Introduction
Hyperhomocysteinemia (OMIM #603174) is a common
metabolic error occurring in infants. A dysfunction in enzymes
involving in the metabolism of methionine i.e., cystathionine
β-synthase, methionine synthase and 5,10-methylenetetrahydrofolate
reductase (MTHFR; also known as 5,10-methyl THR reductase) can lead
to an elevation in plasma homocysteine levels, which can cause
vascular disorders (1). A deficiency
in methionine synthetase and MTHFR caused by deteriorations of
mecobalamin are the most common types (2). The clinical presentation of
hyperhomocysteinemia is highly variable between individuals. Lens
dislocation, vasculopathy, skeletal abnormality and psychomotor
delay are the most common manifestations of MTHFR deficiency
(1,3,4).
Systemic abnormalities and mortality have also been reported
(5). Therefore, the prognosis is
also different among patients.
The MTHFR enzyme encoded by the MTHFR gene
(OMIM #607093) is an essential enzyme which catalyzes the reduction
of 5,10-methyletrahydrofolate (5,10-methyl THF) to
5-methyletrahydrofolate (5-methyl THF), which donates a methyl
group to homocysteine, generating methionine. A deficiency in 5
MTHFR results in a decline in 5-methyl THF, which deteriorates the
conversion of homocysteine to methionine, thereby inducing an
increase in plasma homocysteine. To date, the vascular disorders
caused by hyperhomocysteinemia have been extensively explored
(4). However, the neurological
implications have been less investigated. The present study
describes a case of a patient (infant) with MTHFR deficiency whose
main symptom was hydrocephalus. To the best of our knowledge, this
is the first case of early-onset 5 MTHFR deficiency in a patient
presenting with hydrocephalus to be reported in China.
Case report
The patient was a 4-month-old boy who was admitted
to the Department of Endocrinology and Metabolism at Beijing
Children's Hospital (Beijing, China) with the chief complaint of
‘developmental delay for 4 months with frequent cyanosis induced by
crying for 14 days’. He was the third child of a non-consanguineous
family. The first two children of this family died of unexplained
severe hydrocephalus within their first 2 months of life. The
patient in question was born by cesarean delivery at full-term with
a birth weight of 3.2 kg. Weak crying and cyanosis were observed at
birth, which improved following oxygen inhalation and upper
respiratory tract clearance. However, on the 9th day after birth,
he was admitted to the local hospital again due to feeding
difficulties and high-pitched crying. The results of magnetic
resonance imaging (MRI) results indicated ischemic-anoxic changes
and a hemorrhage at the dura mater and left occipital lobe.
Therefore, mouse nerve growth factor, creatine phosphate sodium and
ganglioside were administered; however, no improvement had been
observed. The patient was then transferred to Peking University
First Hospital due to recurrent seizures at the age of 1 month.
Ventricle dilation was observed by MRI this time; thus,
hypoxic-ischemic encephalopathy was again considered. However,
mouse nerve growth factor combined with hyperbaric oxygen therapy
still failed to lead to any responses. At the age of 4 months, the
boy presented with recurrent transient crying-induced cyanosis,
which autonomously remitted in 2-3 min. Feeding problems and
vomiting were also reported. Delayed developmental milestones were
also manifested. The patient could neither hold his head up nor
roll over. He was also unable to gaze or follow along. He also did
not acquire the ability to smile or recognize individuals. A
cranial CT scan revealed profound hydrocephalus this time.
Therefore, the patient was referred to the Department of
Endocrinology and Metabolism at Beijing Children's Hospital.
Physical examination
The weight of the patient was 4 kg (<-3 SD), his
height was 60 cm (-2 SD) and his head circumference was 41 cm (0±1
SD) upon hospital admission. Apathy, dry skin and acrocyanosis were
observed upon an examination. The patient also presented with
respiratory distress and head bobbing. However, no obvious
abnormalities were found in the heart, lungs, spleen and liver.
Although the patient had no nystagmus or evident abnormalities in
the eyes, a sluggish response to light was still identified.
Hyperreflexia was found in the lower limbs, whereas muscular
tension was normal. Ankle clonus and the Babinski sign were
positive. The patient had no neck stiffness. Neither Kernig's sign
nor Brudzinski's sign was presented. Transverse palmar crease was
found in the right palm.
Laboratory examination
Routine blood and urine tests were conducted to
evaluated liver and renal functions, as well as serum electrolyte,
glucose, lactate and pyruvate levels. The analysis of blood amino
acids and acylcarnitines was performed using an Applied Biosystes
API 3200 MS/MS analyzer and ChemoView software. Urinary organic
acids were analyzed using gas chromatography-mass spectrometry
(GC/MS) using the Shimadzu GC/MS QP2010 analyzer (Shimadzu
Corporation) and the Inborn Errors of Metabolism screening system
software for the differential diagnosis of organic acidurias. A
cranial MRI was also evaluated. No abnormalities were found in the
blood and urine routine examination. A cerebrospinal fluid (CSF)
routine and biochemical test both revealed normal results. The
biochemical blood tests revealed that the alanine aminotransferase
level was 101 U/l (reference value, 5-40 U/l); the aspartate
aminotransferase level was 48.7 U/l (normal range, 5-40 U/l); the
alkaline phosphatase level was 384 U/l (reference range, 5-350
U/l); and the creatine kinase-muscle/brain (CK-MB) level was 77 U/l
(reference, 0-25 U/l). The patient had no viral infections
according to the serum TORCH (Toxoplasma gondii, rubella
virus, cytomegalovirus and herpes simplex virus) IgM and IgG
antibody test results. Gas chromatography-mass spectrometry
analysis of the blood revealed that the blood methionine level was
8.12 µmol/l (reference, 11.0-54.0 µmol/l); the free carnitine level
was 11.06 µmol/l (reference, 20.0-60.0 µmol/l); and the serum
homocysteine level was >50 µmol/l (reference, 1.6-16.0 µmol/l).
Urine organic acid analysis did not reveal any abnormalities
(Table SI).
Imaging analysis
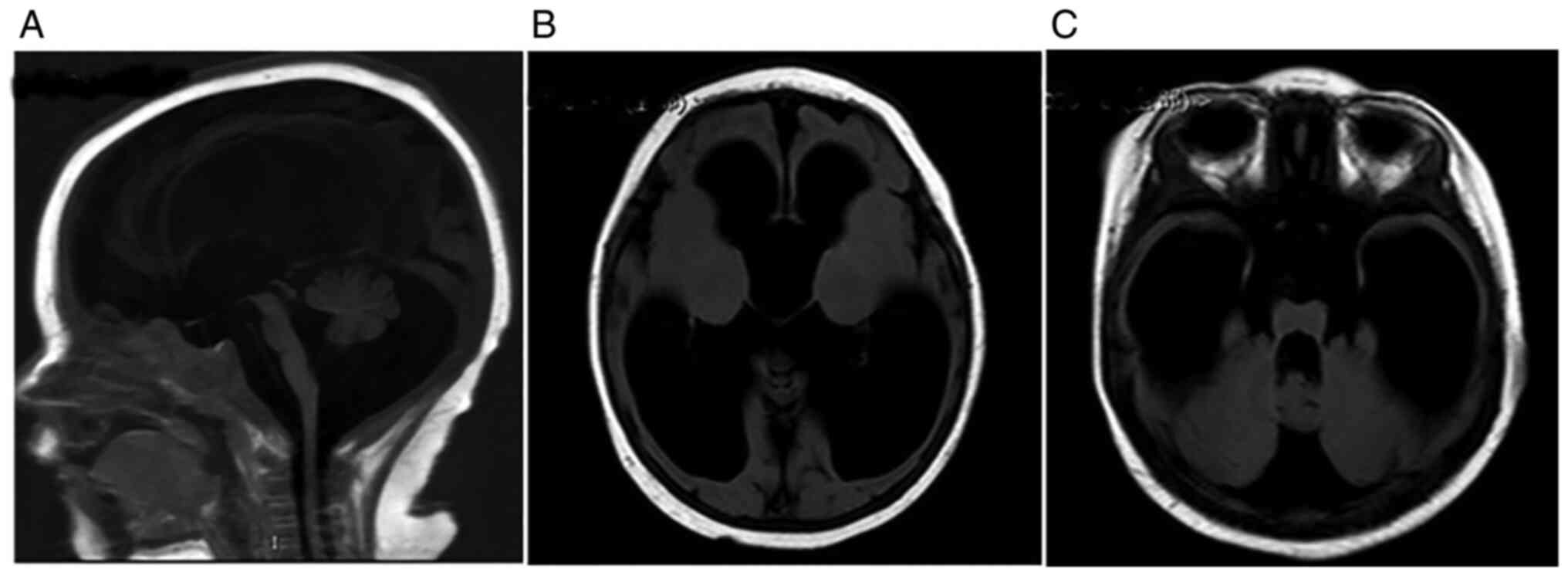
The cranial MRI result revealed that the patient had
severe progressive hydrocephalus (Fig.
1). The echocardiogram revealed asymmetric septal hypertrophy,
pulmonary arterial hypertension, patent foramen ovale (2 mm) and
mild tricuspid regurgitation. A chest X-ray examination revealed
increased lung markings with blurred margins. A video
electroencephalography test demonstrated hypsarrhythmia,
dissociation and epileptic discharges in the brain. Intermittent
rhythmic disorders were also found in the fast waves over posterior
head regions, especially at the left part.
Genetic analysis
The genomic DNA of the patient and his parents were
isolated from peripheral leukocytes using standard methods.
Inherited metabolic disease-related gene analysis of the patient
was performed using gene capture and high-throughput
(next-generation) genomic sequencing (performed by Beijing Kangso
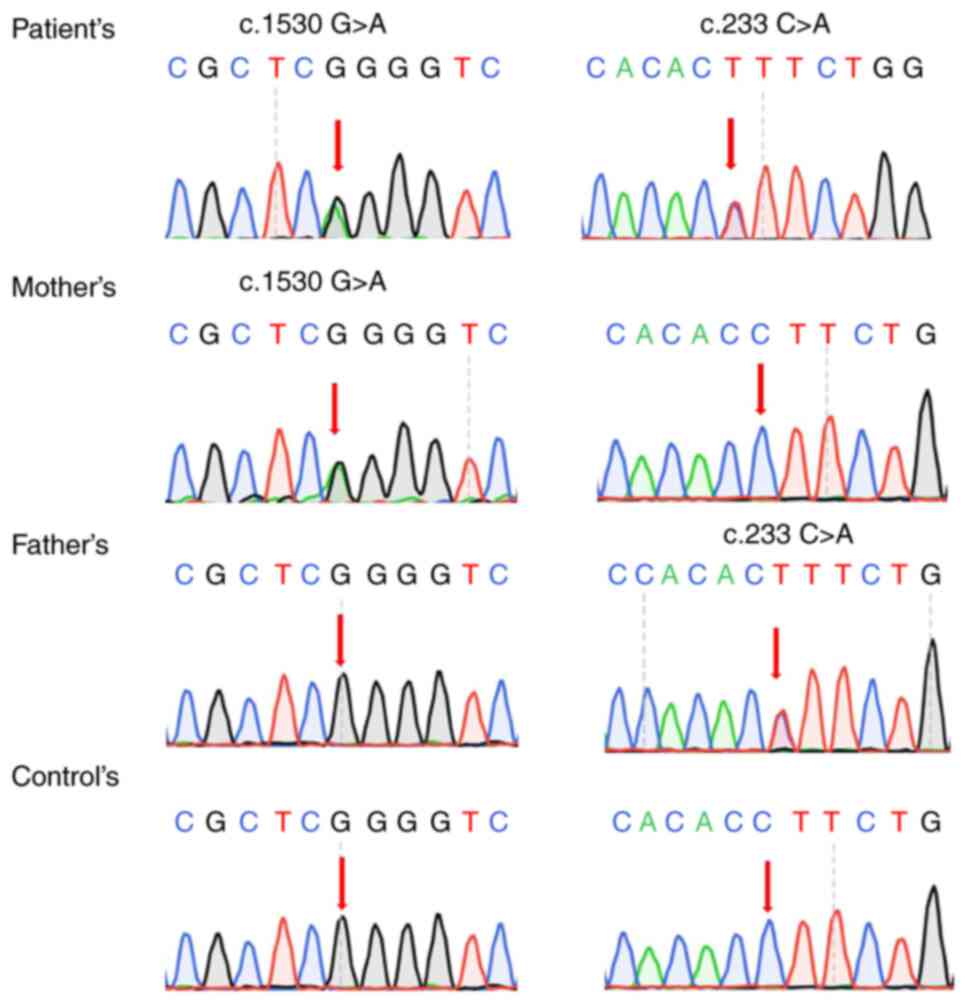
Medical Inspection). The patient was found to carry a paternal
mutation c.1530G>A (p.K510K) and a maternal mutation c.233C>A
(p.S78X) of the MTHFR gene. These two mutations have all
been previously reported (as mentioned in the Discussion section
below). According to the ACMG guidelines, these two mutations can
all be classified as likely pathogenic and pathogenic, respectively
(Fig. 2).
Treatment and follow-up
examinations
A shunt system was surgically inserted to help
divert the CSF. Vitamin B12 (1,000 µg per day) were intramuscularly
injected. A combination of L-carnitine (0.5 g per day), folate (10
mg per day), betaine (500 mg per day) and vitamin B6 (30 mg per
day) was prescribed. The dose of vitamin B12 was reduced to 500 µg
every other day. A follow-up examination after 2 months revealed
that the proband could support his head and follow objects, but
could still not roll over. A physical examination revealed a weight
of 6 kg. The blood carnitine level was 85.93 µmol/l and the
methionine level was 12 µmol/l, which was elevated following
treatment. The serum homocysteine level decreased to 78.1 µmol/l,
although it was still above the normal range. A re-examination of a
head CT scan suggested that the symptoms of cranial hypertension
had improved and the ventricle was slightly retracted. At that
time, the patient was 4 years and 4months old, and he could not
speak, did not recognize individuals and could not walk; however,
his parents have been lost to follow-up (Table SII).
Discussion
An abnormal elevation in homocysteine levels was
first reported as homocystinuria in 1962 (6,7). A
deficiency in vitamin B12 metabolism was first identified as a
cause of homocystinuria and hyperhomocysteinemia (8,9).
Subsequently, hyperhomocysteinemia, including its pathogenesis,
mechanisms, clinical presentations, genotype, diagnosis, screening
and the prevention of the disease have been further researched. The
genes that cause hyperhomocysteinemia include cystathionine
beta-synthase, peroxiredoxin 1, metabolism of cobalamin associated
C (also known as C2orf25), 5-methyltetrahydrofolate-homocysteine
methyltransferase reductase, LMBR1 domain containing
1,5-methyltetrahydrofolate-homocysteine methyltransferase and
MTHFR. A deficiency in the MTHFR gene can cause a
dysfunction in MTHFR. The deficiency in 5 MTHFR destroys the
metabolism of homocysteine, leading to hyperhomocysteinemia. The
clinical presentations of MTHFR deficiency are heterogeneous.
However, hydrocephalus has been rarely observed in patients with
MTHFR deficiency (10). To the best
of our knowledge, the present study presents the first case of
MTHFR deficiency in a patient in China, whose main manifestation
was hydrocephalus. The MTHFR gene is located on 1p36.22 and
encodes MTHFR, a 74.6 kD enzyme composed of 656 amino acids. This
protein contains a methylenetetrahydrofolate reductase domain
(amino acid 38-337) and can catalyze the reduction of 5,10-methyl
THF to 5-methyl THF. In this reaction, the cofactor flavin adenine
dinucleotide functions as a transient carrier of electrons, which
accepts the reducing equivalents from NAD(P)H and donates it to
5,10-methyl THR. In humans, 5,10-methyl THF reductase functions as
a homodimer (11,12).
A MTHFR deficiency is an autosomal recessive
disorder. Only the homozygous carrier or biallelic mutations
carrier may have the disease. To date, there are 131 known
disease-causing mutations in MTHFR listed in the Human Gene
Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). Of these
mutations, >60% are missense. The majority of the pathogenic
mutations are located in exon 5-6 of the gene. This may be due to
the fact that the loci encoding the catalytic domain is located
within the 5th and the 6th exon. A destruction in this domain may
affect the affinity between the enzyme and its substrates (13). In the present study, a synonymous
mutation c.1530G>A (p.K510K) and a nonsense mutation c.233C>A
(p.S78X) were identified. The synonymous mutation was validated to
be a paternal mutation, while the nonsense mutation was validated
to be a maternal mutation by Sanger sequencing. Therefore, the
patient carried a pair of compound-heterozygous variants and this
was in accordance with the inheritance pattern of MTHFR deficiency.
The synonymous mutation c.1530G>A (rs765586205) has been
previously reported. It locates at the consensus splice site (the
last nucleotide) of exon 9. It has been demonstrated that this
alteration can affect splicing, resulting in an actual frame-shift
mutation by skipping of exon 8, to produce a premature termination
of the protein (PS3) (14,15). This mutation is present in population
databases with an extremely low frequency (PM2). In addition, this
mutation is also the most common variant in MTHFR deficiency and
has been reported in various affected patients or families (PP1 in
ACMG but upgrading to moderate evidence due to increasing sample
size) (15-19).
Therefore, this mutation is considered a likely pathogenic
mutation. For the nonsense mutation, p.S78X (PVS1), and has been
previously reported (PP5) (20).
Therefore, this variant is also interpreted as a pathogenic
mutation. The biallelic mutations would result in less catalytic
efficiency of the enzyme, thus causing MTHFR deficiency.
MTHFR deficiency (OMIM #236250) is one of the most
prevalence congenital disorders of folate metabolism characterized
by hyperhomocysteinemia. The clinical manifestations are
heterogeneous, ranging from asymptomatic to severe neurological
symptoms. The onset age also varies from neonate to adolescent even
to adult. Clinical presentations, including neurodegeneration,
psychomotor delay, seizures, psychiatric disorders, vascular
diseases and peripheral neurological disorders have all been
observed. Some patients also have respiratory failure, thus leading
to mortality (15). Early-onset
MTHFR deficiency presenting with neurological symptoms has been
described as severe MTHFR. Patients with late-onset MTHFR
deficiency with severe neurological symptoms have seldom been
observed, apart from one case reported in 2014(21). The typical manifestations of neonatal
patients with severe MTHFR deficiency include hypotonia, feeding
problems, drowsiness, sleep apnea and epilepsy. Patients with
epilepsy usually first present with myoclonic seizures which
progress to head nodding syndrome or complex focal seizures
(22). Patients with MTHFR
deficiency with hydrocephalus have rarely been reported (23).
The main manifestation of the present case was
increased intracranial pressure and hydrocephalus when admitted to
hospital at the age of 4 months. Mild developmental delay with
recurrent seizures was reported when the medical history of the
patient was further inquired upon. The patient did not present
vomiting or other symptoms related to increased intracranial
pressure or ventricle dilation until he was 4 months old. The MRI
indicated that the patient had severe hydrocephalus without
atrophy. The basal ganglia of the patient were not affected. No
invasion or calcification were found. Brain tumor, intraventricular
hemorrhage, head injury and infection-induced meningitis were all
excluded. In addition, the patient also presented with a decreased
blood methionine level and an enhanced homocysteine level, which is
in accordance with the typical presentations of MTHFR
deficiency.
The mechanisms of hydrocephalus in MTHFR deficiency
in patients remain unclarified. However, hyperhomocysteinemia has
been reported to cause vascular disease (1). With its thrombotic tendency,
hyperhomocysteinemia can also cause thrombi in cerebral
capillaries, which may impair the absorption of CSF, causing
hydrocephalus (24). When the CSF
flows into the subarachnoid space, it is diffused and absorbed into
capillaries through intracranial arterial pulsation. The diffusion
and absorption of CSF protects the balance of arterial pulsation in
the brain. However, in metabolic disorders, such as MTHFR
deficiency, the deleterious metabolites can destroy the vessel wall
and affect the diffusion and absorption of the CSF, causing
increased intracranial pressure and hydrocephalus (25).
MTHFR deficiency is clinically characterized by
hyperhomocysteinemia, hypomethioninemia and a decreased level of
folate in serum, erythrocytes and CSF (26). Hypomethioninemia may be considered as
an important indicator for diagnosis. However, biochemical indexes
combined with genetic testing are required for the final diagnosis
(27).
Betaine (trimethylglycine) is an amino acid derivate
which functions as a methyl donor in the alternative pathway of
methionine synthesis. The supplementation of betaine can accelerate
the synthesis of methionine by increasing the substrate
concentration. Clinical trials also support the efficiency of
betaine in preventing disease progression (24,28).
Vitamins B6 and B12 are the essential cofactors in methionine
metabolism. Combined treatment with betaine, and vitamins B6 and
B12 can effectively reduce the deleterious effects of homocysteine
accumulation. The patient described herein also exhibited a notable
improvement following drug administration.
In conclusion, the typical clinical presentation of
MTHFR deficiency includes neurological deterioration and vascular
disorders. The severity also varies, depending on the type of
mutation and the extent of enzyme activity defects. To the best of
our knowledge, the present study is the first to report
hydrocephalus as the main manifestation of MTHFR deficiency. These
findings not only expand the mutation spectrum of severe MTHFR
deficiency, but also highlight the necessity of newborn metabolic
screening and genetic consulting. The majority of the sequelae can
be prevented if patients are diagnosed at an early stage and are
effectively treated. Genetic testing can also provide prenatal
guidance for couples who have a relevant family history.
Supplementary Material
Results of laboratory tests before
surgery and treatment.
Results of laboratory tests after
surgery and treatment.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Beijing Municipal
Science and Technology Commission (grant no. Z201100005520061) and
the Beijing Science and Technology Program (grant no.
2201100005520061).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The original data have been submitted to the NCBI-SRA
database with the submission no. PRJNA801899 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA801899).
Authors' contributions
YD, QW and CXG contributed to the acquisition,
analysis and interpretation of the data; they made substantial
contributions to the conception and design of the work, and
supervised and substantially revised this work. All authors had
equal contributions, equal participation and same rights to this
article. QW and CXG confirm the authenticity of all the raw data.
All authors have read and agreed to the published version of the
manuscript.
Ethics approval and consent to
participate
The Ethics Committee of Beijing Children's Hospital,
Capital Medical University, National Centre for Children's Health
granted approval for this study with the decision no. 2020-k-180
from 20. 07. 2021.
Patient consent for publication
Consent for participation was granted by the
patient's parents and is part of the personal observation
sheet.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McCully KS: Vascular pathology of
homocysteinemia: Implications for the pathogenesis of
arteriosclerosis. Am J Pathol. 56:111–128. 1969.PubMed/NCBI
|
|
2
|
Brustolin S, Giugliani R and Félix TM:
Genetics of homocysteine metabolism and associated disorders. Braz
J Med Biol Res. 43:1–7. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mudd SH, Skovby F, Levy HL, Pettigrew KD,
Wilcken B, Pyeritz RE, Andria G, Boers GH, Bromberg IL and Cerone
R: The natural history of homocystinuria due to cystathionine
beta-synthase deficiency. Am J Hum Genet. 37:1–31. 1985.PubMed/NCBI
|
|
4
|
Kuo HK, Sorond FA, Chen JH, Hashmi A,
Milberg WP and Lipsitz LA: The role of homocysteine in multisystem
age-related problems: A systematic review. J Gerontol A Biol Sci
Med Sci. 60:1190–1201. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Geng B, Chang L, Du JB and Tang CS: A new
strategy to treat hyperhomocysteinemia. Beijing Da Xue Xue Bao Yi
Xue Ban. 37:215–219. 2005.PubMed/NCBI(In Chinese).
|
|
6
|
Carson NA and Neill DW: Metabolic
abnormalities detected in a survey of mentally backward individuals
in Northern Ireland. Arch Dis Child. 37:505–513. 1962.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gerritsen T, Vaughn JG and Waisman HA: The
identification of homocystine in the urine. Biochem Biophys Res
Commun. 9:493–496. 1962.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mudd SH, Finkelstein JD, Irreverre F and
Laster L: Homocystinuria: An enzymatic defect. Science.
143:1443–1445. 1964.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mudd SH, Levy HL, Abeles RH and Jennedy JP
Jr: A derangement in B 12 metabolism leading to homocystinemia,
cystathioninemia and methylmalonic aciduria. Biochem Biophys Res
Commun. 35:121–126. 1969.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Baethmann M, Wendel U, Hoffmann GF,
Göhlich-Ratmann G, Kleinlein B, Seiffert B, Blom H and Voit T:
Hydrocephalus internus in two patients with
5,10-methylenetetrahydrofolate reductase deficiency.
Neuropediatrics. 31:314–317. 2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guenther BD, Sheppard CA, Tran P, Rozen R,
Matthews RG and Ludwig ML: The structure and properties of
methylenetetrahydrofolate reductase from escherichia coli suggest
how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol.
6:359–365. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Shahzad K, Hai A, Ahmed A, Kizilbash N and
Alruwaili J: A structured-based model for the decreased activity of
Ala222Val and Glu429Ala methylenetetrahydrofolate reductase (MTHFR)
mutants. Bioinformation. 9:929–936. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Leclerc D, Sibani S and Rozen R: Molecular
biology of methylenetetrahydrofolate reductase (MTHFR) and overview
of mutations/polymorphisms. MTHFR polymorphisms and disease: CRC
Press; 2005. p.15-3.
|
|
14
|
Buratti E, Chivers M, Královicová J,
Romano M, Baralle M, Krainer AR and Vorechovsky I: Aberrant 5'
splice sites in human disease genes: Mutation pattern, nucleotide
structure and comparison of computational tools that predict their
utilization. Nucleic Acids Res. 35:4250–4263. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Burda P, Schäfer A, Suormala T, Rummel T,
Bürer C, Heuberger D, Frapolli M, Giunta C, Sokolova J, Vlášková H,
et al: Insights into severe 5,10-methylenetetrahydrofolate
reductase deficiency: Molecular genetic and enzymatic
characterization of 76 patients. Hum Mutat. 36:611–621.
2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Broomfield A, Abulhoul L, Pitt W, Jameson
E and Cleary M: Reversal of respiratory failure in both neonatal
and late onset isolated remethylation disorders. JIMD Rep.
16:51–56. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Richard E, Desviat LR, Ugarte M and Pérez
B: Oxidative stress and apoptosis in homocystinuria patients with
genetic remethylation defects. J Cell Biochem. 114:183–191.
2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Knowles L, Morris AA and Walter JH:
Treatment with mefolinate (5-Methyltetrahydrofolate), but not folic
acid or folinic acid, leads to measurable 5-Methyltetrahydrofolate
in cerebrospinal fluid in methylenetetrahydrofolate reductase
deficiency. JIMD Rep. 29:103–107. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Froese DS, Huemer M, Suormala T, Burda P,
Coelho D, Gueant JL, Landolt MA, Kozich V, Fowler B and Baumgartner
MR: Mutation update and review of severe methylenetetrahydrofolate
reductase deficiency. Hum Mutat. 37:427–438. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tonetti C, Saudubray JM, Echenne B,
Landrieu P, Giraudier S and Zittoun J: Relations between molecular
and biological abnormalities in 11 families from siblings affected
with methylenetetrahydrofolate reductase deficiency. Eur J Pediatr.
162:466–475. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kristin E, Bearden D, Watkins D, Hyland K,
Rosenblatt DS and Ficicioglu C: Severe 5,
10-methylenetetrahydrofolate reductase deficiency and two MTHFR
variants in an adolescent with progressive myoclonic epilepsy.
Pediatr Neurol. 51:266–270. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Prasad AN, Rupar CA and Prasad C:
Methylenetetrahydrofolate reductase (MTHFR) deficiency and
infantile epilepsy. Brain Dev. 33:758–769. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Algassim N, Alfadhel M, Nashabat M and
Eyaid W: Clinical presentation of seven patients with
Methylenetetrahydrofolate reductase deficiency. Mol Genet Metab
Rep. 25(100644)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tsuji M, Takagi A, Sameshima K, Iai M,
Yamashita S, Shinbo H, Furuya N, Kurosawa K and Osaka H:
5,10-Methylenetetrahydrofolate reductase deficiency with
progressive polyneuropathy in an infant. Brain Dev. 33:521–524.
2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Greitz D: Radiological assessment of
hydrocephalus: New theories and implications for therapy.
Neuroradiol J. 19:475–495. 2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zittoun J: Congenital errors of folate
metabolism. Baillieres Clin Haematol. 8:603–616. 1995.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lossos A, Teltsh O, Milman T, Meiner V,
Rozen R, Leclerc D, Schwahn BC, Karp N, Rosenblatt DS, Watkins D,
et al: Severe methylenetetrahydrofolate reductase deficiency:
Clinical clues to a potentially treatable cause of adult-onset
hereditary spastic paraplegia. JAMA Neurol. 71:901–904.
2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim YI: 5,10-methylenetetrahydrofolate
reductase polymorphisms and pharmacogenetics: A new role of single
nucleotide polymorphisms in the folate metabolic pathway in human
health and disease. Nut Rev. 63:398–407. 2005.PubMed/NCBI View Article : Google Scholar
|