: Premises,
promises and challenges. Nat Med. 17:313–319. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
O'Brien CA, Kreso A and Jamieson CH:
Cancer stem cells and self-renewal. Clin Cancer Res. 16:3113–3120.
2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Klinge S and Woolford JL Jr: Ribosome
assembly coming into focus. Nat Rev Mol Cell Biol. 20:116–131.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Henras AK, Plisson-Chastang C, O'Donohue
MF, Chakraborty A and Gleizes PE: An overview of pre-ribosomal RNA
processing in eukaryotes. Wiley Interdiscip Rev RNA. 6:225–242.
2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Anderson P and Kedersha N: RNA granules:
Post-transcriptional and epigenetic modulators of gene expression.
Nat Rev Mol Cell Biol. 10:430–436. 2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Beesley J, Sivakumaran H, Moradi Marjaneh
M, Shi W, Hillman KM, Kaufmann S, Hussein N, Kar S, Lima LG, Ham S,
et al: eQTL colocalization analyses identify NTN4 as a candidate
breast cancer risk gene. Am J Hum Genet. 107:778–787.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Meyers RM, Bryan JG, McFarland JM, Weir
BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel
S, et al: Computational correction of copy number effect improves
specificity of CRISPR-Cas9 essentiality screens in cancer cells.
Nat Genet. 49:1779–1784. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pecoraro A, Pagano M, Russo G and Russo A:
Ribosome biogenesis and cancer: Overview on ribosomal proteins. Int
J Mol Sci. 22(5496)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xu X, Xiong X and Sun Y: The role of
ribosomal proteins in the regulation of cell proliferation,
tumorigenesis, and genomic integrity. Sci China Life Sci.
59:656–672. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Elhamamsy AR, Metge BJ, Alsheikh HA,
Shevde LA and Samant RS: Ribosome biogenesis: A central player in
cancer metastasis and therapeutic resistance. Cancer Res.
82:2344–2353. 2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jiao L, Liu Y, Yu XY, Pan X, Zhang Y, Tu
J, Song YH and Li Y: Ribosome biogenesis in disease: New players
and therapeutic targets. Signal Transduct Target Ther.
8(15)2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pelletier J, Thomas G and Volarević S:
Ribosome biogenesis in cancer: New players and therapeutic avenues.
Nat Rev Cancer. 18:51–63. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Bywater MJ, Poortinga G, Sanij E, Hein N,
Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al:
Inhibition of RNA polymerase I as a therapeutic strategy to promote
cancer-specific activation of p53. Cancer Cell. 22:51–65.
2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kato Y, Kunimasa K, Takahashi M, Harada A,
Nagasawa I, Osawa M, Sugimoto Y and Tomida A: GZD824 inhibits GCN2
and sensitizes cancer cells to amino acid starvation stress. Mol
Pharmacol. 98:669–676. 2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Freed EF, Prieto JL, McCann KL, McStay B
and Baserga SJ: NOL11, implicated in the pathogenesis of North
American Indian childhood cirrhosis, is required for pre-rRNA
transcription and processing. PLoS Genet.
8(e1002892)2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hollensen AK, Andersen S, Hjorth K, Bak
RO, Hansen TB, Kjems J, Aagaard L, Damgaard CK and Mikkelsen JG:
Enhanced tailored MicroRNA sponge activity of RNA Pol
II-transcribed TuD hairpins relative to ectopically expressed
ciRS7-derived circRNAs. Mol Ther Nucleic Acids. 13:365–375.
2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang M, Wang X, Zhao J, Yan J, He X, Qin
D, Liang F, Tong K and Wang J: CircRNA-CIRH1A promotes the
development of osteosarcoma by regulating PI3K/AKT and JAK2/STAT3
signaling pathways. Mol Biotechnol. 66:2241–2253. 2024.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Bakhti SZ, Latifi-Navid S, Pahlevan AD,
Sarabi L and Safaralizadeh R: The role of circular RNAs in gastric
cancer: Focusing on autophagy, EMT, and their crosstalk. Biochem
Biophys Rep. 48(102169)2025.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Slominski RM, Raman C, Chen JY and
Slominski AT: How cancer hijacks the body's homeostasis through the
neuroendocrine system. Trends Neurosci. 46:263–275. 2023.PubMed/NCBI View Article : Google Scholar
|