Introduction
Traumatic brain injury (TBI) is the leading cause of
mortality in the young aged population and is a predominant reason
for hospital admission in modern life (1). Mechanical disruption of neurons
triggers a cascade of events leading to neuronal cell death
following TBI (2). Apoptosis has
been attributed to programmed neuronal cell death in TBI (3). Notably, previous studies have also
shown that autophagy is increased following TBI (4). Autophagy is an evolutionarily
conserved pathway that leads to the degradation of proteins and
entire organelles in cells undergoing stress (5). The increased LC3 immunostaining was
located predominantly in neurons 24 h post-TBI (6). However, few studies have addressed
how the autophagy pathway is regulated following traumatic
damage.
Previous studies demonstrated that astrocytes
release ATP, at least in part, by opening connexin 43 (CX43)
hemichannels (7). Connexins are a
family of proteins with dual channel functions (8) involved in forming gap junctions,
which are composed of two docked hemichannels linking the cytosol
of two neighboring cells. Gap junctions allow cell-to-cell passage
of ions and small molecules, including Ca2+, cyclic
adenosine monophosphate, inositol triphosphate, ATP, glutamate and
glucose. In addition, the intracellular condition and
phosphorylation state of Cx affect the intercellular permeability
via gap junctions (9).
Furthermore, TBI results in the rapid loss of astrocytes and
thereafter induces reactive astrocytes in the hippocampus (10). Glial fibrillary acidic protein
(GFAP)-positive astrocytes exist more extensively in
CX43+/+ than in CX43+/−
mice.
Gap junctions were shown to provide neurons, through
astrocytic gap junction channels, with energy-producing compounds,
such as ATP, glucose, glucose-6-phosphate and lactate (11). Conversely, gap junctions also
propagate death signals in astrocytic networks, which may affect
neuronal fate (12). Based on the
results of previous studies, it was hypothesized that CX43
regulates autophagy in TBI-induced damage. To confirm this
hypothesis, using the relatively selective CX43 inhibitor, the
present study aimed to determine whether astrocytic gap
junction-dependent modulation of neuronic LC3 expression occurs
under TBI conditions.
Materials and methods
Animals and TBI model
All experimental procedures were conducted in
accordance with the guidelines of the Chinese Council on Animal
Protection and were approved by the Hebei Medical University Animal
Care and Use Committee (Hebei, China). A total of 126 male
Sprague-Dawley rats (age, 12–16 weeks; weight, 350–375 g; Tangshan,
China) were used in the present study. The rats were housed with a
standard of 12 h light/dark cycle and access to water and food
ad libitum prior to and following surgery or sham operation.
The rat model of TBI was induced using a modified weight-drop
device (Tangshan Railway Vehicle Co., Ltd., Tangshan, China), as
described previously by Marmarou et al(13). Briefly, the rats were anaesthetised
with sodium pentobarbital (Nembutal 60 mg/kg). A midline incision
was made to expose the skull between bregma and lambda suture lines
and a steel disc (10 mm in diameter and 3 mm in thickness) was
adhered to the skull using dental acrylic (Tangshan Railway Vehicle
Co., Ltd., Tangshan, China). Animals were moved onto a foam
mattress underneath a weight-drop device where a weight of 450 g
fell freely through a vertical tube from 1.5 m onto the steel disc.
Sham-operated animals underwent the same surgical procedure without
weight-drop impact. Rats were housed in individual cages following
surgery and placed on heat pads (37°C) for 24 h to maintain normal
body temperature during the recovery period.
Groups and drug administration
The rats were randomly divided into the sham, TBI
and TBI treated with carbenoxolone (CBX) groups. Each sub-group was
composed of five rats and the rats were killed 3, 6, 24 or 48 h
following TBI. CBX (50 μg/kg body mass; Sigma-Aldrich, Yorba Linda,
CA, USA) was administered by right ventricle injection 30 min prior
to sham operation or TBI induction (14). For intracerebroventricular
injection, the animals were fixed in a stereotaxic apparatus
(RWD68025; RWD Life Science Co., Ltd., Shenzhen, China), a midline
incision was made in the skin and a small hole was induced in the
cranial region. Using a Hamilton syringe (RWD62201; RWD Life
science Co., Ltd.), CBX in 5 μl saline was injected into the right
cerebral ventricle according to the following coordinates: bregma:
AP −0.8 mm, L +1.6 mm (midline) and deep 3.4 mm form dura (15).
Western blot analysis
Western blot analysis was conducted as described
previously (16). Briefly, the
rats were deeply anesthetized and underwent an intracardiac
perfusion with 0.1 mol/l phosphate-buffered saline (PBS; pH 7.4).
The hippocampal CA1 was rapidly isolated, total proteins were
extracted and the protein concentration was determined by the
bicinchoninic acid reagent (Beijing Solarbio Science and Technology
Co., Ltd., Beijing, China) method. Samples were subjected to sodium
dodecyl sulfate polyacrylamide gel electrophoresis. Separated
proteins on the gel were transferred onto polyvinylidene fluoride
membranes (Roche Diagnostics, Mannheim, Germany). Blots were
blocked with 5% fat-free dry milk for 1 h at room temperature.
Subsequently, blots were incubated overnight at 4°C with the
following primary antibodies: Rabbit anti-p-CX43 polyclonal
antibodies, rabbit anti-LC3 polyclonal antibody and mouse
anti-β-actin monoclonal antibody (dilution, 1:500; Santa Cruz
Biotechnology, Inc.). The blots were then incubated with
horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse
IgG (dilution,1:5000; Cell Signaling Technology, Inc., Danvers, MA,
USA) for 2 h at room temperature. Subsequent to incubation with a
properly titrated secondary antibody, the immunoblot on the
membrane was visible following development with an enhanced
chemiluminescence (ECL) detection system (ChemiDoc XRS; Bio-Rad,
Hercules, CA, USA) and the densitometric signals were quantified
using an imaging program (Image Lab 4.1; Bio-Rad). Immunoreactive
bands of the protein expression were normalized to the intensity of
the corresponding bands for β-actin. The western blot analysis
results were analyzed with National Institutes of Health Image 1.41
software (Bethesda, MD, USA).
Immunofluorescence analyses
The brain tissues were fixed in 4% paraformaldehyde
for 24 h, and transferred to a 30% sucrose solution (0.1 M PBS, pH
7.4). When the tissues had sunk to the bottom of the solution,
sections, 200 μm apart from anterior to posterior hippocampus
(bregma −1.90 to −3.00) were made from TBI animal and embedded in
OCT. Frozen sections (15 μm) were sliced with a frozen slicer,
treated with 0.4% Triton X-100 for 10 min, and blocked in normal
donkey serum for 1 h. For double labeling, the frozen sections were
incubated with a mixture of rabbit anti-p-CX43 polyclonal antibody
(dilution, 1:100) and mouse anti-GFAP monoclonal antibody
(dilution, 1:100; Santa Cruz Biotechnology), or rabbit anti-LC3
polyclonal antibody (dilution, 1:100) and mouse anti-neuronal
nuclei (NeuN; dilution, 1:100; Santa Cruz Biotechnology) overnight
at 4°C. The following day, the sections were incubated with a
mixture of fluorescein-conjugated anti-rabbit IgG and anti-mouse
IgG (dilution 1:1000; Santa Cruz Biotechnology) for 2 h at 37°C in
the dark. Images were captured in a laser scanning confocal
microscope (Olympus Fluoview™ FV1000; Olympus, Tokyo Japan).
Primary antibodies were replaced with PBS in the negative control
group.
Statistical analysis
Experiments were repeated three times and similar
results were obtained. Statistical analysis was performed using the
SPSS 16.0 statistics software (SPSS, Chicago, IL, USA). Data were
expressed as the mean ± SE. Statistical analysis was performed
using analysis of variance, followed by the Student-Newman-Keuls
post hoc tests or Student’s t-test (two means comparison).
P<0.05 was considered to indicate a statistically significant
difference.
Results
General
There were no significant differences in body weight
or temperature between the TBI and sham-injured groups, and no
differences in injury levels among the 3-, 6-, 24- or 48-h TBI
groups.
P-CX43 colocalizes with astrocyte
markers
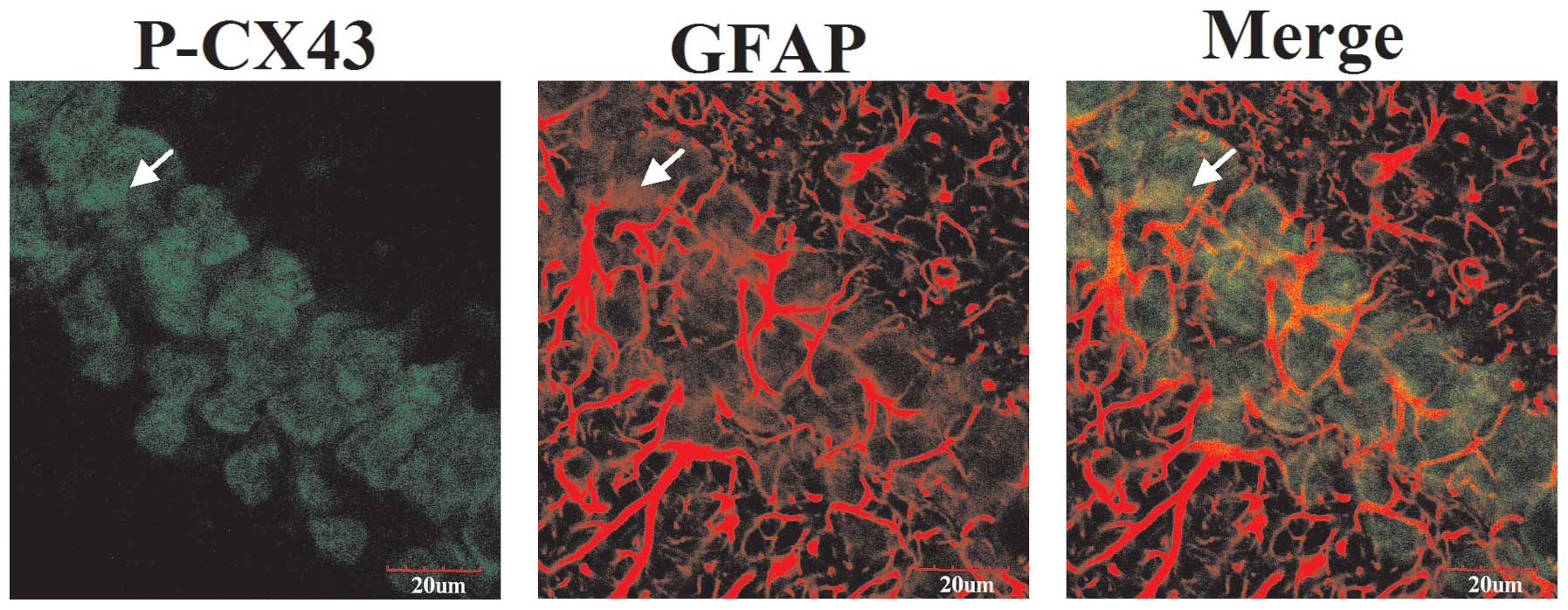
To assess the effect of connexins on astrocytic gap
junctions evoked by TBI, double immunofluorescence staining was
performed to investigate co-localization of p-CX43 and GFAP
expression. As shown in Fig. 1,
p-CX43 was stained with rabbit anti-p-CX43 antibody and secondary
antibodies labeled with green fluorescence. Astrocytes were stained
with mouse anti-GFAP antibodies and secondary antibody labeled with
red fluorescence. The images were merged and yellow was observed
under a laser scanning confocal microscope. These results suggest
that the majority of p-CX43 colocalizes with astrocytes.
LC3 colocalizes with neuron markers
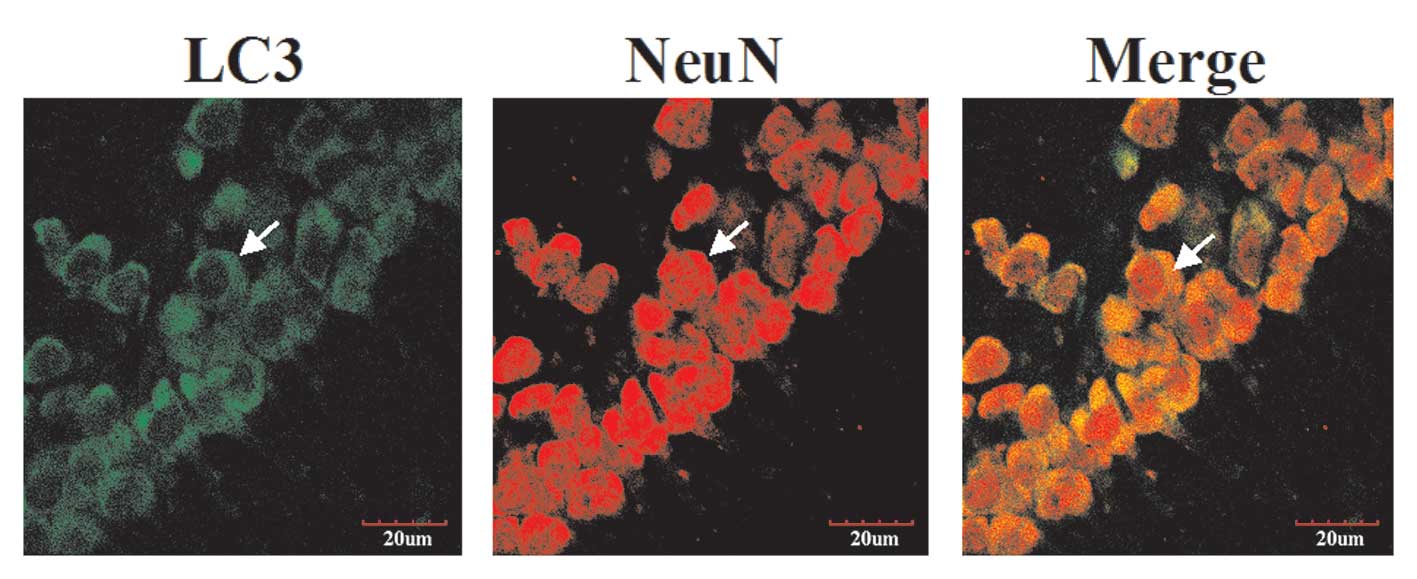
Experiments were performed to investigate the
involvement of autophagy in TBI-induced brain damage. LC3
expression in hippocampal neurons 24 h following TBI was detected
using immunohistochemistry and confocal microscopy (indicated by
arrows, Fig. 2). As shown in
Fig. 2, LC3 was stained with
rabbit anti-LC3 antibody and secondary antibodies labeled with
green fluorescence. The LC3-immunoreactive structures appeared
cup-shaped or circular, which may reflect the different stages of
autophagosome formation (the isolation of membranes prior to and
following closure to form autophagosomal vesicles). In addition,
neurons were stained with mouse anti-NeuN antibody and secondary
antibody labeled with red fluorescence. The images were merged and
yellow was observed under a laser scanning confocal microscope.
These results clarified alterations in the LC3 proteins in neurons
in the hippocampal region following TBI.
CBX treatments suppress p-CX43 protein
expression
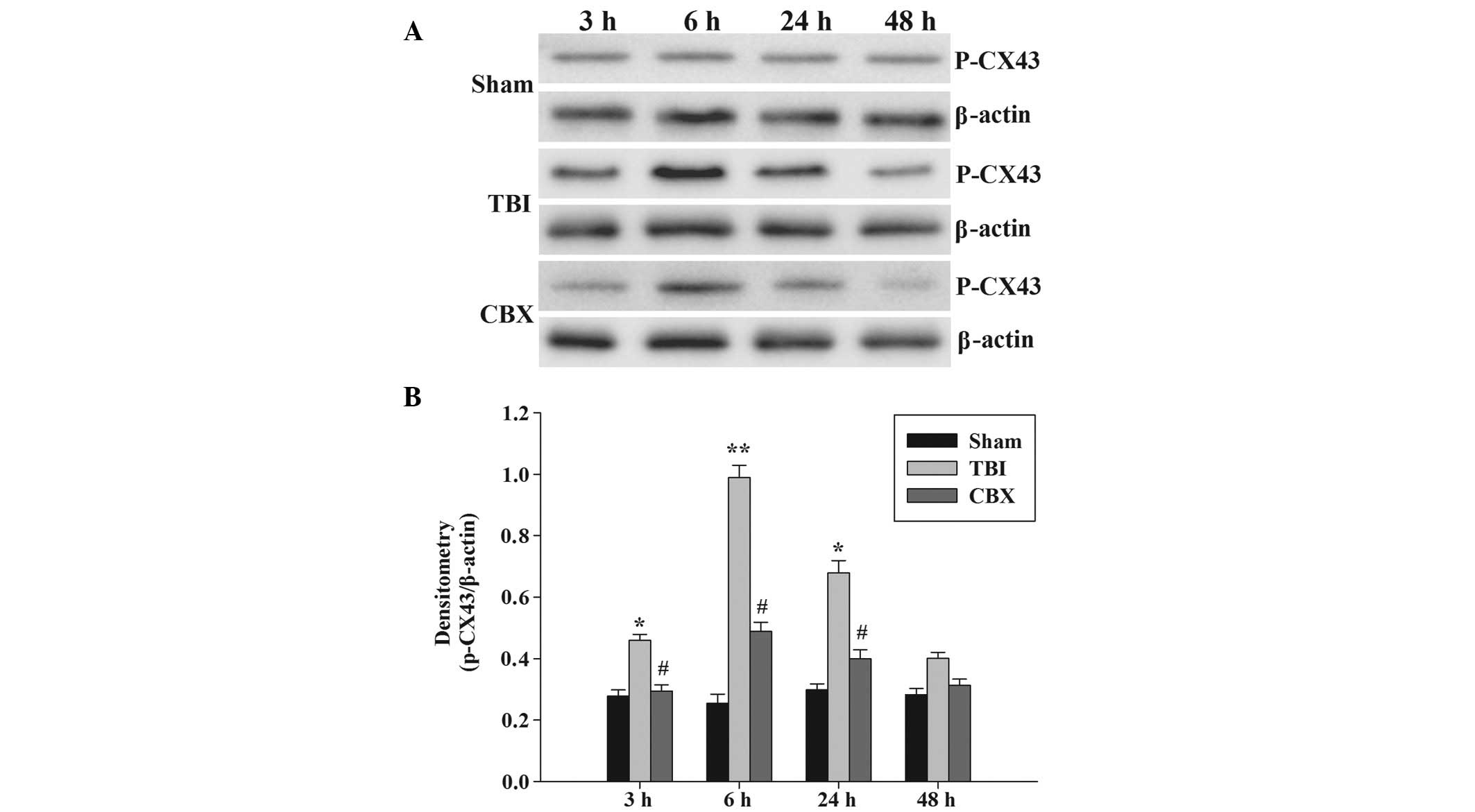
P-CX43 protein expression was analyzed by western
blot analysis (Fig. 3A). The
p-CX43 protein expression was identified at low levels in the
hippocampus in the sham group. The immunoreactivity of p-CX43 in
the hippocampus was significantly induced 3 h following injury,
persisted at a high level until 24 h after injury and thereafter,
gradually decreased. In addition, the p-CX43 protein content
reached a maximum level 6 h following injury. As demonstrated in
Fig. 3B, the p-CX43 protein band
intensity was quantified and the results demonstrated that CBX
pretreatment significantly inhibited the upregulation of p-CX43
protein levels compared with that of the TBI groups at 3, 6 and 24
h.
CBX treatments suppress LC3-II protein
expression
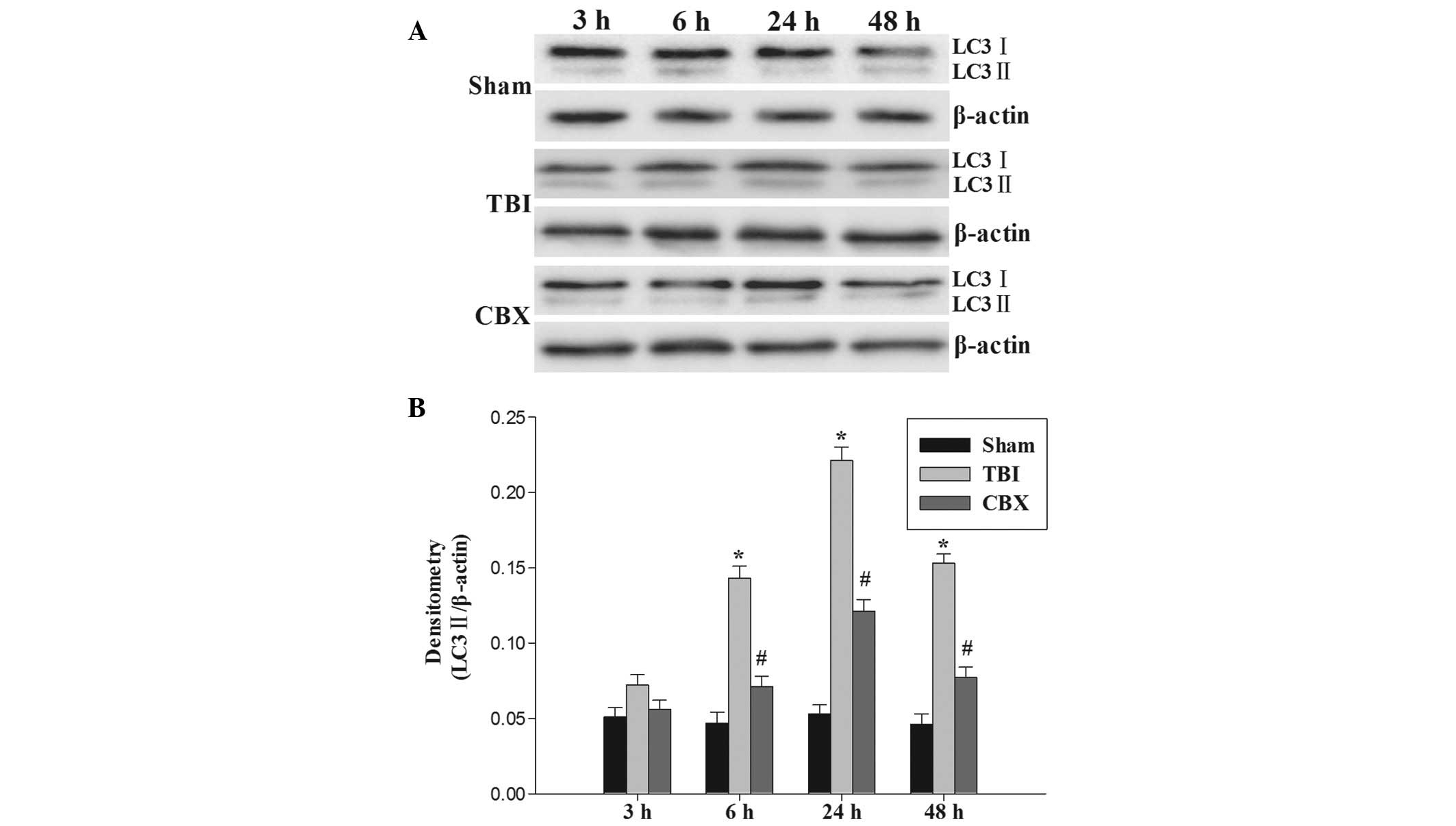
To determine how autophagic activity is altered
following TBI and to confirm the ability of CBX to inhibit
autophagy, the protein levels of LC3-II were determined (Fig. 4A). The expression of LC3-II protein
in the hippocampus was significantly upregulated 6 h following TBI
and persisted at a high level until 48 h following injury. As
demonstrated in Fig. 4B,
pretreatment with CBX significantly reduced the relative protein
expression of LC3-II in the hippocampus.
Discussion
Management of traumatic brain injury poses
considerable challenges to healthcare services (17). Brain injury may result in an energy
crisis and oxidative stress, which induces cell death (18). Cell death is broadly classified
into three types: necrosis, apoptosis [type 1 programmed cell death
(PCD)] and autophagy (type 2 PCD) (19). Autophagy is a process that is
regulated and is key in numerous diseases (20–22).
TBI causes pathophysiologic responses leading to autophagy
activation, cell membrane breakdown, cell loss and motor and
cognitive outcome deficits. Pretreatment with 3-methyladenine
(3-MA), a relatively selective autophagy inbihitor, attenuates
TBI-induced cell death, lesion volume and behavioral outcome
deficits, thereby indicating that inhibition of autophagy may be a
therapeutic target for TBI treatment (23). However, the way in which to
manipulate the autophagy pathway is not clear, therefore, the
probable regulatory mechanism of autophagy in TBI has been
hypothesized based on the studies.
Previous studies using LC3 as an autophagic
biomarker showed that autophagy is detected in the human brain
following trauma and critical illness (24). LC3, an autophagosomal ortholog of
yeast Atg8, is one of the most reliable markers in the study of
autophagy induction (25). LC3 is
synthesized as pro-LC3, which is cleaved by ATG4 protease to form
the 16–18 kDa LC3-I. On activation of autophagy, LC3-I is
conjugated with phosphatidylethanolamine (lipidated). The lipidated
form is referred to as LC3-II (26). Another study has demonstrated
autophagosomal vacuole formation by the observation of a shift from
LC3-I to -II in hippocampal neurons following TBI and has
demonstrated a marked increase in LC3-II levels from 1 to 48 h
post-TBI. Pretreatment with a specific autophagy inhibitor 3-MA
partially inhibited traumatic-elicited induction of LC3-II
(23). Previous studies have
demonstrated that astrocytes, the predominant cell type in the
brain, receive signals from neurons and also release neuroactive
substances (27), provide energy
substrates to neurons (28) and
are important in neuronal support in normal and pathological
conditions. In the central nervous system, astrocytes established a
glial syncytium through intercellular connections via gap junctions
(29). CX43 is the primary
component protein in astrocytic gap junctions (30). Gap junctional intercellular
communication (GJIC) mediates electronic coupling and permits rapid
propagation among cell networks (31). GJIC between astrocytes may regulate
the concentration of extracellular K+ and distribute
neurotransmitters (32). Certain
studies have suggested that astroglial cells may participate in
neuronal apoptosis through their gap junctions under ischemic
conditions (33). However, no
studies have focused on astrocytic gap junction-dependent
modulation of neuronal autophagy following TBI in vivo.
In the present study, coupling of labeled LC3 with
NeuN and p-CX43 with GFAP was conducted to demonstrate the
correlation between p-CX43 and autophagy following TBI. P-CX43 was
colocalized with GFAP immunoreactivity in the hippocampal
astrocytes (Fig. 1). Thus, p-CX43
is expressed by astrocytes. LC3 immunoreactivity was located
predominantly in living hippocampal neurons under confocal
microscopy (Fig. 2). Autophagy may
occur in cells other than astrocytes, particularly in neurons.
Astrocytes may communicate through their gap junctions and be able
to affect neurons (27). In the
present study, astrocytic gap junction proteins were phosphorylated
in the hippocampus following TBI. An increase in p-CX43 protein
expression was observed in the hippocampus of injured brain at 3, 6
and 24 h, with peak relative abundance at 6 h (Fig. 3). These results suggested that
phosphorylation of CX43 induces cell injury in the post-traumatic
region. Therefore, it was hypothesized that phosphorylation of CX43
may contribute to hippocampal dysfunction through astrocytic gap
junction communication in the early phases following TBI. The
results also indicated that TBI activates autophagy, which may
begin at 6 h or earlier, and lasts at least 48 h in the hippocampus
following TBI (Fig. 4). This
result is concurrent with that of previous studies (23). Carbenoxolone is related to
glycyrrhetinic acid and is thought to bind directly to connexins,
inducing a conformational change and results in a closure of gap
junctions (34). Pretreatment with
a specific CX43 inhibitor, CBX, partially inhibited
traumatic-elicited induction of p-CX43 (Fig. 3). The results of the present study
demonstrated that inhibition of p-CX43 suppressed TBI-induced
autophagy (determined by the expression of LC3-II detected by
western blot analysis) (Fig. 4).
Thus, this identified astrocytic gap junctions/connexins as part of
the regulatory network controlling in vivo autophagy in
neurons of the hippocampus.
Astrocytes exchange inositol 1,4,5-triphosphate,
lactate, glutamate and other smaller molecules through gap
junctions and provide energy substrates such as ATP to neurons
(35). It is well known that
astrocytes also modulate extracellular glutamate concentrations,
thereby contributing to extracellular neurotransmitter homeostasis
and astrocyte-neuron signaling (36). Disturbance of extracellular
glutamate levels, acting on N-methyl-D-aspartate receptors, is a
primary cause of neuronal cell death following acute damage, which
is observed following stroke, trauma and seizure (37). Furthermore, Rami et
al(38) demonstrated that the
inhibition of gap junction permeability effectively decreases
neuronal death. A large number of intracellular/extracellular
stimuli, including amino acid starvation and invasion of
microorganisms, are able to induce the autophagic response
(39). The autophagy pathway
triggers a cascade of events leading to tissue edema, neuronal cell
death and impaired motor and cognitive functions following TBI
(23). Elucidation of the
molecular mechanisms by which astrocytic gap junctions regulate
neuronic autophagy is an important area of investigation and aims
to develop novel therapeutic interventions for the prevention of
autophagy formation following traumatic brain injury.
In conclusion, the correlation between gap junctions
and autophagy following TBI was shown and it was determined that
astrocytic gap junctions/connexins act as a regulatory factor
controlling neuronal autophagy in the hippocampus. As connexin
expression and/or astrocytic gap junction coupling is affected in
various forms of brain injury (40), this regulatory mechanism may be
prominent in the diseased brain.
Acknowledgements
The present study was supported by a grant from the
Natural Science Foundation of Hebei Province (grant no.
H2012401071).
Abbreviations:
|
CX43
|
connexin 43
|
|
CBX
|
carbenoxolone
|
|
TBI
|
traumatic brain injury
|
|
LC3
|
light chain 3
|
|
GFAP
|
glia fibrillary acidic protein
|
|
NeuN
|
neuronal nuclei
|
|
GJIC
|
gap junctional intercellular
communication
|
References
|
1
|
Mammis A, McIntosh TK and Maniker AH:
Erythropoietin as a neuroprotective agent in traumatic brain injury
Review. Surgical Neurol. 71:527–531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luo CL, Chen XP, Yang R, et al: Cathepsin
B contributes to traumatic brain injury-induced cell death through
a mitochondria-mediated apoptotic pathway. J Neurosci Res.
88:2847–2858. 2010.PubMed/NCBI
|
|
3
|
Tehranian R, Rose ME, Vagni V, et al:
Disruption of Bax protein prevents neuronal cell death but produces
cognitive impairment in mice following traumatic brain injury. J
Neurotrauma. 25:755–767. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lai Y, Hickey RW, Chen Y, et al: Autophagy
is increased after traumatic brain injury in mice and is partially
inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J
Cereb Blood Flow Metab. 28:540–550. 2008. View Article : Google Scholar
|
|
5
|
Pozuelo-Rubio M: Regulation of autophagic
activity by 14-3-3ζ proteins associated with class III
phosphatidylinositol-3-kinase. Cell Death Differ. 18:479–492.
2011.
|
|
6
|
Liu CL, Chen S, Dietrich D and Hu BR:
Changes in autophagy after traumatic brain injury. J Cereb Blood
Flow Metab. 28:674–683. 2008. View Article : Google Scholar
|
|
7
|
Cotrina ML and Nedergaard M: Physiological
and pathological functions of P2X7 receptor in the spinal cord.
Purinergic Signal. 5:223–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bennett MV, Contreras JE, Bukauskas FF and
Sáez JC: New roles for astrocytes: gap junction hemichannels have
something to communicate. Trends Neurosci. 26:610–617. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF
and Burt JM: Mechanism of v-Src-and mitogen-activated protein
kinase-induced reduction of gap junction communication. Am J
Physiol Cell Physiol. 284:C511–C520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao X, Ahram A, Berman RF, Muizelaar JP
and Lyeth BG: Early loss of astrocytes after experimental traumatic
brain injury. Glia. 44:140–152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giaume C, Tabernero A and Medina JM:
Metabolic trafficking through astrocytic gap junctions. Glia.
21:114–123. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin JH, Weigel H, Cotrina ML, et al:
Gap-junction-mediated propagation and amplification of cell injury.
Nature Neurosci. 1:494–500. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats. J Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Khorasani MZ, Hosseinzadeh SA and Vakili
A: Effect of central microinjection of carbenoxolone in an
experimental model of focal cerebral ischemia. Pak J Pharm Sci.
22:349–354. 2009.PubMed/NCBI
|
|
15
|
Milad MR, Vidal-Gonzalez I and Quirk GJ:
Electrical stimulation of medial prefrontal cortex reduces
conditioned fear in a temporally specific manner. Behav Neurosci.
118:389–394. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song SX, Gao JL, Wang KJ, et al:
Attenuation of brain edema and spatial learning deficits by the
inhibition of NADPH oxidase activity using apocynin following
diffuse traumatic brain injury in rats. Mol Med Rep. 327–331.
2012.
|
|
17
|
Eghwrudjakpor PO and Allison AB: Oxidative
stress following traumatic brain injury: enhancement of endogenous
antioxidant defense systems and the promise of improved outcome.
Niger J Med. 19:14–21. 2010. View Article : Google Scholar
|
|
18
|
Uryu K, Laurer H, McIntosh T, et al:
Repetitive mild brain trauma accelerates Abeta deposition, lipid
peroxidation, and cognitive impairment in a transgenic mouse model
of Alzheimer amyloidosis. J Neurosci. 22:446–454. 2002.
|
|
19
|
Werner C and Engelhard K: Pathophysiology
of traumatic brain injury. Br J Anaesth. 99:4–9. 2007. View Article : Google Scholar
|
|
20
|
Geng J and Klionsky DJ: The Atg8 and Atg12
ubiquitin-like conjugation systems in macroautophagy. ‘Protein
modifications: beyond the usual suspects’ review series. EMBO Rep.
9:859–864. 2008.
|
|
21
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meijer AJ and Codogno P: Autophagy:
regulation and role in disease. Crit Rev Clin Lab Sci. 46:210–240.
2009. View Article : Google Scholar
|
|
23
|
Luo CL, Li BX, Li QQ, et al: Autophagy is
involved in traumatic brain injury-induced cell death and
contributes to functional outcome deficits in mice. Neuroscience.
184:54–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Clark RS, Bayir H, Chu CT, Alber SM,
Kochanek PM and Watkins SC: Autophagy is increased in mice after
traumatic brain injury and is detectable in human brain after
trauma and critical illness. Autophagy. 4:88–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maiuri MC, Criollo A, Tasdemir E, et al:
BH3-only proteins and BH3 mimetics induce autophagy by
competitively disrupting the interaction between Beclin 1 and
Bcl-2/Bcl-X(L). Autophagy. 3:374–376. 2007. View Article : Google Scholar
|
|
26
|
Kabeya Y, Mizushima N, Yamamoto A,
Oshitani-Okamoto S, Ohsumi Y and Yoshimori T: LC3, GABARAP and
GATE16 localize to autophagosomal membrane depending on form-II
formation. J Cell Sci. 117:2805–2812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Araque A, Parpura V, Sanzgiri RP and
Haydon PG: Tripartite synapses: glia, the unacknowledged partner.
Trends Neurosci. 22:208–215. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsacopoulos M and Magistretti PJ:
Metabolic coupling between glia and neurons. J Neurosci.
16:877–885. 1996.PubMed/NCBI
|
|
29
|
Dermietzel R and Spray DC: From neuro-glue
(‘Nervenkitt’) to glia: a prologue. Glia. 24:1–7. 1998.PubMed/NCBI
|
|
30
|
Giaume C, Fromaget C, el Aoumari A,
Cordier J, Glowinski J and Gros D: Gap junctions in cultured
astrocytes: single-channel currents and characterization of
channel-forming protein. Neuron. 6:133–143. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Paul DL: New functions for gap junctions.
Curr Opin Cell Biol. 7:665–672. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hansson E, Muyderman H, Leonova J, et al:
Astroglia and glutamate in physiology and pathology: aspects on
glutamate transport, glutamate-induced cell swelling and
gap-junction communication. Neurochem Int. 37:317–329. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nakase T, Fushiki S and Naus CC:
Astrocytic gap junctions composed of connexin 43 reduce apoptotic
neuronal damage in cerebral ischemia. Stroke. 34:1987–1993. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rozental R, Srinivas M and Spray DC: How
to close a gap junction channel. Efficacies and potencies of
uncoupling agents. Methods Mol Biol. 154:447–476. 2001.PubMed/NCBI
|
|
35
|
Magistretti PJ: Cellular bases of
functional brain imaging: insights from neuron-glia metabolic
coupling. Brain Res. 886:108–112. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Anderson CM and Swanson RA: Astrocyte
glutamate transport: review of properties, regulation, and
physiological functions. Glia. 32:1–14. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hardingham GE and Bading H: The Yin and
Yang of NMDA receptor signalling. Trends Neurosci. 26:81–89. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rami A, Volkmann T and Winckler J:
Effective reduction of neuronal death by inhibiting gap junctional
intercellular communication in a rodent model of global transient
cerebral ischemia. Exp Neurol. 170:297–304. 2001. View Article : Google Scholar
|
|
39
|
Zhang YB, Li SX, Chen XP, et al: Autophagy
is activated and might protect neurons from degeneration after
traumatic brain injury. Neurosci Bull. 24:143–149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chew SS, Johnson CS, Green CR and
Danesh-Meyer HV: Role of connexin43 in central nervous system
injury. Exp Neurol. 225:250–261. 2010. View Article : Google Scholar : PubMed/NCBI
|