Introduction
Colorectal cancer (CRC), the second leading cause of
global cancer-related death and the third most common cancer
worldwide, exhibits a concerning rise in incidence among younger
populations in mainland China, with an annual increase of 1-2% in
individuals <55 years (1,2).
Although pathological stage at diagnosis remains the strongest
prognostic factor, >50% of patients present with advanced or
intermediate-stage disease (3),
underscoring the urgent need to elucidate CRC pathogenesis for
early detection and improved therapies.
CRC primarily arises from adenomatous colonic
polyps, the most prevalent type, constituting 60-70% of all colonic
polyps. Conventional adenomatous polyps are histologically
classified as tubular, villous or tubulovillous. Over time, some
adenomas may progress from low-grade dysplasia to high-grade
dysplasia, carcinoma in situ or invasive adenocarcinoma,
with <90% of CRC cases being adenocarcinoma of colonic/rectal
mucosal origin (4). Furthermore,
70-90% of CRC cases develop into carcinoma from adenoma, driven by
mutations in tumor suppressor genes such as Adenomatous polyposis
coli and TP53 as well as in oncogenes such as KRAS,
while a distinct subset (~10%) follows the serrated neoplasia
pathway, marked by epigenetic dysregulation (such as MLH1
promoter hypermethylation) and DNA repair defects (5).
Microsomal glutathione S-transferase (MGST) belongs
to the microsomal subfamily of the glutathione S-transferase (GST)
family and plays a pivotal role in cellular detoxification
(6). Specifically, it catalyzes
the conjugation of reduced glutathione to xenobiotic substrates,
thereby protecting cells against oxidative stress-induced damage.
Furthermore, these enzymes effectively neutralize toxic compounds
such as lipid peroxidation products and prostaglandins,
contributing to the overall maintenance of cellular health and
homeostasis.
In the present study, to gain a comprehensive
understanding of the molecular signature underlying CRC
progression, a cohort of patients representing diverse stages of
colorectal diseases was assembled, aiming to map the progression
from normal colonic epithelium to adenomatous lesions and invasive
carcinoma. Using RNA-sequencing (RNA-seq) analysis of clinical
samples, the present study systematically identified and
characterized differentially expressed genes (DEGs) in CRC,
providing a comprehensive gene expression profile. The glutathione
metabolic pathway is crucial for maintaining cellular homeostasis
and preventing oncogenic transformation. Key components of this
pathway include microsomal glutathione S-transferase 3
(MGST3), peroxiredoxin 6 (PRDX6), hematopoietic
prostaglandin D synthase (HPGDS), γ-glutamyltransferase 1
(GGT1), glutathione peroxidase 3 (GPX3), glutathione
S-transferase µ4 (GSTM4), glutathione S-transferase α1
(GSTA1) and glutathione reductase (GSR). Therefore,
the present study aimed to investigate the functional role of
MGST3 in colon carcinogenesis, with a specific focus on its
potential link to ferroptosis. To achieve this, short hairpin RNA
(shRNA) was employed to knock down MGST3 expression in cell
models and the resulting susceptibility to ferroptosis was
assessed. These discoveries may provide insights into the molecular
mechanisms of CRC, particularly colon cancer, and highlight the
potential of MGST3 and related genes as biomarkers for early
detection and as therapeutic targets. By targeting these pathways,
novel strategies for CRC prevention and treatment can be developed,
ultimately improving patient outcomes.
Materials and methods
Clinical samples
A retrospective analysis was performed on samples
from patients with colorectal tumors treated at the Department of
Gastroenterology, The Seventh Medical Center of Chinese PLA General
Hospital (Beijing, China) between October 1, 2016 and October 31,
2016. Patient selection was based on the following inclusion
criteria: i) Pathological confirmation of colorectal adenoma or
adenocarcinoma; ii) availability of fresh-frozen tumor tissue with
paired adjacent normal tissue (within 5 cm of the tumor margin);
and iii) complete clinical and pathological data. The exclusion
criteria were: i) Prior neoadjuvant chemotherapy or radiotherapy;
and ii) diagnosis of synchronous multiple cancers or hereditary CRC
syndromes. Based on these criteria, a total of 10 patients were
included. The cohort comprised 2 cases of tubular adenoma, 3 of
villous adenoma, 3 with early-stage cancer and 2 with advanced
tumors. In addition, 4 adjacent normal tissue samples from these 10
patients were collected as controls.
Cell culture
The normal human colon cell line NCM460 (cat. no.
IM-H445; cell batch, IM-H445031603) was purchased from the Xiamen
Yimo Biotechnology Co, Ltd. These cells were cultured in Dulbecco's
Modified Eagle's Medium (DMEM; Procell Life Science &
Technology Co., Ltd.) and supplemented with 10% (v/v)
heat-inactivated fetal bovine serum (FBS; Shanghai ExCell Biology,
Inc.), 1% (v/v) penicillin-streptomycin and 1% (v/v) L-glutamine.
All cell lines were maintained in a humidified incubator at 37˚C
with 5% CO2. All cell lines tested negative for
mycoplasma contamination.
Plasmid construction
To construct shRNA-expressing vectors, annealed
complementary shRNA oligonucleotides were ligated into the
PiggyBac-H1-2O2-shRNA-CopGFP-puro vector, which was constructed
from the PiggyBac-CMV-CNR (human)-EF1a-CopGFP-T2A-Puro plasmid
(cat. no. P31653; MiaoLingBio) by replacing the CMV promoter with
the H1-2O2 promoter. The shRNA sequence was
5'-GCAAGAAGTACAAAGTGGAGT-3' and the sequence for the negative
control group was 5'-TTCTCCGAACGTGTCACGT-3'. NCM460 cells were
seeded into 6-well plates and transfected with a total of 4 µg of
plasmid DNA using Lipofectamine 3000 reagent (cat. no. L3000008;
Invitrogen; Thermo Fisher Scientific, Inc.). The plasmid mixture
consisted of the shRNA-expressing PiggyBac vector and Super
PiggyBac Transposase (cat. no. P0179; MiaoLingBio) at a mass ratio
of 2.5:1. The DNA-lipid complexes were incubated at room
temperature for 15 min before being added to the cells. After 24 h
of incubation, the cells were selected with 1 µg/ml puromycin for 3
days, after which almost all surviving cells exhibited green
fluorescence. Single clones were then obtained by serial dilution
and maintained under a lower dose of puromycin (0.1 µg/ml) for 1 to
2 weeks to establish stable cell lines. Protein knockdown was
finally confirmed by western blotting.
Clinical sample RNA-seq
RNA was extracted from clinical samples using RNAiso
Plus (cat. no. 9109; Takara Bio, Inc.). The integrity of the RNA
samples was assessed by agarose gel electrophoresis. RNA
concentrations were quantified using a Qubit fluorometer (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Subsequent RNA-seq library preparation and deep
sequencing were performed by BGI Genomics (Shenzhen, China). The
samples were subjected to paired-end (2x150 bp) sequencing on an
Illumina NovaSeq 6000 platform (cat. no. 20012850; Illumina, Inc.).
Sequencing data were trimmed using Trimmomatic (v.0.39) (7) and aligned to the reference genome
hg38 using STAR (v.2.7.7a) (8)
software. Subsequently, read counting and differential gene
expression analysis were performed using Cufflinks (v.2.2.1)
(9). The data generated from the
present study have been deposited in the NCBI Gene Expression
Omnibus database under the accession number GSE316039.
Cell viability assays
Cell viability assays were performed as previously
described (10). For cell
viability assays, cells were seeded into 96-well plates
(1x103/well) and then incubated at 37˚C under 5%
CO2. The viability was measured at four different time
points: 24, 48, 72 and 96 h. At each designated time point, 10 µl
of Cell Counting Kit-8 (CCK-8) reagent (cat. no. HY-K0301;
MedChemExpress) was added to each well, followed by incubation for
an additional 2 h (the incubation duration was determined by a
pre-experimental optimization to ensure the OD values were within
the linear range). The optical density was then measured using a
microplate spectrophotometer (BioTek Synergy H1; Agilent
Technologies, Inc.).
Colony formation assay
The colony formation assay was performed as
previously described (11). A
total of 1x103 shMGST3 and shNC transfected cells were
seeded into 6-well plates. After 14 days, colonies were fixed with
4% paraformaldehyde (room temperature, 30 min), stained with 0.1%
crystal violet (room temperature, 20 min) and imaged. Colonies
containing ≥50 cells were quantified.
Scratch healing assay
The scratch healing assay was performed to assess
the migration capacity of cells in vitro (12). Cells were seeded in 6-well plates
and cultured in medium containing 3% FBS. A scratch was created
across the monolayer using a 200 µl pipette tip. Cell migration was
observed at 0 and 12 h post-scratch using a fluorescence microscope
(magnification, x4) to monitor wound closure. Images were captured
to evaluate the migration levels of the transfected cell groups.
The wound healing rate was calculated using the formula: Percentage
change=([width at 0 h - width at 12 h]/width at 0 h) x100.
Transwell assay
The Transwell assay was performed as previously
described (13). The Transwell
chamber contains a porous membrane with a pore size of 8 µm. To
assess invasion capability, the bottom membrane of the chamber was
coated with Matrigel at a ratio of 1:40 (Matrigel: serum-free
medium; 100 µl). The lower chamber was filled with 500 µl of
serum-free medium. The plates were then incubated at 37˚C for 2 h
to allow the Matrigel to solidify. After 2 h, 5x104
cells transfected with shMGST3 or shNC were seeded into the upper
chamber, while the lower chamber was supplemented with 500 µl of
DMEM containing 10% FBS. After a 24 h incubation at 37˚C in a 5%
CO2 incubator, cells on the membrane were fixed with 4%
paraformaldehyde for 30 min at 25˚C, followed by staining with 0.1%
crystal violet at 25˚C for 10 min. Following three PBS washes,
non-migrated cells on the upper side of the membrane were removed
with a cotton swab. In total, five random fields (magnification,
x20) were selected under a fluorescence microscope using the
brightfield mode and the number of stained cells was counted to
quantify invasion.
Protein extraction and western
blotting analysis
Protein extraction and western blotting assays were
performed as previously described (14). Cells were lysed on ice using RIPA
buffer (cat. no. 9806; Cell Signaling Technology, Inc.)
supplemented with PMSF and protease inhibitors. The supernatant was
collected and boiled for 10 min after adding 5x SDS protein loading
Buffer (cat. no. 30215ES; Shanghai Yeasen Biotechnology Co., Ltd.).
A total of 10 µg of protein per lane was resolved on 4-20% SDS-PAGE
gels (cat. no. ET12420Gel; ACE Biotechnology Co., Ltd.) and
subsequently transferred to a PVDF membrane (cat. no. ISEQ00010;
MilliporeSigma). The membrane was blocked with 5% skim milk at room
temperature for 1 h, followed by overnight incubation at 4˚C with
primary antibodies against MGST3 (cat. no. ab192254; Abcam) and
GAPDH (cat. no. GB15002-100; Wuhan Servicebio Technology Co., Ltd.)
diluted to 1:1,000. Following incubation with the primary antibody,
the membrane was washed three times with TBST (containing 0.1%
Triton X-100) and then incubated for 1 h at room temperature with
horseradish peroxidase-conjugated goat anti-rabbit IgG (H+L) (cat.
no. A0208; Beyotime Biotechnology) and goat anti-mouse IgG (H+L)
(cat. no. A0216; Beyotime Biotechnology), each at a dilution of
1:5,000. After another three TBST washes, protein bands were
visualized using an ultrasensitive ECL chemiluminescent substrate
(cat. no. P0018FM; Beyotime Biotechnology) and imaged with a Tanon
system equipped with Tanon Bio Imaging Analysis Software (version
1.0.0000; Tanon Science & Technology Co., Ltd.).
Colon cancer data sets
Clinical variables including tumor node metastasis
staging and the corresponding gene expression matrices profiles of
patients with colon adenocarcinoma (COAD) were downloaded from The
Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov).
Enrichment analysis
Enrichment analysis were conducted on Metascape
(https://metascape.org/) with the following
screening criteria: A minimum overlap of 3 genes, P≤0.01 and a
minimum enrichment factor of 1.5. The signals of the 14 pathways in
the high and low expression samples of the glutathione gene set
were calculated using the PROGENy package (15) in R software (version 4.2.3;
https://www.R-project.org/), with
samples stratified into high- and low-expression groups based on
the median expression of the glutathione gene set derived from TCGA
colon cancer data.
Protein-protein interaction (PPI)
molecular analysis
Search Tool for the Retrieval of Interacting Genes
(https://cn.string-db.org) (16) was employed for constructing PPI
networks using the gene set from Cluster 3. The resulting network
file was imported into Cytoscape software (version 3.9.1) (17), which generated multiple
subnetworks. The MCODE plugin (version 2.0.3) was applied to
identify the most significant subnetwork based on the node score
and number of genes, defined as a module with a node score >5
and containing >10 genes. Subsequently, the hub genes were
selected from this subnetwork using the cytoHubba plugin (version
0.1).
Tumor mutation burden (TMB)
TMB was defined as the total number of somatic
coding base substitutions, insertions and deletions per megabase of
examined genome (18). Using the
TCGA-COAD cohort, stratified by the expression level of the
glutathione gene set, TMB analysis and distribution of tumor
mutation types were calculated by R package maftools (version
2.14.0) (19).
Computation of EMT score
The EMT gene sets in both the study by Tan et
al (20) and the EMTome
database (http://www.emtome.org) were searched and
the enrichment scores of the samples with high and low expression
of the glutathione gene set were calculated using the single-sample
Gene Set Enrichment Analysis (GSEA) algorithm (21).
Tumor immune infiltration
analysis
The tumor immune infiltration analysis was performed
using the CIBERSORT (version 0.1.0) (22) and ESTIMATE packages (version
1.0.13) (23) within the R
(version 4.2.3) programming environment and based on gene
expression matrices obtain from TCGA database.
Statistical analyses
Statistical analyses were performed as follows:
Fisher's exact test was used for Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway enrichment analysis of DEGs from the RNA-seq
data; gene expression across different tumor stages in TCGA
database were analyzed by Kruskal-Wallis test followed by Dunn's
multiple comparisons test; a two-tailed unpaired t-test was applied
to analyze the following metrics: TMB, immune score and the results
of the Transwell and scratch healing assays, comparison of
ferroptosis-related gene expression and pathway signals (14
pathways) between samples stratified by high and low expression
levels of the glutathione gene set; two-way ANOVA test followed by
Bonferroni's multiple comparisons test was employed for
proliferation analysis of shMGST3 and shNC cells; and the
Kolmogorov-Smirnov test was used for GSEA of the glutathione gene
set. P<0.05 was considered to indicate a statistically
significant difference.
Results
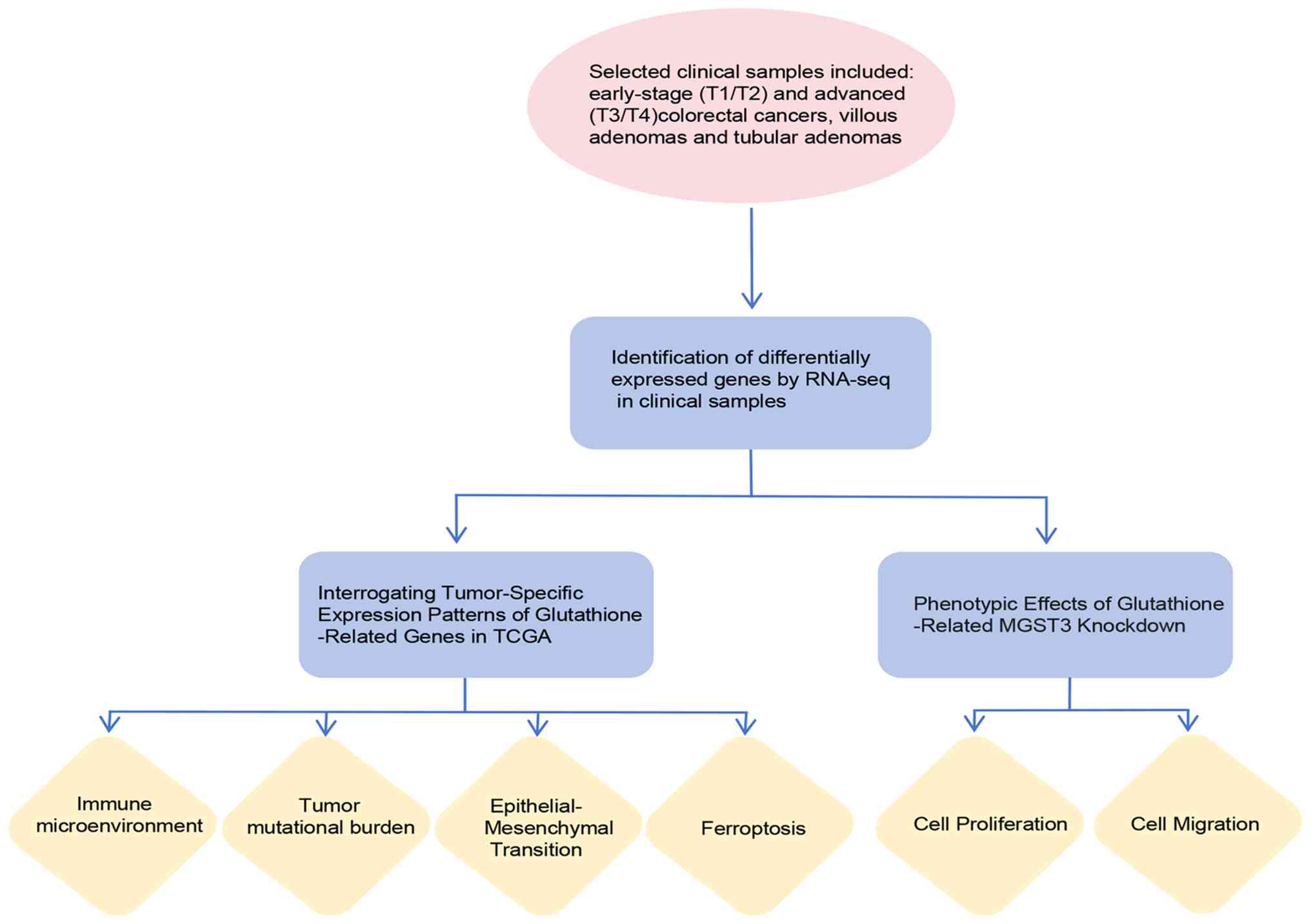
Determination of a gene cluster that
has low expression in CRC
Colorectal tumor samples from 5 male and 5 female
patients, all aged 40-60 years were classified into early-stage
tumors (pathological T1/T2), advanced tumors (pathological T3/T4),
villous adenomas and tubular adenomas. This analytical approach
included RNA-seq of patient samples, functional phenotypic
validation in cell models and bioinformatics analyses of
tumor-related indicators using data from TCGA database (TCGA-COAD
cohort) (Fig. 1). Specifically,
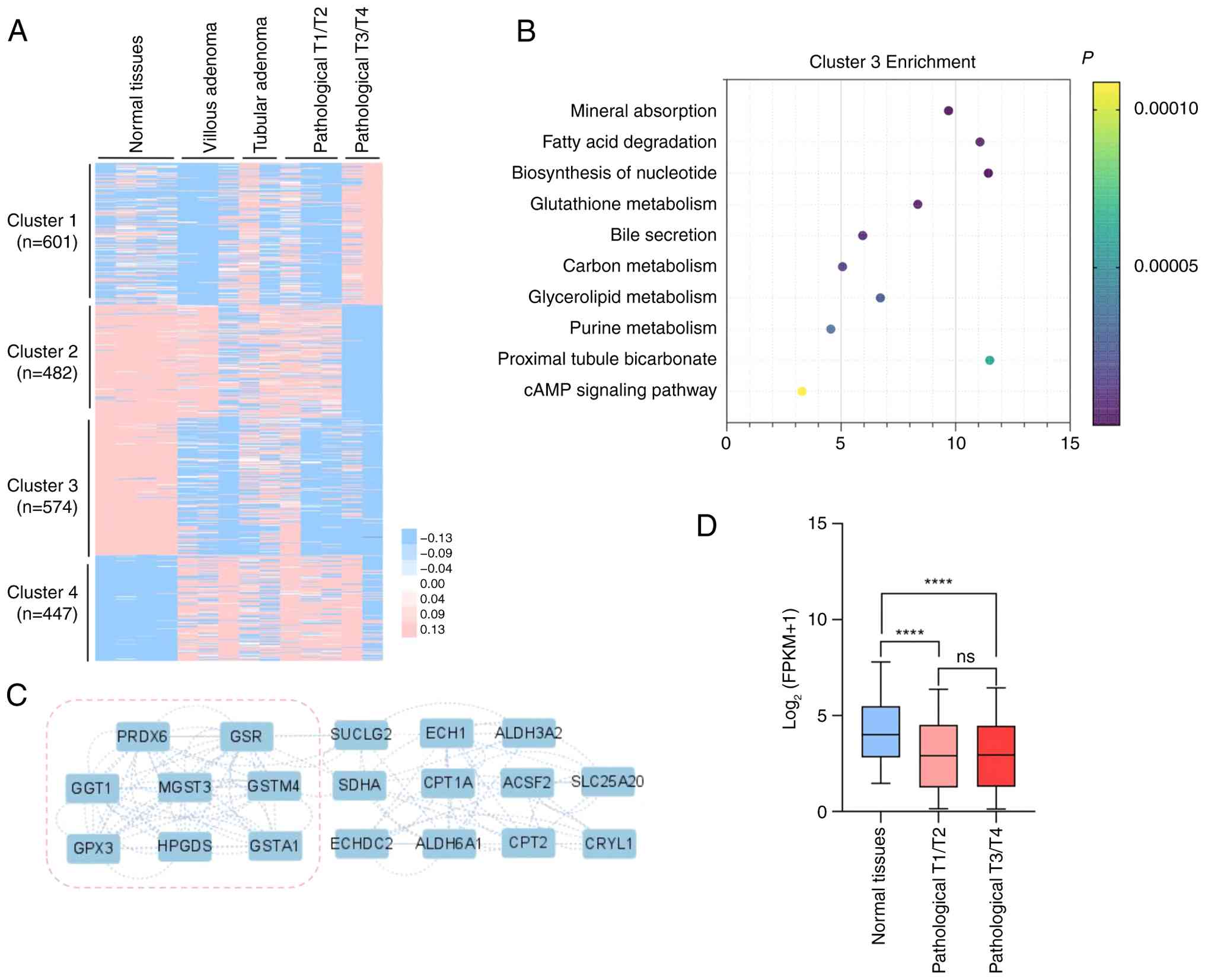
differential gene expression analysis [P<0.01,
|log2(fold change)| ≥1] was performed comparing adjacent
normal tissues to the defined tumor stages (Fig. 2A). Through clustering of
significant DEGs, four distinct expression patterns were
identified: i) Cluster 1 (601 genes) showed specific upregulation
in advanced tumors; ii) Cluster 2 (482 genes) exhibited high
expression in normal tissues but progressive downregulation across
precancerous lesions (villous/tubular adenomas) and tumors, with
maximal suppression in advanced cases; iii) Cluster 3 (574 genes)
demonstrated elevated expression in normal tissues that sharply
declined in tubular adenomas and showed further reduction in
villous adenomas and tumors; and iv) Cluster 4 (447 genes)
displayed consistently lower expression in normal tissues vs. all
pathological groups.
Among these clusters, Cluster 3 exhibited the most
uniform and distinct expression patterns in CRC, differing markedly
from both adjacent normal tissues and adenomas. Given this clear
differentiation, Cluster 3 was selected for further investigation.
KEGG pathway enrichment analysis revealed that the top enriched
pathways in this cluster were predominantly metabolic-related
(Fig. 2B). PPI network analysis
using Cytoscape identified the ‘Glutathione metabolism’ pathway as
containing the highest proportion of hub genes (Fig. 2C), suggesting its potential central
role in CRC pathogenesis. Glutathione metabolism has been
functionally implicated in CRC pathogenesis (24). Based on this network analysis, a
glutathione metabolism gene set comprising eight key enzymes:
MGST3, PRDX6, HPGDS, GGT1, GPX3,
GSTM4, GSTA1 and GSR was identified. These
genes were selected based on their hub status in the PPI network
and their enrichment in glutathione-related pathways.
To validate these findings, transcriptomic data from
TCGA-COAD were analyzed, stratifying the samples by clinical stage
(early and advanced tumors). The glutathione gene set showed
significantly higher expression in adjacent normal tissues compared
with both tumor stages (Fig. 2D).
This consistent downregulation across tumor progression suggests
that glutathione pathway dysregulation may contribute to colon
carcinogenesis. To further investigate the significance of the
glutathione gene set, its expression patterns were analyzed across
multiple malignancies. Consistently, reduced expression in tumor
vs. normal adjacent tissue was observed in several tumor types,
including breast invasive carcinoma, lung squamous cell carcinoma
and stomach adenocarcinoma (Fig.
S1). These findings were consistent with existing literature
reports (25-29)
and collectively demonstrated that glutathione pathway
downregulation represents an early and conserved event in
tumorigenesis across diverse cancer types, including CRC.
Differences in tumor characteristics
and the immune microenvironment in the glutathione gene set
Given the established interplay between CRC
progression and the tumor microenvironment (TME) (30), the potential role of the
glutathione gene set was investigated in this context. Using
TCGA-COAD data, the expression profiles of these genes were
extracted and their mean expression levels in each sample were
calculated. Samples were then stratified into high and low
expression groups based on median gene set expression, enabling
comparative analysis of some characteristics between these
subgroups. Next, differences in TMB, EMT markers (31) and immune cell infiltration
(32) between the high and low
expression groups were evaluated. Notably, it was found that tumors
with low expression of the glutathione gene set exhibited
significantly elevated TMB compared with tumors with high
expression (Fig. 3A). Conversely,
tumors with high expression showed significantly enhanced EMT
activity (Fig. 3B and C). These results indicated that elevated
expression of glutathione pathway genes may be associated with
tumor aggressiveness, as previously reported (33).
![Differences in tumor characteristics
and the immune microenvironment in glutathione gene set expression
profiles in The Cancer Genome Atlas-colon adenocarcinoma cohort.
(A) Differences in TMB between the high (n=178) and low (n=179)
glutathione gene set expression samples (two-tailed unpaired
t-test; *P<0.05). (B) Distribution of high and low
glutathione gene set expression across lymphocyte, macrophage,
dendritic cell, mast cell, neutrophil and eosinophil populations
(two-tailed unpaired t-test; ns, not significant;
**P<0.01, ****P<0.0001). (C) Difference
in the EMT scores (gene set: EMTome) between the high (n=236) and
low (n=235) glutathione gene set expression samples (two-tailed
unpaired t-test; ***P<0.001). (D) EMT scores [gene
set: Tan et al (20)]
between high (n=236) and low (n=235) glutathione gene set
expression samples (two-tailed unpaired t-test; ns, not
significant; **P<0.01, ****P<0.0001).
(E) Differences in immune score, estimate comprehensive score and
stromal score between the high (n=236) and low (n=235) glutathione
gene set expression samples. (two-tailed unpaired t-test;
*P<0.05, ***P<0.001). EMT,
epithelial-mesenchymal transition; TMB, tumor mutation burden.](/article_images/mco/24/5/mco-24-05-02938-g02.jpg) | Figure 3Differences in tumor characteristics
and the immune microenvironment in glutathione gene set expression
profiles in The Cancer Genome Atlas-colon adenocarcinoma cohort.
(A) Differences in TMB between the high (n=178) and low (n=179)
glutathione gene set expression samples (two-tailed unpaired
t-test; *P<0.05). (B) Distribution of high and low
glutathione gene set expression across lymphocyte, macrophage,
dendritic cell, mast cell, neutrophil and eosinophil populations
(two-tailed unpaired t-test; ns, not significant;
**P<0.01, ****P<0.0001). (C) Difference
in the EMT scores (gene set: EMTome) between the high (n=236) and
low (n=235) glutathione gene set expression samples (two-tailed
unpaired t-test; ***P<0.001). (D) EMT scores [gene
set: Tan et al (20)]
between high (n=236) and low (n=235) glutathione gene set
expression samples (two-tailed unpaired t-test; ns, not
significant; **P<0.01, ****P<0.0001).
(E) Differences in immune score, estimate comprehensive score and
stromal score between the high (n=236) and low (n=235) glutathione
gene set expression samples. (two-tailed unpaired t-test;
*P<0.05, ***P<0.001). EMT,
epithelial-mesenchymal transition; TMB, tumor mutation burden. |
To characterize the tumor immune microenvironment,
immune cell infiltration profiling was performed using CIBERSORT.
Infiltrating immune cells were characterized into six functional
groups, including lymphocytes, macrophages, dendritic cells, mast
cells, eosinophils and neutrophils, using the method described by
Wellenstein and de Visser (34).
Comparative analysis revealed significantly elevated infiltration
of lymphocytes, macrophages and neutrophils in tumors with high
expression of the glutathione gene set (Fig. 3D). These findings were validated
using the ESTIMATE package, which similarly demonstrated enhanced
overall immune activity in tumors with high expression (Fig. 3E). Collectively, these results
demonstrated that elevated glutathione pathway activity may be
associated with a more immunologically active TME, suggesting these
genes may serve as potential biomarkers for immunotherapy response
(35).
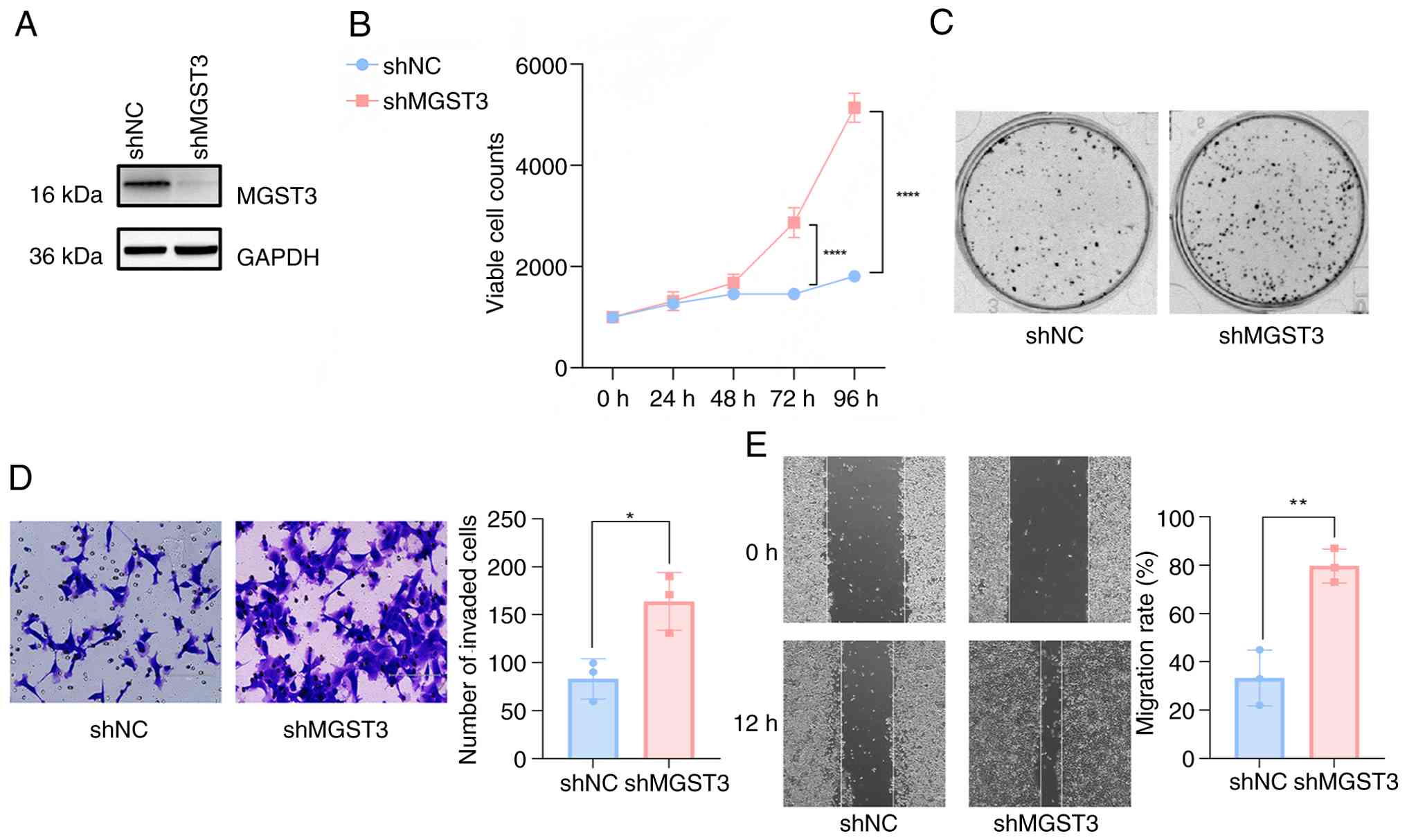
Knockdown of MGST3 promotes cell
proliferation and migration
Within the glutathione-related genes, decreased
MGST3 expression may contribute to compromised carcinogen
detoxification processes and further facilitate cellular
transformation into a malignant state (36). To elucidate the functional
consequences of MGST3 dysregulation in CRC, with a specific focus
on colon carcinogenesis, stable MGST3 knockdown was established in
the NCM460 normal human colon mucosal epithelial cell line
(37). Western blot analysis
confirmed efficient MGST3 depletion (Fig. 4A). Subsequent functional
characterization revealed that shMGST3 transfection significantly
enhanced cellular proliferation compared with shNC, as demonstrated
by both CCK-8 viability and colony formation assays (Fig. 4B). These results indicated that
MGST3 deficiency promoted proliferative advantages in colonic
epithelial cells, potentially representing an early event in
malignant transformation.
To further evaluate the malignant potential
conferred by MGST3 depletion, the invasive and migratory capacities
were assessed. MGST3-knockdown cells demonstrated a significantly
increased invasion and migration rate in compared with the shNC
cells (Fig. 4D and E). These functional assays collectively
established that knocking down MGST3 expression enhanced both the
invasive and migratory properties of colon epithelial cells.
Relationship between MGST3 dysfunction
and ferroptosis
Given the established connection between glutathione
metabolism and ferroptosis regulation (38), key ferroptosis-associated genes in
CRC (glutathione peroxidase 4, heat shock protein B1; solute
carrier family 40 member 1 and lipocalin 2) were investigated
(39-42).
Comparative analysis revealed significantly reduced expression of
these ferroptosis regulators in tumors with low expression of the
glutathione gene set compared with tumors with high expression of
the glutathione gene set (Fig.
5A). This consistent downregulation of key ferroptosis
inhibitors suggests enhanced susceptibility to ferroptosis in
tumors with lower expression of the glutathione gene set. Given
that ferroptosis is closely related to the levels of reactive
oxygen species (ROS) and fatty acid metabolism, GSEA was performed
to systematically evaluate these pathways in the expression groups.
Analysis of the glutathione gene set revealed a significantly
stronger enrichment of both ROS-related and fatty acid metabolic
gene signatures in the low expression group compared with the high
expression group.(Fig. 5B and
C). These results demonstrated
that reduced glutathione pathway activity may be associated with
molecular profiles characteristic of ferroptosis susceptibility,
marked by enhanced ROS production and dysregulated lipid
metabolism. Furthermore, analysis revealed a significant enrichment
of the p53 pathway in samples where the glutathione gene set was
highly expressed compared with samples with low expression
(Fig. S2).
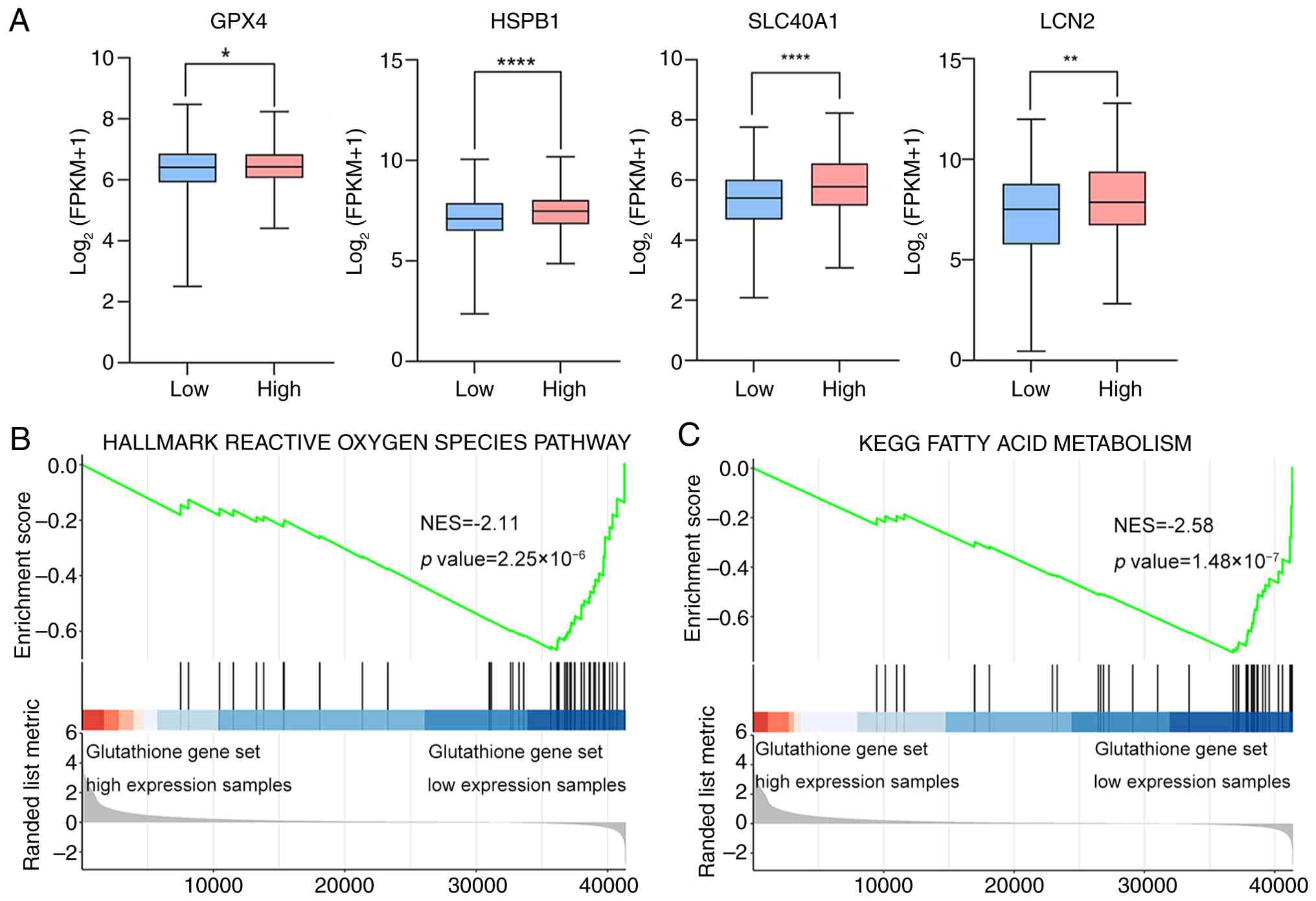 | Figure 5Relationship between the glutathione
gene set dysfunction and ferroptosis. (A) The expression
differences of ferroptosis-related genes in the glutathione gene
set between the low (n=235) and high (n=236) expression samples in
TCGA database (TCGA-COAD cohort) (two-tailed unpaired t-test;
*P<0.05, **P<0.01,
****P<0.0001). (B) The enrichment status of the
glutathione gene set between the low (n=235) and high (n=236)
expression samples in the reactive oxygen species-related gene set
by GSEA analysis. (C) The enrichment status of the glutathione gene
set between the low (n=235) and high (n=236) expression samples in
the fatty acid metabolism gene set by GSEA analysis
(Kolmogorov-Smirnov test). FPKM, fragments per kilobase of exon
model per million mapped fragments; GPX4, glutathione peroxidase 4;
HSPB1, heat shock protein family B (small) member 1; SLC40A1,
solute carrier family 40 member 1; LCN2, lipocalin-2; GSEA, Gene
Set Enrichment Analysis; NES, normalized enrichment score; KEGG,
Kyoto Encyclopedia of Genes and Genomes; TCGA, The Cancer Genome
Atlas; COAD, colon adenocarcinoma. |
These integrated analyses demonstrated that elevated
glutathione gene set expression confers resistance to ferroptosis
in colon cancer cells, which potentially implies a more unfavorable
prognosis for patients. This effect, potentially mediated through
fatty acid metabolism and ROS regulation, suggests that glutathione
gene set expression may serve as a potential therapeutic
target.
Discussion
In the present study, gene expression analysis of
patients with CRC, particularly in the colon, revealed a specific
role for MGST3 in the early phases of tumor development.
While MGST3 expression was significantly reduced in
early-stage tumors compared with normal tissue, its expression
levels remained stable across different stages of progression. This
indicates that the reduction in MGST3 levels is a key early event
in the adenoma-carcinoma pathway, rather than a factor involved in
the ongoing progression of the disease. This suggests that the loss
of MGST3 function is a key driver of early colorectal
tumorigenesis. Therefore, MGST3 not only represents a promising
tumor suppressor but also a potential biomarker for the early
detection of colon cancer specifically.
In the present study, based on PPI network analysis
of DEGs, a highly interconnected core functional module and
selected hub genes including MGST3, PRDX6,
HPGDS, GGT1, GPX3, GSTM4, GSTA1
and GSR were identified. These genes are primarily involved
in glutathione metabolism and oxidative stress pathways. Notably,
MGST3, GSTM4 and GSTA1 belong to the GST
family, which comprises detoxifying enzymes that conjugate
glutathione to mitigate oxidative damage from ROS and neutralize
toxic compounds such as lipid peroxidation products, thereby
maintaining cellular homeostasis and influencing colorectal tumor
development (43). Other hub genes
also play significant roles in tumor biology. For example,
targeting GGT1 can alleviate immunosuppressive functions (44). Targeting GPX3, a key player in
cholesterol-mediated T-cell immune responses in CRC, could be a
viable strategy (45). Conversely,
inhibiting PRDX6 expression diminishes the malignant potential of
colon cancer cells (46). In
summary, the identified hub genes are closely associated with key
tumor-related pathways and processes, underscoring their importance
in tumor progression.
In the present study these genes were termed the
glutathione gene set. Considering the differences in their
expression profiles in tumor samples, the present study focused on
alterations in TMB, immune invasion and EMT in this gene cluster.
The findings revealed that tumors with low expression of this gene
cluster exhibited significantly lower TMB compared with those with
high expression. In colon cancer, patients with increased TMB tend
to have favorable prognoses. Additionally, numerous studies suggest
that TMB can serve as a tumor biomarker (47-50)
that can be used to identify patients who are most likely to
respond to immune checkpoint inhibitors (ICIs). Therefore, patients
with colon cancer who exhibit high expression of the glutathione
gene set may be more suitable for ICI therapy (51).
Furthermore, in the present study, samples with high
expression of the glutathione gene set demonstrated significantly
higher EMT scores and immune infiltration levels of lymphocytes,
macrophages and neutrophils compared with those with low
expression. Originally described during embryonic development, EMT
involves the transformation of epithelial cells into cells with a
mesenchymal phenotype (52). This
process not only enhances the motility and invasiveness of cancer
cells but also enables them to evade apoptosis, oncogene addiction,
cellular senescence and general immune defense mechanisms (53). Therefore, EMT is an integral
component of colon cancer progression and its analysis can provide
novel targets for prognosis and therapy. The number and types of
tumor-infiltrating immune cells have been proven to be crucial
indicators of effective antitumor immune responses, ultimately
determining the prognosis of cancer (54). Additionally, tumor-associated
macrophages and neutrophils have been strongly associated with poor
prognosis in patients with CRC (55). Based on these observations, we
consider that patients with higher expression of this gene cluster
may be more prone to tumor metastasis and have a less favorable
prognosis.
A previous study has indicated a close association
between glutathione redox status and the progression of CRC, where
the induction of glutathione synthesis has been shown to promote
liver metastasis of CRC (56). The
present study showed that colon cancer samples with low expression
of the glutathione gene set were significantly enriched in ROS
response and fatty acid metabolism-related gene sets compared with
those with high expression of the glutathione gene set. Research
has shown that ROS are involved in lipid peroxidation.
Specifically, this process generates compounds such as
4-hydroxynonenal and the peroxidation of the phospholipid bilayer
may facilitate the occurrence of ferroptosis (57). This suggests that colon cancer with
low expression of the glutathione gene set is more prone to
ferroptosis than colon cancer with high expression. Typically,
tumors experiencing ferroptosis exhibit a more favorable prognosis
(58).
In the present study, a significant enrichment of
the p53 pathway was observed in samples with high expression of the
glutathione gene set compared with those with low expression. Upon
activation, p53 can mediate a diverse array of cellular responses,
encompassing ferroptosis, stem cell reprogramming, invasion and
metastasis as well as metabolic regulation (59). p53 inhibits dipeptidyl peptidase 4
(DPP4) activity and negatively regulates ferroptosis in CRC cells.
Conversely, the absence of p53 promotes the interaction between
DPP4 and NADPH oxidase 1 (NOX1), leading to the formation of a
NOX1-DPP4 complex that mediates plasma membrane lipid peroxidation
and subsequently triggers ferroptosis (60). Therefore, in samples with low
expression of the glutathione gene set, the inhibition of p53 may
be responsible for the induction of ferroptosis. Meanwhile, in drug
treatments targeting tumors with a high expression profile of the
glutathione gene set, modulation of ferroptosis occurrence through
targeting p53, as primarily investigated in colon cancer models,
may hold notable implications for the progression of CRC, offering
significance for targeted therapies in CRC (61). The present study identified changes
and influences of MGST3 and a set of genes (including
PRDX6, HPGDS, GGT1, GPX3, GSTM4, GSTA1 and GSR) whose
products interact within a PPI network in the occurrence and
progression of CRC, adding a significant contribution to the
research and treatment of CRC.
In the present study, although the knockdown
experiments demonstrated that MGST3 deficiency activated
glutathione metabolism and induces tumorigenic phenotypes, the
precise molecular mechanisms by which MGST3 may regulate this
pathway, such as through specific protein interactions or
transcriptional regulation, remain incompletely elucidated.
Furthermore, the present study did not include overexpression
experiments or in vivo animal model validation, which limits
the generalizability of the potential tumor-suppressive function of
MGST3. In addition, while low MGST3 expression was associated with
increased TMB, altered immune infiltration, suppressed EMT and
enhanced ferroptosis, the causal relationships among these
phenotypes have not been established. For instance, it remains
unclear whether the potentiation of ferroptosis and the observed
immunosuppressive microenvironment are directly linked or merely
concurrent events. Finally, independent validation of the key
findings in a separate cohort represents an essential future
objective, as it fell beyond the scope and resources of the present
study.
Supplementary Material
Expression profile of the glutathione
gene set was analyzed across various cancer types and their stages
using publicly available transcriptomic data from The Cancer Genome
Atlas database, with a focus on comparing normal tissues, early
stage tumors (pathological T1/T2) and advanced stage tumors
(pathological T3/T4). The expression of the glutathione gene set
was significantly higher in normal tissues than in tumor tissues
across multiple cancer types, including BRCA, LUSC, READ, STAD,
HNSC and KICH, when compared with both pathological T1/T2 and
pathological T3/T4 stages. However, in KIRP, this elevated
expression in normal tissues was only significant when compared
with pathological T1/T2 stage tumors. However, no significant
differences were observed in other cancer types (Kruskal Wallis
test followed by Dunn's test; ns, not significant;
**P<0.01, ***P<0.001,
****P<0.0001). BRCA, breast invasive carcinoma; LUSC,
lung squamous cell carcinoma; READ, rectal adenocarcinoma; STAD,
stomach adenocarcinoma; HNSC, head and neck squamous cell
carcinoma; KICH, kidney chromophobe; KIRP, kidney renal papillary
cell carcinoma; CESC, cervical squamous cell carcinoma and
endocervical adenocarcinoma; ESCA, esophageal carcinoma; CHOL,
cholangiocarcinoma; LIHC, liver hepatocellular carcinoma; PAAD,
pancreatic adenocarcinoma; THCA, thyroid carcinoma; FPKM, fragments
per kilobase of exon model per million mapped fragments.
Enrichment differences of the
glutathione gene set between high and low expression samples in 14
pathways within The Cancer Genome Atlas.Colon Adenocarcinoma
dataset show distinct patterns. In the EGFR, estrogen, p53 and
Trail pathways, the enrichment of the high expression samples was
significantly higher than that of the low expression samples.
Conversely, in the PI3K and VEGF pathways, the opposite trend was
observed, with low expression samples exhibiting higher enrichment.
No significant differences were detected in the remaining pathways.
(two.tailed unpaired t.test; *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001). ns, not significant.
Acknowledgements
Not applicable.
Funding
Funding: This research was financially supported by the National
Natural Science Foundation of China for Young Scholars (grant no.
32100460).
Availability of data and materials
The RNA-seq data generated in the present study may
be found in the NCBI Gene Expression Omnibus database under the
accession number GSE316039 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE316039.
Authors' contributions
YW was responsible for the original concept, data
collection, figure creation, experimental design and writing the
manuscript. CF and FH contributed to data collection and the
acquisition of research materials. JZ and YZ made substantial
contributions to the conception and design of the study and
performed critical revision of the manuscript for important
intellectual content. JW and YH were responsible for the analysis
and interpretation of data and provided project supervision. All
authors read and approved the final version of the manuscript. YW
and JW confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
This study was performed in line with the principles
of the Declaration of Helsinki. Approval was granted by the Ethics
Committee of the Chinese People's Liberation Army General Hospital
(approval no. 2016-70). Informed consent for participation in the
study was obtained from all patients. Additionally, separate
consent was obtained for the publication of the data.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript or to generate images, and subsequently,
the authors revised and edited the content produced by the
artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Siegel RL, Giaquinto AN and Jemal A:
Cancer statistics, 2024. CA Cancer J Clin. 74:12–49.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Qu R, Ma Y, Zhang Z and Fu W: Increasing
burden of colorectal cancer in China. Lancet Gastroenterol Hepatol.
7(700)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dantas AAG, de Oliveira NPD, Costa GAB,
Martins LFL, Dos Santos JEM, Migowski A, de Camargo Cancela M and
de Souza DLB: Multilevel analysis of social determinants of
advanced stage colorectal cancer diagnosis. Sci Rep.
14(9667)2024.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Myers DJ and Arora K: Villous adenoma
(Archived). In: StatPearls. StatPearls Publishing, Treasure Island,
FL, 2025.
|
|
5
|
Nguyen LH, Goel A and Chung DC: Pathways
of colorectal carcinogenesis. Gastroenterology. 158:291–302.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Uno Y, Murayama N, Kunori M and Yamazaki
H: Characterization of microsomal glutathione S-transferases MGST1,
MGST2, and MGST3 in cynomolgus macaque. Drug Metab Dispos.
41:1621–1625. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ghosh S and Chan CK: Analysis of RNA-seq
data using tophat and cufflinks. Methods Mol Biol. 1374:339–361.
2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Adan A, Kiraz Y and Baran Y: Cell
proliferation and cytotoxicity assays. Curr Pharm Biotechnol.
17:1213–1221. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Justus CR, Marie MA, Sanderlin EJ and Yang
LV: Transwell in vitro cell migration and invasion assays. Methods
Mol Biol. 2644:349–359. 2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mahmood T and Yang PC: Western blot:
Technique, theory, and trouble shooting. N Am J Med Sci. 4:429–434.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Schubert M, Klinger B, Klünemann M, Sieber
A, Uhlitz F, Sauer S, Garnett MJ, Blüthgen N and Saez-Rodriguez J:
Perturbation-response genes reveal signaling footprints in cancer
gene expression. Nat Commun. 9(20)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Szklarczyk D, Nastou K, Koutrouli M,
Kirsch R, Mehryary F, Hachilif R, Hu D, Peluso ME, Huang Q, Fang T,
et al: The STRING database in 2025: protein networks with
directionality of regulation. Nucleic Acids Res. 53:D730–D737.
2025.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo X, Liang X, Wang Y, Cheng A, Zhang H,
Qin C and Wang Z: Significance of tumor mutation burden combined
with immune infiltrates in the progression and prognosis of
advanced gastric cancer. Front Genet. 12(642608)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tan TZ, Miow QH, Miki Y, Noda T, Mori S,
Huang RY and Thiery JP: Epithelial-mesenchymal transition spectrum
quantification and its efficacy in deciphering survival and drug
responses of cancer patients. EMBO Mol Med. 6:1279–1293.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen B, Khodadoust MS, Liu CL, Newman AM
and Alizadeh AA: Profiling tumor infiltrating immune cells with
CIBERSORT. Methods Mol Biol. 1711:243–259. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yoshihara K, Shahmoradgoli M, Martínez E,
Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW,
Levine DA, et al: Inferring tumour purity and stromal and immune
cell admixture from expression data. Nat Commun.
4(2612)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yan H, Talty R and Johnson CH: Targeting
ferroptosis to treat colorectal cancer. Trends Cell Biol.
33:185–188. 2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jardim BV, Moschetta MG, Leonel C,
Gelaleti GB, Regiani VR, Ferreira LC, Lopes JR and Zuccari DAPDC:
Glutathione and glutathione peroxidase expression in breast cancer:
An immunohistochemical and molecular study. Oncol Rep.
30:1119–1128. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Inoue T, Ishida T, Sugio K, Maehara Y and
Sugimachi K: Glutathione S transferase Pi is a powerful indicator
in chemotherapy of human lung squamous-cell carcinoma. Respiration.
62:223–227. 1995.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Xu H, Hu C, Wang Y, Shi Y, Yuan L, Xu J,
Zhang Y, Chen J, Wei Q, Qin J, et al: Glutathione peroxidase 2
knockdown suppresses gastric cancer progression and metastasis via
regulation of kynurenine metabolism. Oncogene. 42:1994–2006.
2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xiao Y and Meierhofer D: Glutathione
metabolism in renal cell carcinoma progression and implications for
therapies. Int J Mol Sci. 20(3672)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sobhakumari A, Love-Homan L, Fletcher EV,
Martin SM, Parsons AD, Spitz DR, Knudson CM and Simons AL:
Susceptibility of human head and neck cancer cells to combined
inhibition of glutathione and thioredoxin metabolism. PLoS One.
7(e48175)2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Schmitt M and Greten FR: The inflammatory
pathogenesis of colorectal cancer. Nat Rev Immunol. 21:653–667.
2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bethmann D, Feng Z and Fox BA:
Immunoprofiling as a predictor of patient's response to cancer
therapy-promises and challenges. Curr Opin Immunol. 45:60–72.
2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wellenstein MD and de Visser KE:
Cancer-cell-intrinsic mechanisms shaping the tumor immune
landscape. Immunity. 48:399–416. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mukherjee AG, Wanjari UR, Namachivayam A,
Murali R, Prabakaran DS, Ganesan R, Renu K, Dey A, Vellingiri B,
Ramanathan G, et al: Role of immune cells and receptors in cancer
treatment: An immunotherapeutic approach. Vaccines (Basel).
10(1493)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mazari AMA, Zhang L, Ye ZW, Zhang J, Tew
KD and Townsend DM: The multifaceted role of glutathione
S-transferases in health and disease. Biomolecules.
13(688)2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Moyer MP, Manzano LA, Merriman RL,
Stauffer JS and Tanzer LR: NCM460, a normal human colon mucosal
epithelial cell line. In Vitro Cell Dev Biol Anim. 32:315–317.
1996.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Xue X, Wang M, Cui J, Yang M, Ma L, Kang
R, Tang D and Wang J: Glutathione metabolism in ferroptosis and
cancer therapy. Cancer Lett. 621(217697)2025.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ma T, Du J, Zhang Y, Wang Y, Wang B and
Zhang T: GPX4-independent ferroptosis-a new strategy in disease's
therapy. Cell Death Discov. 8(434)2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X,
Wang H, Cao L and Tang D: HSPB1 as a novel regulator of ferroptotic
cancer cell death. Oncogene. 34:5617–5625. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang Y, Zou L, Li X, Guo L, Hu B, Ye H
and Liu Y: SLC40A1 in iron metabolism, ferroptosis, and disease: A
review. WIREs Mech Dis. 16(e1644)2024.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chaudhary N, Choudhary BS, Shah SG,
Khapare N, Dwivedi N, Gaikwad A, Joshi N, Raichanna J, Basu S,
Gurjar M, et al: Lipocalin 2 expression promotes tumor progression
and therapy resistance by inhibiting ferroptosis in colorectal
cancer. Int J Cancer. 149:1495–1511. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Klusek J, Głuszek S and Klusek J: GST gene
polymorphisms and the risk of colorectal cancer development.
Contemp Oncol (Pozn). 18:219–221. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Xie Z, Kawasaki T, Zhou H, Okuzaki D,
Okada N and Tachibana M: Targeting GGT1 eliminates the
tumor-promoting effect and enhanced immunosuppressive function of
myeloid-derived suppressor cells caused by G-CSF. Front Pharmacol.
13(873792)2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chen J, Wu Y, Zhou Q, Song Y, Zhuang J, Lu
K and Yang X: GPX3 is a key cholesterol-related gene associated
with prognosis and tumor-infiltrating T cells in colorectal cancer.
Neoplasma. 70(230704N348)2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Lagal DJ, Montes-Osuna AM, Ortiz-Olivencia
A, Arribas-Parejas C, Ortiz-Alcántara Á, Pescuezo-Castillo C,
Bárcena JA, Padilla CA and Requejo-Aguilar R: Tumoral malignancy
decreases coupled with higher ROS and lipid peroxidation in HCT116
Colon cancer cells upon loss of PRDX6. Antioxidants (Basel).
13(881)2024.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Merino DM, McShane LM, Fabrizio D, Funari
V, Chen SJ, White JR, Wenz P, Baden J, Barrett JC, Chaudhary R, et
al: Establishing guidelines to harmonize tumor mutational burden
(TMB): In silico assessment of variation in TMB quantification
across diagnostic platforms: Phase I of the friends of cancer
research TMB harmonization project. J ImmunoTher Cancer.
8(e000147)2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Sha D, Jin Z, Budczies J, Kluck K,
Stenzinger A and Sinicrope FA: Tumor mutational burden as a
predictive biomarker in solid tumors. Cancer Discov. 10:1808–1825.
2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zgura A, Chipuc S, Bacalbasa N, Haineala
B, Rodica A and Sebastian V: Evaluating tumour mutational burden as
a key biomarker in personalized cancer immunotherapy: A Pan-cancer
systematic review. Cancers (Basel). 17(480)2025.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Kim ES, Velcheti V, Mekhail T, Yun C,
Shagan SM, Hu S, Chae YK, Leal TA, Dowell JE, Tsai ML, et al:
Blood-based tumor mutational burden as a biomarker for atezolizumab
in non-small cell lung cancer: The phase 2 B-F1RST trial. Nat Med.
28:939–945. 2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Addeo A, Friedlaender A, Banna GL and
Weiss GJ: TMB or not TMB as a biomarker: That is the question. Crit
Rev Oncol/Hematol. 163(103374)2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134.
2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kim DH, Xing T, Yang Z, Dudek R, Lu Q and
Chen YH: Epithelial mesenchymal transition in embryonic
development, tissue repair and cancer: A comprehensive overview. J
Clin Med. 7(1)2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Atreya I and Neurath MF: Immune cells in
colorectal cancer: Prognostic relevance and therapeutic strategies.
Expert Rev Anticancer Ther. 8:561–572. 2008.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ye L, Zhang T, Kang Z, Guo G, Sun Y, Lin
K, Huang Q, Shi X, Ni Z, Ding N, et al: Tumor-infiltrating immune
cells act as a marker for prognosis in colorectal cancer. Front
Immunol. 10(2368)2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Nguyen A, Loo JM, Mital R, Weinberg EM,
Man FY, Zeng Z, Paty PB, Saltz L, Janjigian YY, de Stanchina E and
Tavazoie SF: PKLR promotes colorectal cancer liver colonization
through induction of glutathione synthesis. J Clin Investig.
126:681–694. 2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Endale HT, Tesfaye W and Mengstie TA: ROS
induced lipid peroxidation and their role in ferroptosis. Front
Cell Dev Biol. 11(1226044)2023.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Li S, Tao K, Yun H, Yang J, Meng Y, Zhang
F and Ma X: Ferroptosis is a protective factor for the prognosis of
cancer patients: A systematic review and meta-analysis. BMC Cancer.
24(604)2024.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liebl MC and Hofmann TG: The role of p53
signaling in colorectal cancer. Cancers (Basel).
13(2125)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J,
Zhong M, Yuan H, Zhang L, Billiar TR, et al: The tumor suppressor
p53 limits ferroptosis by blocking DPP4 activity. Cell Rep.
20:1692–1704. 2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Yang L, WenTao T, ZhiYuan Z, Qi L, YuXiang
L, Peng Z, Ke L, XiaoNa J, YuZhi P, MeiLing J, et al: Cullin-9/p53
mediates HNRNPC degradation to inhibit erastin-induced ferroptosis
and is blocked by MDM2 inhibition in colorectal cancer. Oncogene.
41:3210–3221. 2022.PubMed/NCBI View Article : Google Scholar
|