Introduction
Cerebrovascular disease-related stroke affects ≤300
of every 100,000 members of the Chinese population (1) and the most common form of stroke,
ischemic stroke, remains a leading cause of mortality and long-term
disability in aging patients (2).
Over the course of as little as 30 min, ischemia begins to produce
oxygen-glucose deprivation (OGD)-induced neuronal apoptosis that
may progress rapidly with increasing ischemia duration (3). Thus, the primary goal of the majority
of contemporary medicines for stoke is the mediation of OGD-induced
neuronal apoptosis during cerebral ischemia/reperfusion (4). Acetylpuerarin is a novel compound
that has been reported to effectively attenuate the morphological
changes leading to apoptosis in hippocampal cells following
cerebral ischemia/reperfusion, with potential benefits even when
applied a number of hours following the initial ischemic event
(5). Unlike the extremely small
treatment window (~3 h) of current United States Food and Drug
Administration-approved therapies for stroke, including tissue
plasminogen activator (6–8), acetylpuerarin may provide an
alternative for stroke treatment at advanced stages (9). Prior to clinical implementation, a
greater understanding of the dose-effects and mechanistic action of
acetylpuerarin on hippocampal cell apoptosis during
ischemia/reperfusion is required.
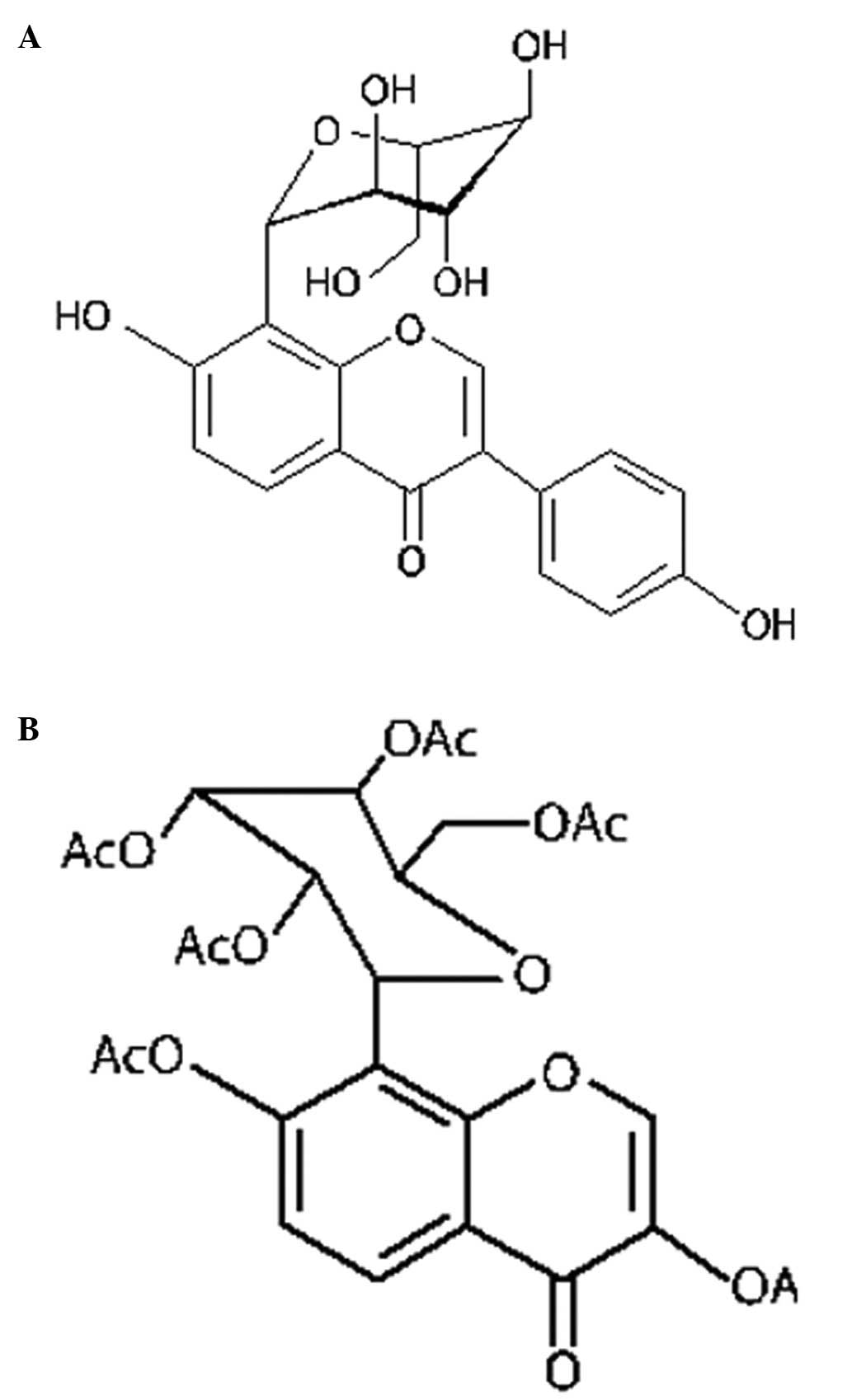
Acetylpuerarin is a modern synthetic derivative of
puerarin (daidzein-8-C-glucoside) with higher lipid solubility than
the naturally occurring compound, allowing it to better penetrate
the blood-brain barrier (Fig. 1).
Puerarin is an isoflavonoid compound used for treating
cardiovascular disorders derived from the Chinese medical herb
Radix puerariae, known as ‘Ge Gen’, from the root of the
kudzu plant. Treatment of puerarin has been reported to effectively
reduce symptoms following acute ischemic stroke with few side
effects in a number of preliminary in vivo and in
vitro studies (10–12). A number of studies have reported
that acetylpuerarin exerts protective effects against
ischemia-reperfusion injury in the hippocampus by mediating the
cascade of events leading to OGD-induced damage (5,13,14).
Thus, acetylpuerarin may be capable of mediating the degree of
irreversible injury caused by neuronal cell death following
OGD/reperfusion (OGD/R), a complex, synergistic process in which
proliferation of reactive oxygen species and free radicals,
induction of tumor necrosis factor-α (TNF-α) and other inflammatory
factors, release of cytokines, excitotoxicity, unbalanced activites
of caspase-8 and -3, and abnormalities in calcium levels contribute
to apoptosis (9). The
neuroprotective effects of puerarin and acetylpuerarin on cerebral
ischemic damage are associated with the inhibition of inflammatory
factor TNF-α and apoptosis (active caspase-3), free radical
scavenging or antioxidative activity (15). However, the full mechanism of
acetylpuerarin action by dosage has not been characterized.
The current study examines the detailed mechanisms
underlying the neuroprotective effects of acetylpuerarin in
hippocampal neurons by dose, including the effects on caspase-8 and
-3 activities associated with apoptosis, Fas-ligand (Fas-L),
Fas-associated death domain (FADD) and TNF-α inflammatory factors
and neuronal cell viability in OGD/R-induced hippocampal neurons.
The observations of the resultant injury and neuroprotective
mechanisms lead to an improved mechanistic understanding of
acetylpuerarin that provides an essential basis for potential
clinical applications of this drug in ischemic stroke patients.
Materials and methods
Animal subjects and study design
Hippocampal cells were isolated from Wistar rat
embryos (day 18, E18) purchased from the Laboratory Animal Center
of the Shandong University (Shandong, China; Grade II, Certificate
No. 20021015) and cultured for 8 days.
Cells were subjected to 3 h OGD treatment followed
by reperfusion for 12, 24 or 36 h. For each time interval, a group
of cells was left untreated (OGD/R groups) and treated with 0.1,
0.4 and 1.6 μM acetylpuerarin (OGD/R+acetylpuerarin) [13 groups:
control (I); OGD/R 12, 24 or 36 h -only (II, VI, X) and OGD/R 12,
24 or 36 h +0.1, 0.4 and 1.6 μM acetylpuerarin (III–V, VII–IX and
XI–XIII), respectively]. The effects and mechanisms of
acetylpuerarin on hippocampal neuron apoptosis in OGD/R and
normoxic cells were observed. The study was approved by Shandong
University Ethical Committee and animals were used in accordance
with all relevant guidelines for animal use and care.
Primary embryonic hippocampal neuron
cultures
Hippocampi were dissected from E18 rat brains and
sampled primary embryonic hippocampal neuron cells were seeded onto
96-well tissue culture plates coated with poly-D-Lysine
(Sigma-Aldrich, St. Louis, MO, USA) at 5×105 cells/ml or
on 6-well tissue culture plates at 1×106 cells/ml, as
previously described (16).
Neurons were cultured in a medium containing neurobasal medium
(Gibco-BRL, Carlsbad, CA, USA), 2% B27 (Gibco-BRL), 2 mM glutamine
(Sigma -Aldrich), 50 U/ml penicillin G and 50 μg/ml streptomycin
(Gibco-BRL) in a humidified incubator with 5% CO2 at
37°C. Half of the cultured medium was changed at 3 day intervals
and cells were sampled for use in the following experiments at 8
days.
OGD/R treatment
Cells in the OGD/R-only and OGD/R+acetylpuerarin
groups underwent OGD, as previously described (17). Briefly, experimental cell culture
medium was replaced with glucose-free Hanks’ balanced salt solution
(Invitrogen Life Technologies, Carlsbad, CA, USA) and cells were
placed in an anaerobic chamber (95% N2/5% CO2) for 180
min, following which OGD was terminated by removing the cultures
from the anaerobic chamber, replacing deoxygenated and glucose-free
Hanks’ BSS with pre-OGD culture medium without B27 supplement and
returning cells to normoxic conditions. Reperfusion was conducted
for 12, 24 or 36 h. Control group cells were similarly incubated in
Hanks’ BSS with 10 mM glucose in a normoxic incubator.
Acetylpuerarin administration
Acetylpuerarin was dissolved in DMSO ≤0.1% and
administered at the onset of the ischemic period in concentrations
of 0.1, 0.4 and 1.6 μM in the OGD/R+acetylpuerarin 9 groups III–V,
VII–IX and XI–XIII, respectively, according to the dosage
concentrations suggested by Liu et al(5). Each experiment was performed in
cultures from three E18 rats and was repeated in triplicate.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Upon completion of reperfusion, 20 μl/well of MTT
was added at a final concentration of 0.5 mg/ml. Cultures were
allowed to incubate at 37°C and 5% CO2 for 4 h. The
culture medium was aspirated and replaced with 150 μl/well of DMSO.
The optical density (OD) was measured at a test wavelength of 492
nm in a microplate reader (Thermo MK3; Thermo Fisher Scientific,
Shanghai, China). The following formula (18) was used to calculate cell viability
as percentage: (absorbance of treated cells / absorbance of normal
cells) × 100%.
4′,6-diamidino-2-phenylindole (DAPI)
staining
Upon completion of reperfusion, cultured neurons
were incubated with 5 μg/ml fluorescent DNA-binding dye DAPI
(Sigma-Aldrich) at 37°C for 1 min. Staining solution was removed
and apoptotic cells were visualized by fluorescence microscopy
(Olympus DP-72; Olympus Corporation, Tokyo, Japan). Apoptotic cells
were morphologically identified by cytoplasmic or nuclear
shrinkage, chromatin condensation and DNA fragmentation. The
percentage of apoptotic cells was determined in 5 randomly selected
fields of ~500 neurons per field (magnification, ×100).
Experimental data was pooled from three coverslips and each
experiment was conducted three times.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
Neuronal apoptosis was detected using a TUNEL assay
24 h following OGD initiation. TUNEL-positive (apoptotic) cells
appeared dark brown. These cells were counted in 5 randomly
selected microscopic fields (Olympus DP-71; Olympus Corporation)
(magnification, ×100). Apoptotic index (AI) was calculated as:
(apoptotic neurons / total neurons) × 100%.
Western blot analysis for Fas-L, FADD and
TNF-α expression
Total protein (50 μg) was extracted from samples
from each group, separated by 12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis and blotted onto polyvinylidene
fluoride membranes (Beijing Solarbio Science and Technology Co.
Ltd., Beijing, China). Membranes were probed with primary
antibodies against Fas-L (1:400), FADD (1:500) and TNF-α (1:300;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and
peroxidase-conjugated secondary antibody (1:5,000; ZSGB-Bio,
Beijing, China). β-actin (1:2,000; ZSGB-Bio) was used as a control.
Band intensities were quantitated using AlphaEaseFC software
(Genetic Technologies, Miami, FL, USA).
Caspase-8 and -3 activity assay
Caspase-8 and -3 activities were assessed using
FLICE/caspase-8 and caspase-3/CPP32 colorimetric assay kits,
respectively, (BioVision, Inc., Milpitas, CA, USA), according to
the instructions suggested by the manufacturer. Briefly, 100 or 50
μg of total protein were incubated with 10 mM dithiothreitol and
IETD-pNA for caspase-8 and -3 activity quantification (caspase-8
substrate; 200 μM final concentration) or MDEVD-pNA (caspase-3
substrate; 200 μM final concentration) at 37°C for 2 h. Samples
were determined at 405 nm in a microplate reader (Therm MK3;
Finland). The final reading did not include the background reading
of cell lysates and buffers. Enzymatic activity was expressed in
arbitrary units of OD per mg protein.
Statistical analysis
All data were analyzed using SPSS version 16 (SPSS,
Inc., Chicago, IL, USA) and expressed as means ± SEM. Between group
and multiple group differences were analyzed using Student’s
t-tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
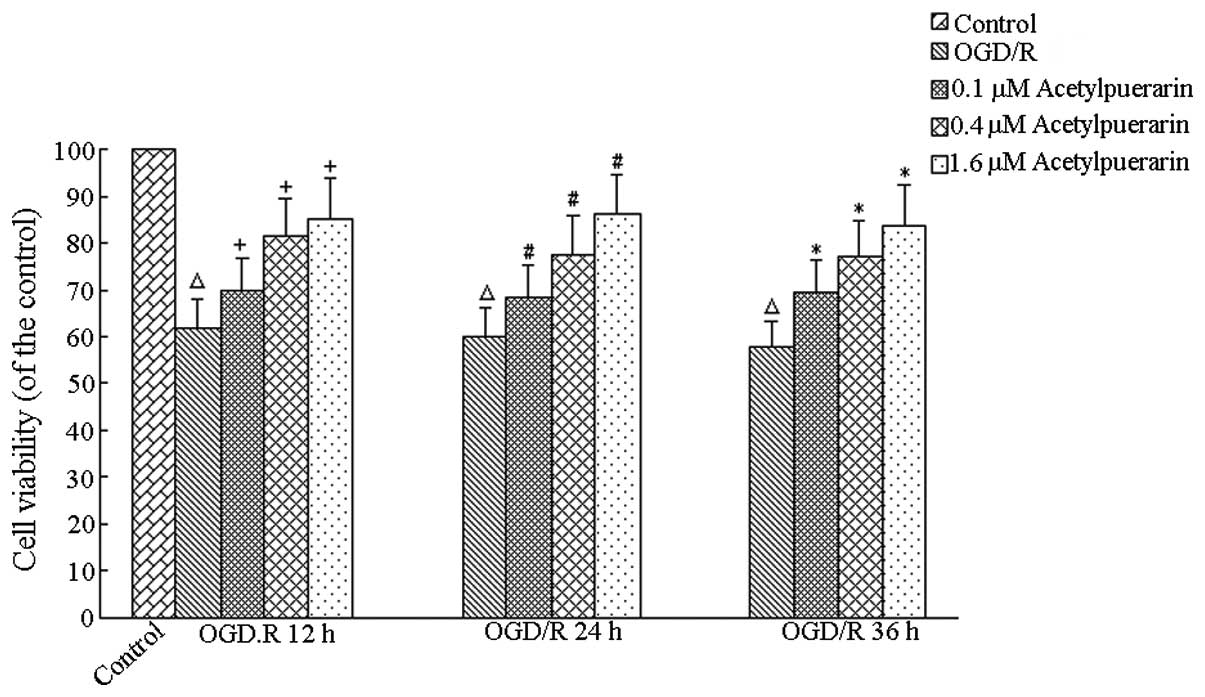
Acetylpuerarin increases neuron viability
following OGD/R
The MTT assay revealed no significant changes in
viability in the control and normoxic control groups treated with
acetylpuerarin treatment at all times (P>0.05, data not shown).
Compared with the OGD/R-only group, cell survival was 61.94±2.73%
(OGD/R, 12 h), 60.61±3.29% (OGD/R, 24 h) and 57.77±0.66% (OGD/R, 36
h). Compared with the control group, OGD/R+acetylpuerarin groups
treated with acetylpuerarin doses of 0.1, 0.4 and 1.6 μM increased
cell survival and viability by 69.93±2.28%, 81.49±2.13% and
85.28±2.38% at 12 h, respectively; 68.59±3.02%, 77.85±2.84% and
85.64±4.39% at 24 h, respectively and 69.70±1.70%, 77.21±3.21% and
83.90±2.12% at 36 h, respectively (P<0.05; Fig. 2). Higher acetylpuerarin
concentrations enhanced neuron survival more efficiently in a
concentration-dependent manner. Similar results were obtained by
morphological analysis. The control group exhibited round cell
bodies with clear edges and fine dendritic network. OGD/R groups
exhibited a decreasing number of neurons, shrinkage of cell bodies
and disruption of dendritic networks. Acetylpuerarin mitigated the
morphological manifestations of cell damage (data not shown).
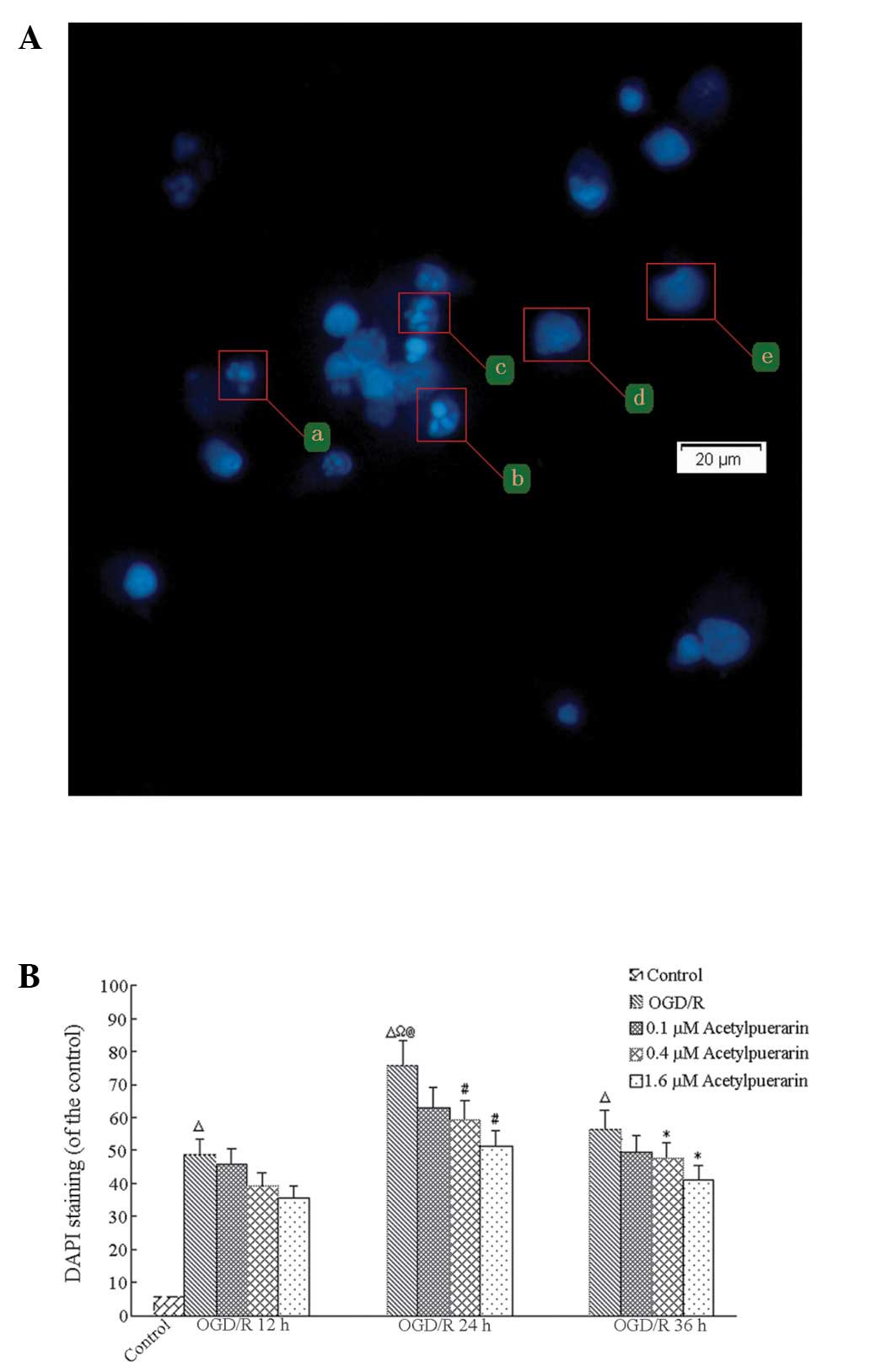
Acetylpuerarin reduces apoptosis
following ODG/R
DAPI staining revealed that <8% of control-only
cells showed signs of apoptosis at any time point; however, OGD/R
treatment resulted in significant time-dependent increases in
apoptotic cell numbers, peaking at 24 h (P<0.01; Fig. 3). In the OGD/R-only group at 24 h,
75.85±7.59% of remaining cells showed signs of apoptosis.
Comparatively, OGD/R+acetylpuerarin groups treated with 0.4 μM
acetylpuerarin doses exhibited reductions in DAPI positive neurons
to 63.01±7.35%, 59.06±5.98% and 51.05±5.98%, respectively (all
P<0.05). Thus, increasing the concentration of acetylpuerarin
exhibited no significant effects on apoptosis rates in cells
treated with OGD/R following 24 h reperfusion (all P>0.05).
These results were confirmed with TUNEL staining,
indicating that 5% of the control-only group cells exhibited
apoptosis under basal conditions. TUNEL-positive cells were
markedly increased in the OGD/R-only group at 24 h and the AI was
68.79±10.01%. Acetylpuerarin treatment decreased the number of
TUNEL-positive cells to 52.30±9.73%, 46.08±13.42% and 39.04±7.29%
in all OGD/R+acetylpuerarin groups treated with 24 h reperfusion
(all P<0.05; Table I).
 | Table IEffects of acetylpuerarin on AI (24-h
reperfusion). |
Table I
Effects of acetylpuerarin on AI (24-h
reperfusion).
| Group | Acetylpuerarin
concentration, μM | AI, % |
|---|
| Control | 0 | 4.57±2.48 |
| OGD/R | 0 | 68.79±10.06a |
|
OGD/R+acetylpuerarinb |
| VII | 0.1 | 52.30±9.73c |
| VIII | 0.4 | 46.08±13.42c |
| IX | 1.6 | 39.04±7.29c |
Acetylpuerarin inhibited OGD/R-induced
caspase-8 and -3 activation
Caspase-8 was significantly increased in hippocampal
neurons following OGD/R treatments at 24 h and acetylpuerarin
treatment led to a concentration-dependent decrease in the
expression of enzymatic activity of caspase-8 (Table II). As caspase-3 is considered to
be a prototypical caspase and a key effector of programmed cell
death (19), enzymatic activity of
caspase-3 was also determined. As shown in Table II, exposure to OGD significantly
enhanced caspase-3 proteolytic activity by 24 h, while
acetylpuerarin significantly diminished the activation of caspase-3
activity in a concentration-dependent manner.
| Table IIEffects of acetylpuerarin on caspase-3
and caspase-8 activities (24-h reperfusion). |
Table II
Effects of acetylpuerarin on caspase-3
and caspase-8 activities (24-h reperfusion).
| | OD, mg |
|---|
| |
|
|---|
| Group | Acetylpuerarin
concentration, μM | Caspase-3 | Caspase-8 |
|---|
| OGD/R | 0 | 4.36±0.26 | 4.27±0.29 |
| OGD/R +
acetylpuerarina |
| VII | 0.1 | 3.78±0.13b | 3.40±0.28c |
| VIII | 0.4 | 3.14±0.26b | 2.80±0.32c |
| IX | 1.6 | 2.28±0.27b | 2.55±0.23c |
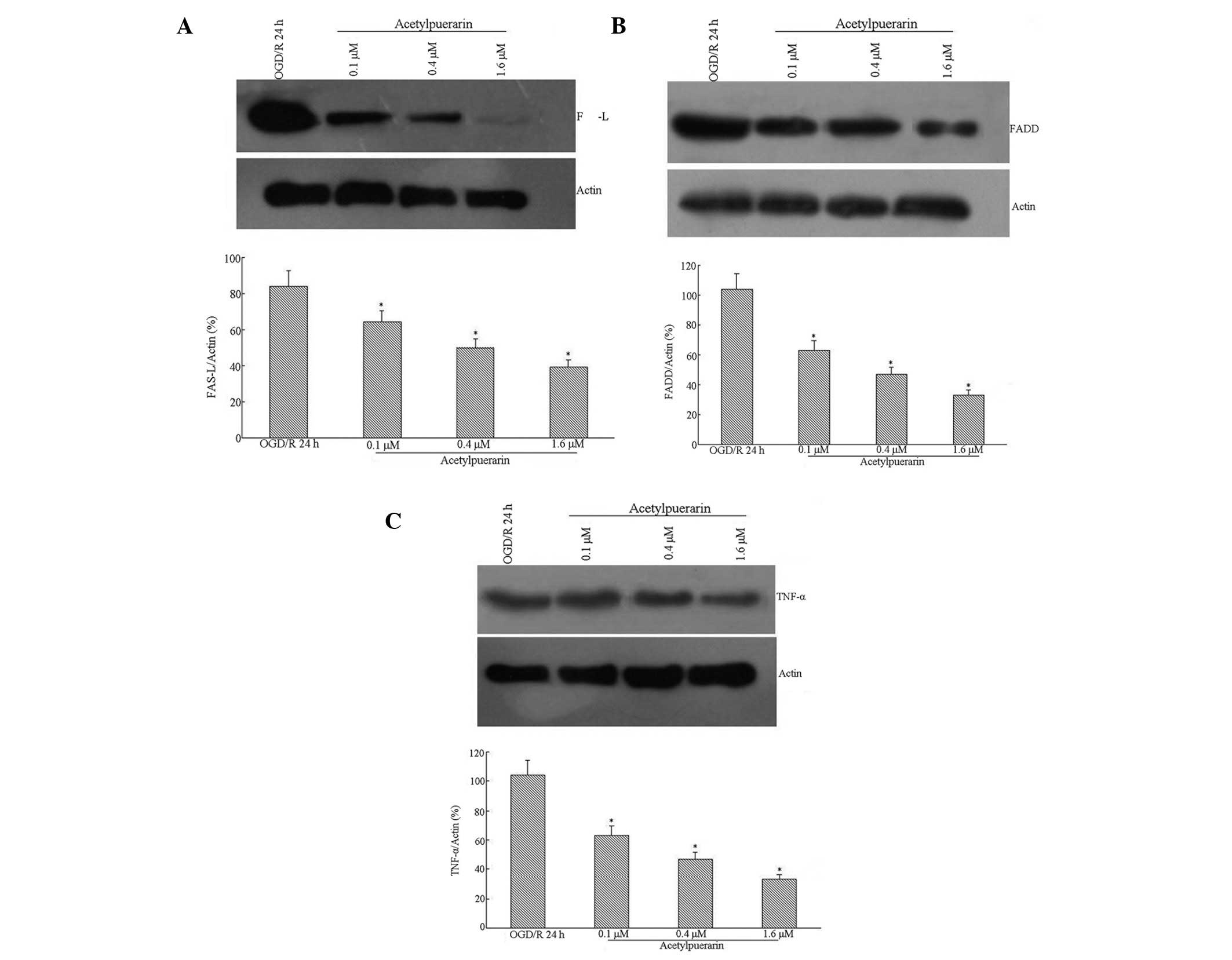
Acetylpuerarin inhibited OGD/R-induced
Fas-L, FADD and TNF-α protein expression
Western blot analysis indicated that Fas-L levels
were induced by OGD treatment in hippocampal neurons by 24 h
post-reperfusion (Fig. 4A). This
Fas-L induction was significantly attenuated by acetylpuerarin
treatment in a concentration-dependent manner. High levels of FADD
protein were detected in neurons following OGD/R 24 h. However,
FADD expression was significantly downregulated by acetylpuerarin
(Fig. 4B). OGD/R-induced apoptosis
was accompanied by a significant increase in TNF-α (Fig. 4C). Western blot analysis also
revealed that acetylpuerarin inhibited TNF-α expression induced by
OGD/R 24 h in a dose-dependent manner.
Discussion
A model of ischemic OGD/R in hippocampal neurons was
used to assess the potential protective role of acetylpuerarin in
ischemic injury. Acetylpuerarin not only increased neuron
viability, but also decreased DAPI and TUNEL-positive cells,
caspase-8 and -3 activity and Fas-L protein expression, FADD and
TNF-α induced by OGD/R. Thus, acetylpuerarin may be capable of
mediating the protection to hippocampal cells by decreasing
neuronal apoptosis during acute ischemia and may be useful in
clinical treatment development, pending further assessment of the
clinical effects of the drug.
The cell-extrinsic apoptosis pathway Fas→FADD→
caspase-8→caspase-3 is activated by binding of pro-apoptotic
ligands, including Fas/CD95L (TNFSF6) or Apo2L/TRAIL (TNFSF10) to
their cognate death domain-containing receptors on the surface of
target cells (20–25). In a previous study, acetylpuerarin
was shown to alleviate morphological damage and increase neuron
viability, consistent with the current findings (5). The current study is also consistent
with reports indicating that TNF-α activation, followed by the
inhibition of subsequent inflammatory responses, apoptosis
formation (active caspase-3) and neutrophil activation are central
to the neuroprotective action of puerarin, which may be
hypothesized to be similar to those of its derivative,
acetylpuerarin (15). Furthermore,
the patterns of cell loss following OGD/R are likely to depend on
the severity of injury (19), a
hypothesis supported by the current observations that changes in
DAPI and TUNEL-positive cells (apoptosis) at 24 h occurred
independent of acetylpuerarin treatment concentrations. Thus,
acetylpuerarin acts on several of the key mechanisms that affect
apoptosis rate during cerebral ischemia.
In the case of clinical ischemic stroke, the total
ischemic lesion consists of a penumbral field, which is the most
common site of cell death and a core that is the central site to
necrosis (20). While rapid damage
to the core may not be easily avoided (21), subsequent cell death in the
penumbral field over following days and weeks may be mediated by
medications, which prevent apoptosis (22). Increasing inflammatory cytokine
levels following OGD/R are mechanistically associated with cell
death following the ischemic episode. Specifically, upregulation of
OGD-induced TNF-α may increase binding to the TNF receptor I
(TNFRI) and TNFRII, inducing receptor oligomerization and
upregulating recruitment of other cytoplasmic signaling proteins
(23). Since acetylpuerarin
effectively and consistently reduces hippocampal neuron apoptosis
in the current study and in a previous study (5), it may therefore, present an effective
neuroprotection strategy, meriting further clinical investigation.
In addition, the current study demonstrated that Fas and its only
endogenous ligand, Fas-L, factors which are linked to the reduction
of apoptosis signaling in ischemic stroke patients, were
downregulated, (24,25). Thus, acetylpuerarin may have
additional downstream effects on the TNFRI-associated death domain
protein (TRADD). Notably, reductions in the signaling molecules Fas
and FADD, and reduced subsequent activation of caspase-8, further
suggested that TRADD was downregulated by acetylpuerarin treatment
following OGD (23,26). Thus, acetylpuerarin may confer
neuroprotection by mediating OGD-induced TNF-α and thus,
downregulating a number of downstream inflammatory signaling
molecules, ultimately reducing apoptosis.
Since acetylpuerarin treatment inhibited capsase-8
activation, it may also reduce mitochondrial-dependent and
-independent apoptosis, characterized, respectively, by conversion
of the inactive cytosolic form of Bid, a ‘BH3-only’ pro-apoptosis
Bcl-2 family protein, into a truncated fragment (tBid), followed by
mitochondrial translocation (27)
and activation of caspase-3, the ‘executioner caspase’ by caspase-8
(28). Truncated tBid also appears
to have increased affinity for anti-apoptotic Bcl-2, as well as for
Bax/Bak-like proteins, causing Bax/Bak oligomerization and
cytochrome c release. Released cytochrome c binds to
apoptotic protease-activating factor-1 and in turn triggers
caspase-9 activation. This apical caspase activates one or more
downstream caspases, including caspases-3, -6 and -7, causing
apoptotic cell death (29). It has
also been suggested that activation of the apoptosis-triggering
enzyme caspase-8 reaches a maximum at 6 h following the OGD/R
insult (19), consistent with the
current increased caspase-8 spectrophotometric detection assay
findings. Thus, by inhibiting caspase-8 and -3 activities,
acetylpuerarin prevents neuronal apoptosis through two distinct and
well-described mechanisms.
In considering these results, it is important to
consider that in vivo results may vary and that systemic
side effects may be associated with treatment. Thus, the effects of
acetylpuerarin in vivo merit further clinical investigation.
Since the effects of acetylpuerarin were examined over a relatively
short 24 h period following reperfusion, it is also important to
determine, in future studies, whether acetylpuerarin is capable of
mediating neuronal death and promoting functional neurological
recovery over longer periods, a current topic of research in our
laboratory.
The current findings demonstrated that
acetylpuerarin treatment induced dose-dependent neuroprotection
against OGD/R-induced neuronal cell death by inhibiting Fas-L, FADD
and TNF-α pathway-mediated apoptosis. Furthermore, caspase-3 and -8
activities were inhibited, providing further evidence that
acetylpuerarin impacts a number of pathways known to induce
apoptosis following OGD/R. Thus, acetylpuerarin may be a promising
future candidate for the clinical treatment of acute ischemic
stroke, pending further mechanistic verification and clinical
investigations.
Acknowledgements
This study was supported by a grant from the Nature
Science Foundation of Shandong province (no. ZR2010HM132).
References
|
1
|
Truelsen T, Begg S and Mathers C: The
global burden of cerebrovascular disease. Global Burden of Disease
2000. World Health Organization; Geneva: 2000
|
|
2
|
Zhao DX, Feng J, Cong SY and Zhang W:
Association of E-selectin gene polymorphisms with ischemic stroke
in a Chinese Han population. J Neurosci Res. 90:1782–1787. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wise-Faberowski L, Raizada MK and Sumners
C: Oxygen and glucose deprivation-induced neuronal apoptosis is
attenuated by halothane and isoflurane. Anesth Analg. 93:1281–1287.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu Y, Zhang J, Ma B, et al: Glycine
attenuates cerebral ischemia/reperfusion injury by inhibiting
neuronal apoptosis in mice. Neurochem Int. 61:649–658. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu R, Wei XB and Zhang XM: Effects of
acetylpuerarin on hippocampal neurons and intracellular free
calcium subjected to oxygen-glucose deprivation/reperfusion in
primary culture. Brain Res. 1147:95–104. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
No authors listed. Tissue plasminogen
activator for acute ischemic stroke. The National Institute of
Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl
J Med. 333:1581–1587. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elijovich L and Chong JY: Current and
future use of intravenous thrombolysis for acute ischemic stroke.
Curr Atheroscler Rep. 12:316–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wardlaw JM, Warlow CP and Counsell C:
Systematic review of evidence on thrombolytic therapy for acute
ischaemic stroke. Lancet. 350:607–614. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hou L, Wei XB, Li XM, Zhong Y, Zuo CX and
Zhang XM: Protective effects of acetylpurarin on focal brain
ischemia-reperfusion injury in rats. Chin Pharm J. 42:1469–1472.
2007.(In Chinese).
|
|
10
|
Tan Y, Liu M and Wu B: Puerarin for acute
ischemic stroke. Cochrane Database Syst Rev.
2008:CD0049552008.PubMed/NCBI
|
|
11
|
Wu B, Liu M, Liu H, et al: Meta-analysis
of traditional Chinese patent medicine for ischemic stroke. Stroke.
38:1973–1979. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu X, Zhang S, Zhang L, Yan W and Zheng X:
The neuroprotection of puerarin against cerebral ischemia is
associated with the prevention of apoptosis in rats. Planta Med.
71:585–591. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hou L, Zhang XM and Wei XB: Protective
effects of Compounds N-2211 on focal brain ischemia-reperfusion
injury in rats. Qilu Pharm Affairs. 23:52–54. 2004.(In
Chinese).
|
|
14
|
Li XM, Wei XB, Zhang XM, Hou L, Zhong Y
and Zuo CX: Effect of acetylpuerarin on NO level and NOS activity
in brain tissue and serum of focal cerebral ischemia reperfusion
injury rats. Chin Pharm J. 11:829–832. 2005.
|
|
15
|
Lou HY, Wei XB, Zhang B, Sun X and Zhang
XM: Hydroxyethylpuerarin attenuates focal cerebral
ischemia-reperfusion injury in rats by decreasing TNF-alpha
expression and NF-kappaB activity. Yao Xue Xue Bao. 42:710–715.
2007.PubMed/NCBI
|
|
16
|
Yao YY, Liu DM, Xu DF and Li WP: Memory
and learning impairment induced by dexamethasone in senescent but
not young mice. Eur J Pharmacol. 574:20–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ziu M, Fletcher L, Rana S, Jimenez DF and
Digicaylioglu M: Temporal differences in microRNA expression
patterns in astrocytes and neurons after ischemic injury. PLoS One.
6:e147242011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sreelatha A, Jeyachitra A and Padma PR:
Antiproliferation and induction of apoptosis by Moringa
oleifera leaf extract on human cancer cells. Food Chem Toxicol.
49:1270–1275. 2011. View Article : Google Scholar
|
|
19
|
Badiola N, Malagelada C, Llecha N, et al:
Activation of caspase-8 by tumour necrosis factor receptor 1 is
necessary for caspase-3 activation and apoptosis in oxygen-glucose
deprived cultured cortical cells. Neurobiol Dis. 35:438–447. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao H, Takasawa R, Fukuda K, et al: DNA
fragmentation in ischemic core and penumbra in focal cerebral
ischemia in rats. Brain Res Mol Brain Res. 91:112–118. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barone FC and Feuerstein GZ: Inflammatory
mediators and stroke: new opportunities for novel therapeutics. J
Cereb Blood Flow Metab. 19:819–834. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang Y, Wu J, Keep RF, Hua Y, Hoff JT and
Xi G: Hypoxia-inducible factor-1alpha accumulation in the brain
after experimental intracerebral hemorrhage. J Cereb Blood Flow
Metab. 22:689–696. 2002. View Article : Google Scholar
|
|
23
|
Chen G and Goeddel DV: TNF-R1 signaling: a
beautiful pathway. Science. 296:1634–1635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu L, Kim JY, Koike MA, et al: FasL
shedding is reduced by hypothermia in experimental stroke. J
Neurochem. 106:541–550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ruan W, Lee CT and Desbarats J: A novel
juxtamembrane domain in tumor necrosis factor receptor superfamily
molecules activates Rac1 and controls neurite growth. Mol Biol
Cell. 19:3192–3202. 2008. View Article : Google Scholar
|
|
26
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome
c release from mitochondria in response to activation of
cell surface death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Varfolomeev EE and Ashkenazi A: Tumor
necrosis factor: an apoptosis JuNKie? Cell. 116:491–497. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garrido C, Gurbuxani S, Ravagnan L and
Kroemer G: Heat shock proteins: endogenous modulators of apoptotic
cell death. Biochem Biophys Res Commun. 286:433–442. 2001.
View Article : Google Scholar : PubMed/NCBI
|