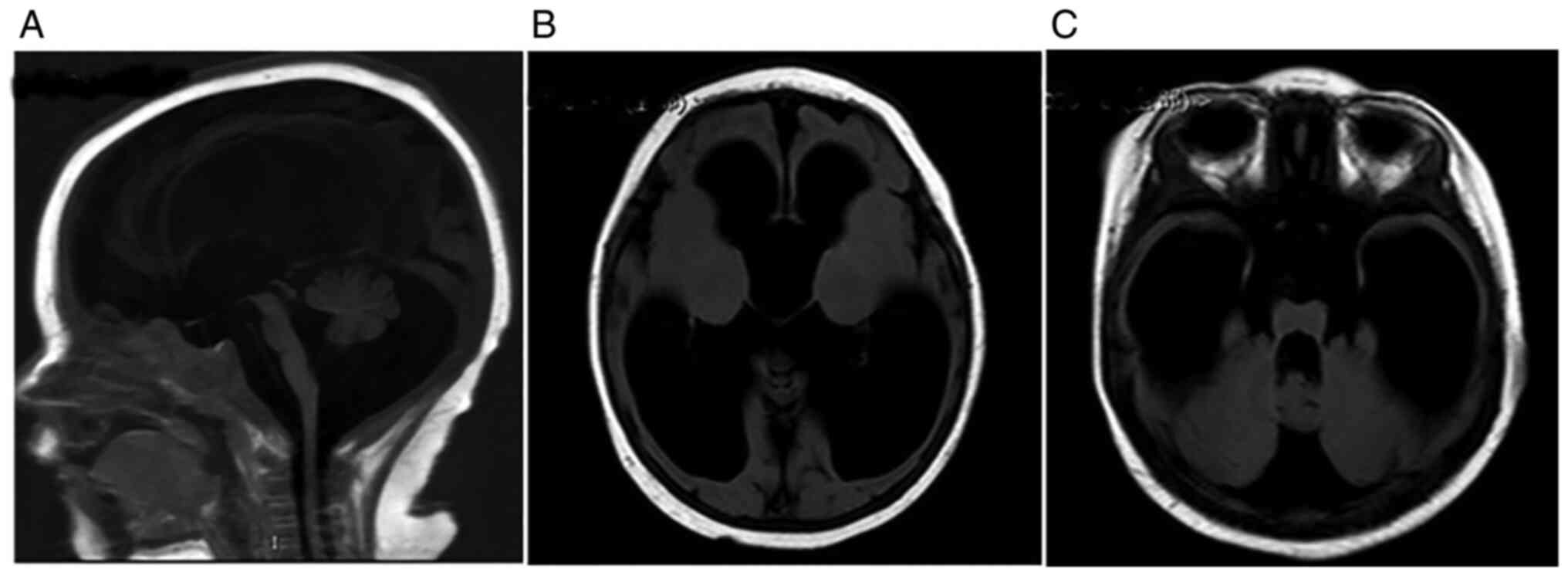
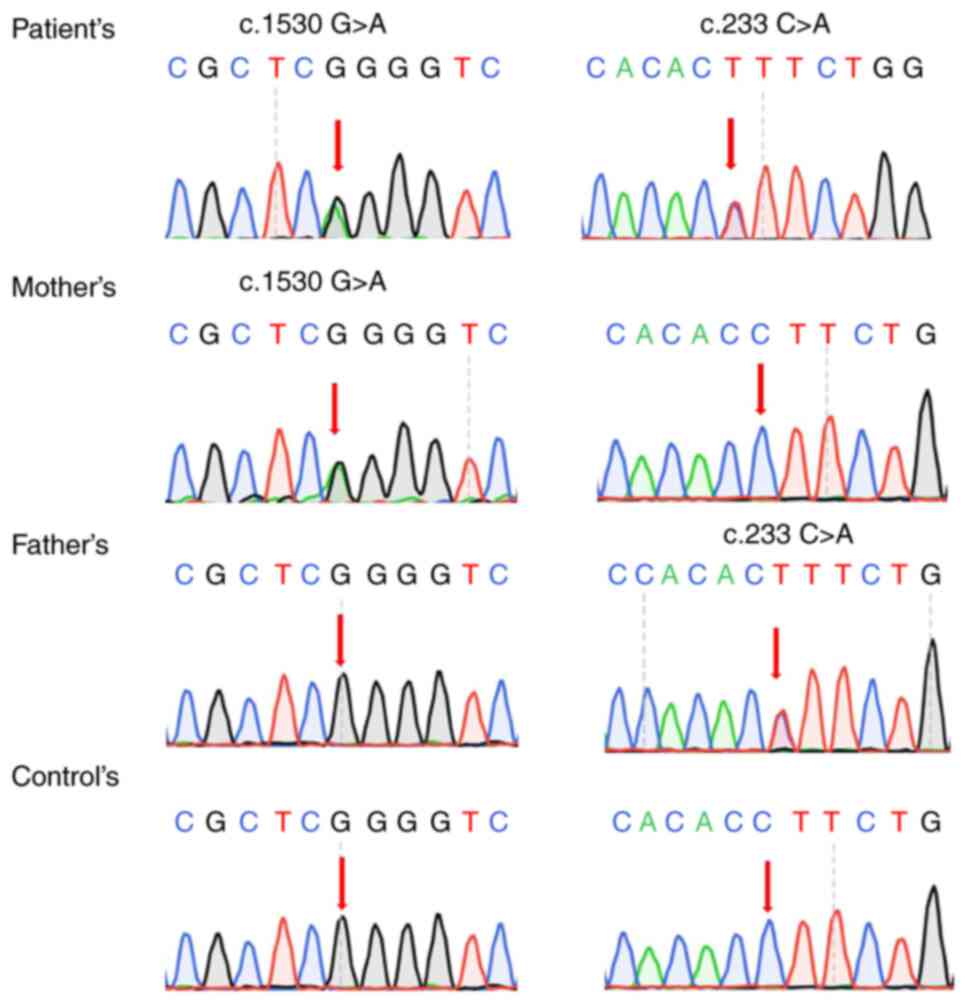
|
1
|
McCully KS: Vascular pathology of
homocysteinemia: Implications for the pathogenesis of
arteriosclerosis. Am J Pathol. 56:111–128. 1969.PubMed/NCBI
|
|
2
|
Brustolin S, Giugliani R and Félix TM:
Genetics of homocysteine metabolism and associated disorders. Braz
J Med Biol Res. 43:1–7. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mudd SH, Skovby F, Levy HL, Pettigrew KD,
Wilcken B, Pyeritz RE, Andria G, Boers GH, Bromberg IL and Cerone
R: The natural history of homocystinuria due to cystathionine
beta-synthase deficiency. Am J Hum Genet. 37:1–31. 1985.PubMed/NCBI
|
|
4
|
Kuo HK, Sorond FA, Chen JH, Hashmi A,
Milberg WP and Lipsitz LA: The role of homocysteine in multisystem
age-related problems: A systematic review. J Gerontol A Biol Sci
Med Sci. 60:1190–1201. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Geng B, Chang L, Du JB and Tang CS: A new
strategy to treat hyperhomocysteinemia. Beijing Da Xue Xue Bao Yi
Xue Ban. 37:215–219. 2005.PubMed/NCBI(In Chinese).
|
|
6
|
Carson NA and Neill DW: Metabolic
abnormalities detected in a survey of mentally backward individuals
in Northern Ireland. Arch Dis Child. 37:505–513. 1962.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gerritsen T, Vaughn JG and Waisman HA: The
identification of homocystine in the urine. Biochem Biophys Res
Commun. 9:493–496. 1962.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mudd SH, Finkelstein JD, Irreverre F and
Laster L: Homocystinuria: An enzymatic defect. Science.
143:1443–1445. 1964.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mudd SH, Levy HL, Abeles RH and Jennedy JP
Jr: A derangement in B 12 metabolism leading to homocystinemia,
cystathioninemia and methylmalonic aciduria. Biochem Biophys Res
Commun. 35:121–126. 1969.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Baethmann M, Wendel U, Hoffmann GF,
Göhlich-Ratmann G, Kleinlein B, Seiffert B, Blom H and Voit T:
Hydrocephalus internus in two patients with
5,10-methylenetetrahydrofolate reductase deficiency.
Neuropediatrics. 31:314–317. 2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guenther BD, Sheppard CA, Tran P, Rozen R,
Matthews RG and Ludwig ML: The structure and properties of
methylenetetrahydrofolate reductase from escherichia coli suggest
how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol.
6:359–365. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Shahzad K, Hai A, Ahmed A, Kizilbash N and
Alruwaili J: A structured-based model for the decreased activity of
Ala222Val and Glu429Ala methylenetetrahydrofolate reductase (MTHFR)
mutants. Bioinformation. 9:929–936. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Leclerc D, Sibani S and Rozen R: Molecular
biology of methylenetetrahydrofolate reductase (MTHFR) and overview
of mutations/polymorphisms. MTHFR polymorphisms and disease: CRC
Press; 2005. p.15-3.
|
|
14
|
Buratti E, Chivers M, Královicová J,
Romano M, Baralle M, Krainer AR and Vorechovsky I: Aberrant 5'
splice sites in human disease genes: Mutation pattern, nucleotide
structure and comparison of computational tools that predict their
utilization. Nucleic Acids Res. 35:4250–4263. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Burda P, Schäfer A, Suormala T, Rummel T,
Bürer C, Heuberger D, Frapolli M, Giunta C, Sokolova J, Vlášková H,
et al: Insights into severe 5,10-methylenetetrahydrofolate
reductase deficiency: Molecular genetic and enzymatic
characterization of 76 patients. Hum Mutat. 36:611–621.
2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Broomfield A, Abulhoul L, Pitt W, Jameson
E and Cleary M: Reversal of respiratory failure in both neonatal
and late onset isolated remethylation disorders. JIMD Rep.
16:51–56. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Richard E, Desviat LR, Ugarte M and Pérez
B: Oxidative stress and apoptosis in homocystinuria patients with
genetic remethylation defects. J Cell Biochem. 114:183–191.
2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Knowles L, Morris AA and Walter JH:
Treatment with mefolinate (5-Methyltetrahydrofolate), but not folic
acid or folinic acid, leads to measurable 5-Methyltetrahydrofolate
in cerebrospinal fluid in methylenetetrahydrofolate reductase
deficiency. JIMD Rep. 29:103–107. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Froese DS, Huemer M, Suormala T, Burda P,
Coelho D, Gueant JL, Landolt MA, Kozich V, Fowler B and Baumgartner
MR: Mutation update and review of severe methylenetetrahydrofolate
reductase deficiency. Hum Mutat. 37:427–438. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tonetti C, Saudubray JM, Echenne B,
Landrieu P, Giraudier S and Zittoun J: Relations between molecular
and biological abnormalities in 11 families from siblings affected
with methylenetetrahydrofolate reductase deficiency. Eur J Pediatr.
162:466–475. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kristin E, Bearden D, Watkins D, Hyland K,
Rosenblatt DS and Ficicioglu C: Severe 5,
10-methylenetetrahydrofolate reductase deficiency and two MTHFR
variants in an adolescent with progressive myoclonic epilepsy.
Pediatr Neurol. 51:266–270. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Prasad AN, Rupar CA and Prasad C:
Methylenetetrahydrofolate reductase (MTHFR) deficiency and
infantile epilepsy. Brain Dev. 33:758–769. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Algassim N, Alfadhel M, Nashabat M and
Eyaid W: Clinical presentation of seven patients with
Methylenetetrahydrofolate reductase deficiency. Mol Genet Metab
Rep. 25(100644)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tsuji M, Takagi A, Sameshima K, Iai M,
Yamashita S, Shinbo H, Furuya N, Kurosawa K and Osaka H:
5,10-Methylenetetrahydrofolate reductase deficiency with
progressive polyneuropathy in an infant. Brain Dev. 33:521–524.
2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Greitz D: Radiological assessment of
hydrocephalus: New theories and implications for therapy.
Neuroradiol J. 19:475–495. 2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zittoun J: Congenital errors of folate
metabolism. Baillieres Clin Haematol. 8:603–616. 1995.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lossos A, Teltsh O, Milman T, Meiner V,
Rozen R, Leclerc D, Schwahn BC, Karp N, Rosenblatt DS, Watkins D,
et al: Severe methylenetetrahydrofolate reductase deficiency:
Clinical clues to a potentially treatable cause of adult-onset
hereditary spastic paraplegia. JAMA Neurol. 71:901–904.
2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim YI: 5,10-methylenetetrahydrofolate
reductase polymorphisms and pharmacogenetics: A new role of single
nucleotide polymorphisms in the folate metabolic pathway in human
health and disease. Nut Rev. 63:398–407. 2005.PubMed/NCBI View Article : Google Scholar
|