Introduction
Signaling by members of the wingless-type MMTV
integration site (Wnt) family of proteins controls various cellular
and biological processes, ranging from cell adhesion, cell
migration, and cancer development to differentiation of multiple
cell lineages, cell polarity, and stem cell self-renewal (1). The majority of research on Wnts has
focused on the Wnt/β-catenin pathway, previously referred to as the
canonical Wnt pathway. Activation of the canonical Wnt pathway
leads to nuclear translocation of β-catenin, which binds to
lymphoid enhancer-binding factor/T cell-specific transcription
factor (LEF/TCF) and activates transcription of target genes
(1–3). The stability of β-catenin is also
regulated by integrin-linked kinase (ILK), a serine/threonine
protein kinase that interacts with the cytoplasmic domains of β1
and β3 integrins. Integrins composed of α and β transmembrane
subunits are bidirectional signaling molecules. Inside-out
signaling by integrins occurs when direct interactions between the
cytoplasmic tail of β-subunits and the cytoskeletal protein, talin,
regulate the affinity for the ligand. Following ligand binding,
integrins engage in outside-in signaling by recruiting signaling
and adaptor proteins to the cytoplasmic tail of the α and/or β
subunits. This recruitment results in actin reorganization and
modulation of various intracellular signaling pathways (4,5).
ILK-integrin interactions organize the connections of the
extracellular matrix (ECM) to the cytoskeleton and, by doing so,
regulate cell migration and adhesion. In addition, ILK activity
links integrin function to Wnt signaling (6–9). At
present, in vitro and in vivo evidence supports the
hypothesis that phosphorylation by ILK stimulates β-catenin to
translocate into the nucleus and form a complex with LEF. In
addition, ILK phosphorylates and inactivates glycogen synthase
kinase-3β (GSK-3β). Inactivation of GSK-3β eventually results in
activation of canonical Wnt target genes (6–10).
However, little is known about how Wnt/β-catenin signaling
activates ILK or integrin signaling. In the current study, we
activated the canonical Wnt signaling by recombinant Wnt3a to
investigate the migration and adhesion ability of vascular smooth
muscle cells (VSMCs) and the role of ILK and β1-integrin.
Materials and methods
Cell culture and reagents
Primary VSMCs were isolated from the thoracic aorta
of male Sprague Dawley rats (weight, 100–150 g), and then cultured
in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal
bovine serum (FBS; Hyclone, Logan, UT, USA), 100 U/ml penicillin
and 100 μg/ml streptomycin (Beyotime, Shanghai, China). The cells
were incubated at 37°C in a humidified atmosphere of 95% air and 5%
CO2. The purity of the VSMCs was estimated to be ~90%,
based on cell morphology and the results of immunostaining with
monoclonal mouse anti-α-actin antibodies (Sigma-Aldrich, St. Louis,
MO, USA). The VSMCs used for all experiments were between the 3rd
and 6th passages. Recombinant Wnt3a was purchased from R&D
Systems (Minneapolis, MN, USA). In this study, anti-β-catenin
antibody (polyclonal, rabbit) and anti-phospho-β-catenin (Ser675)
antibody (polyclonal, rabbit; both from Cell Signaling Technology
Inc., Danvers, MA, USA), anti-GSK-3β antibody (polyclonal, rabbit),
anti-phospho-GSK-3β (Ser9) antibody (polyclonal, goat) and
anti-β-actin antibody (polyclonal, rabbit; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), anti-ILK antibody
(polyclonal, rabbit; Sigma-Aldrich, St. Louis, MO, USA),
anti-β1-integrin antibody (monoclonal, rabbit) and
anti-active-β1-integrin antibody (monoclonal, mouse; both from
Millipore, Billerica, MA, USA) were used.
Transwell migration assay
The VSMC migration assay was performed using
transwell cell culture inserts (Transwell Assay system; Corning
Inc., Acton, MA, USA) for 24-well plates. A total of 100 μl VSMCs
(3×105 cells/ml) suspended in serum-free DMEM was added
to the upper polycarbonate membrane insert (pore size, 8 μm), Wnt3a
(final concentration, 100 ng/ml) was added to the upper chamber and
600 μl culture medium containing 10% FBS was added to the lower
chamber. The cells were allowed to migrate for 24 h while the
plates were incubated in a humidified incubator in a 5%
CO2 atmosphere at 37°C. After 24 h, the cells that
remained on the upper surface of the membrane were removed with a
cotton swab. The membrane was fixed with anhydrous methanol for 20
min at room temperature and then stained with 0.1% crystal violet
for 15 min. A microscope (Nikon, Tokyo, Japan) was used to
determine the number of migratory cells by counting the cells in
five randomly selected fields of view. All experiments were
performed in triplicate.
Wound healing assay
VSMCs were plated in 6-well plates and grown to
70–80% confluency. The experimental wounds were created by dragging
200 μl pipette tips across the bottom of the cell culture wells.
The cells were rinsed with phosphate-buffered saline (PBS) and the
culture medium was replaced with fresh maintenance medium
supplemented with Wnt3a (final concentration; 100 ng/ml). The wound
healing was recorded after 48 h using bright field microscopy. The
wound gap was measured and the percentage of wound repair was
determined.
Cell adhesion assay
The cell adhesion assay was performed as described
previously (11,12). Ninety-six-well tissue culture
plates (Costar, Corning Inc.) were coated overnight at 4°C with
collagen type I (Sigma-Aldrich; 20 μg/ml). After washing with PBS,
the plates were blocked with PBS containing 1% heat-denatured
bovine serum albumin for 1 h at room temperature. The plates were
washed extensively with serum-free DMEM, and 3×104 VSMCs
were plated and incubated with Wnt3a (final concentration, 100
ng/ml) for 1 h at 37°C. After washing with PBS, the cells were
fixed with 4% paraformaldehyde for 30 min at room temperature and
stained with 0.5% toluidine blue for 15 min. The plates were washed
extensively with double distilled water. A microscope (Nikon) was
used to determine the number of adherent cells by counting the
cells in five randomly selected fields of view. The cells were
solubilized in 1% sodium dodecyl sulfate (SDS) and quantified using
a microtiter plate reader set at 590 nm (Tecan, Männedorf,
Switzerland).
Western blot analysis
VSMCs were harvested following treatment with Wnt3a
(final concentration;100 ng/ml) or PBS (control) for three days.
The cells were lysed by incubation with radioimmunoprecipitation
(RIPA) buffer supplemented with proteinase inhibitors for 30 min on
ice. Equal quantities of protein were separated by
SDS-polyacrylamide gel electrophoresis (PAGE) and subjected to
immunoblot analysis according to standard protocols. The separated
proteins were blotted onto nitrocellulose membranes and incubated
with antibodies that recognize total β-catenin, phospho-β-catenin
(Ser675), total GSK-3β, phospho-GSK-3β (Ser9), β1-integrin, ILK and
β-actin overnight at 4°C. Following three washes, the blots were
incubated with peroxidase-conjugated secondary antibodies (Boster,
Wuhan, China) for 1 h at room temperature and subsequently analyzed
using an enhanced chemiluminescence detection system (Clinx,
Shanghai, China).
Flow cytometric analysis
VSMCs were serum starved for 24 h and supplemented
with Wnt3a (final concentration, 100 ng/ml) for three days.
β1-integrin expression on the surface of the VSMCs was evaluated by
using indirect immunofluorescence and flow cytometry. Following
washing with PBS, the cells were incubated with a rabbit anti-rat
β1-integrin antibody (dilution; 1:70) and a mouse anti-rat active
β1-integrin antibody (dilution; 1:200) for 30 min at room
temperature in the dark. The cells were washed again and incubated
with phycoerythrin-conjugated goat anti-rabbit IgG (dilution, 1:50;
Bioss, Beijing, China) and Cy3-conjugated goat anti-mouse IgG
(dilution, 1:50; Bioss) secondary antibodies for 45 min. Following
incubation with secondary antibodies, the cells were analyzed by
flow cytometry with a Becton-Dickinson FACS Calibur and Cell Quest
software (BD, Franklin Lakes, NJ, USA).
Statistical analysis
Paired or independent t-tests were performed for
statistical analyses of all data. P<0.05 was considered to
indicate a statistically significant difference. The data are
expressed as the mean ± standard error of the mean.
Results
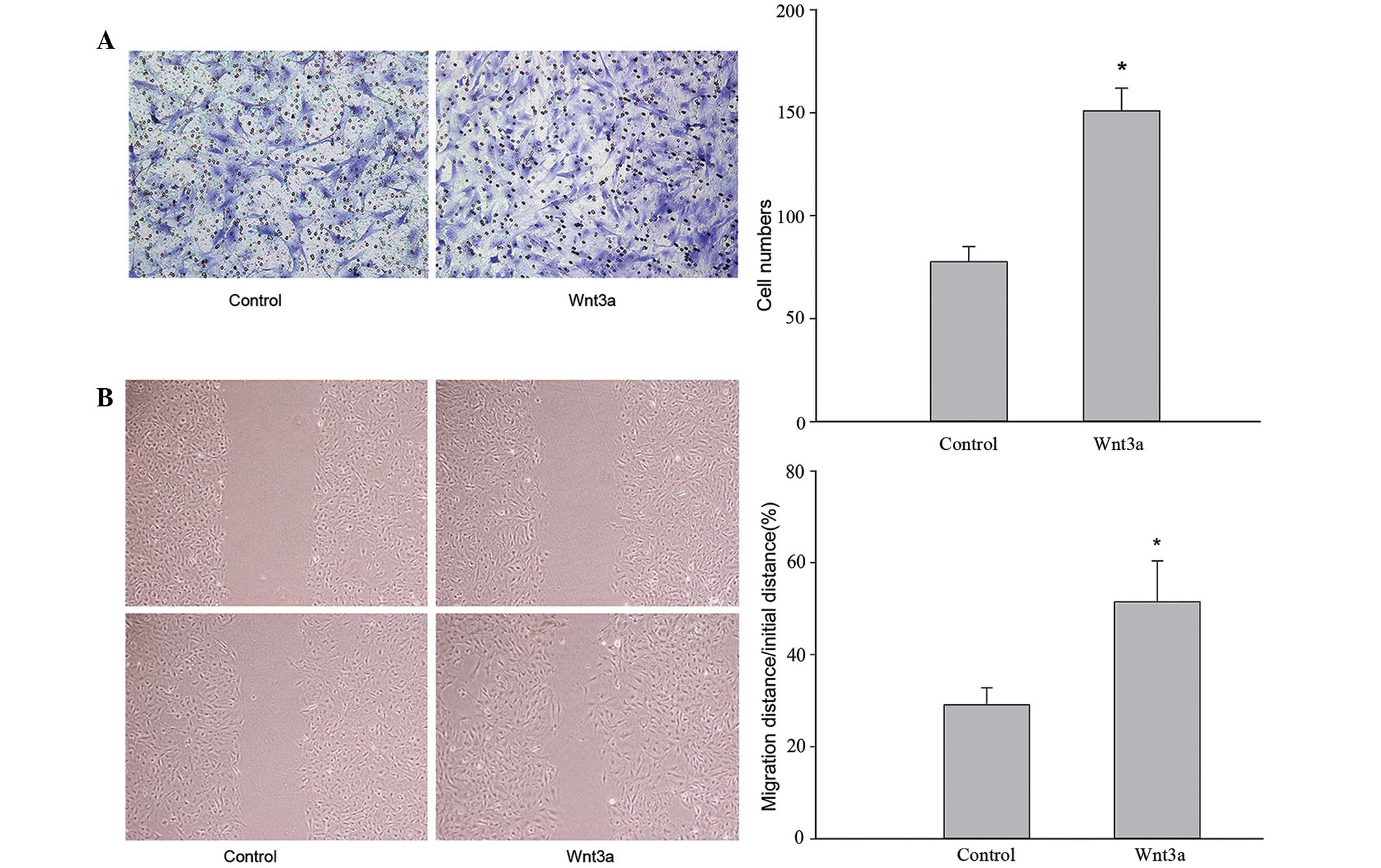
Wnt3a promotes VSMC migration
Transwell migration and wound healing assays were
performed to investigate the effect of Wnt3a treatment on VSMC
migration. The migratory ability of VSMCs treated with Wnt3a was
significantly increased in the transwell migration assay (Fig. 1A). Following 24 h incubation, the
number of cells that migrated across the polycarbonate membrane was
higher in the Wnt3a group compared with the control group
(P<0.05). As shown in Fig. 1B,
VSMCs treated with Wnt3a moved faster than the control cells. These
results demonstrate that Wnt3a treatment induces VSMC
migration.
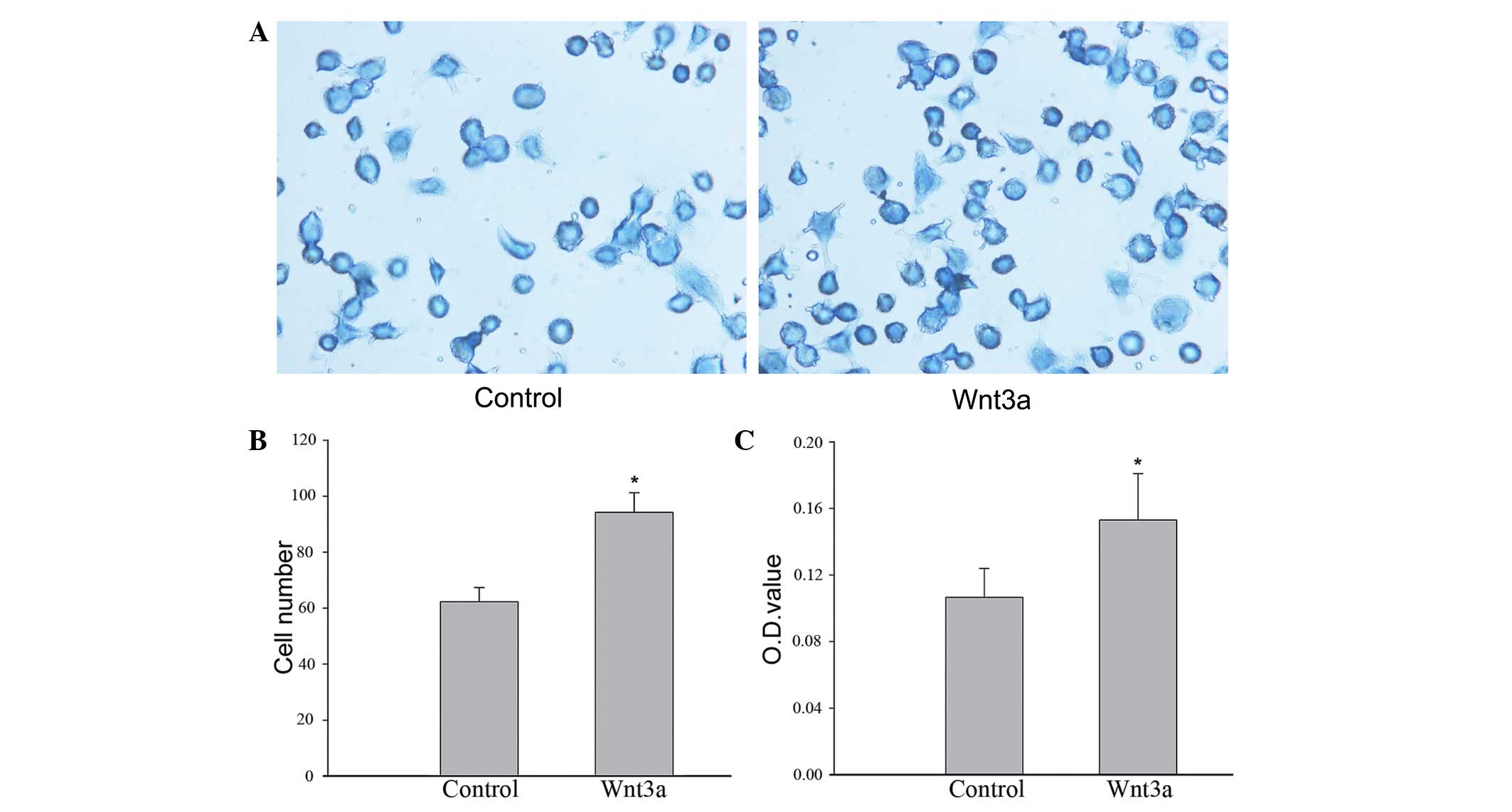
Adhesion of VSMCs to collagen type I
To investigate the influence of Wnt3a on cell-matrix
interactions, adhesion assays were performed. Cells were stained
with toluidine blue and the microplate reader method was used to
monitor cell adhesion. The results demonstrated that the number of
VSMCs that adhered to collagen type I was significantly greater in
the Wnt3a-treated group compared with the control group. In
addition, the optical density value detected by the microplate
reader was higher in the samples from the Wnt3a-treated group
compared with those from the control group (Fig. 2). These results indicate that Wnt3a
treatment significantly improves the adhesion of VSMCs to collagen
type I.
Wnt3a regulates protein expression
Wnt3a is a prominent member of the Wnt family and
may induce the accumulation of β-catenin and activate the canonical
Wnt pathway (13). To understand
the mechanisms by which Wnt3a affects VSMCs, the protein expression
of β-catenin, GSK-3β, ILK, and β1-integrin were analyzed, all of
which are either components of, or targets of the canonical Wnt
signaling pathway. Treatment of VSMCs with Wnt3a upregulated the
expression of phospho-β-catenin (Ser675), phospho-GSK-3β (Ser9),
and ILK; however, the expression of total β-catenin, total GSK-3β,
and β1-integrin was not significantly different between the two
groups (Fig. 3).
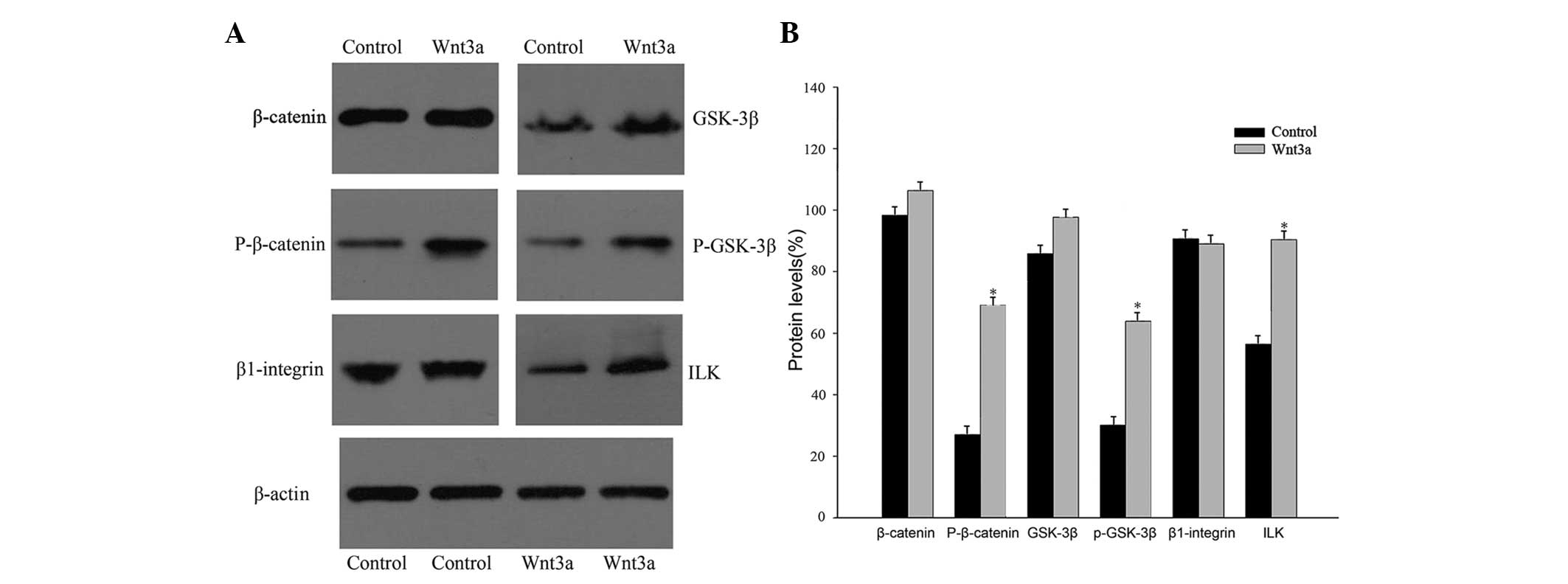 | Figure 3Protein expression was measured in
cultured rat VSMCs that were treated with recombinant Wnt3a or
phosphate-buffered saline (control) for three days. (A) Western
blot analysis showing total β-catenin, phospho-β-catenin (Ser675),
total GSK-3β, phospho-GSK-3β (Ser9), β1-integrin, and ILK protein
expression in control and Wnt3a-treated cells. (B) The
phospho-β-catenin and phospho-GSK-3β protein signals were
normalized to those of total β-catenin and total GSK-3β,
respectively. The β1-integrin and ILK protein signals were
quantified and normalized to that of β-actin
(*P<0.05, vs. control). GSK-3β, glycogen synthase
kinase 3β; ILK, integrin-linked kinase; P-, phosphorylated; VSMCs,
vascular smooth muscle cells; Wnt3a, wingless-type MMTV integration
site family, member 3a. |
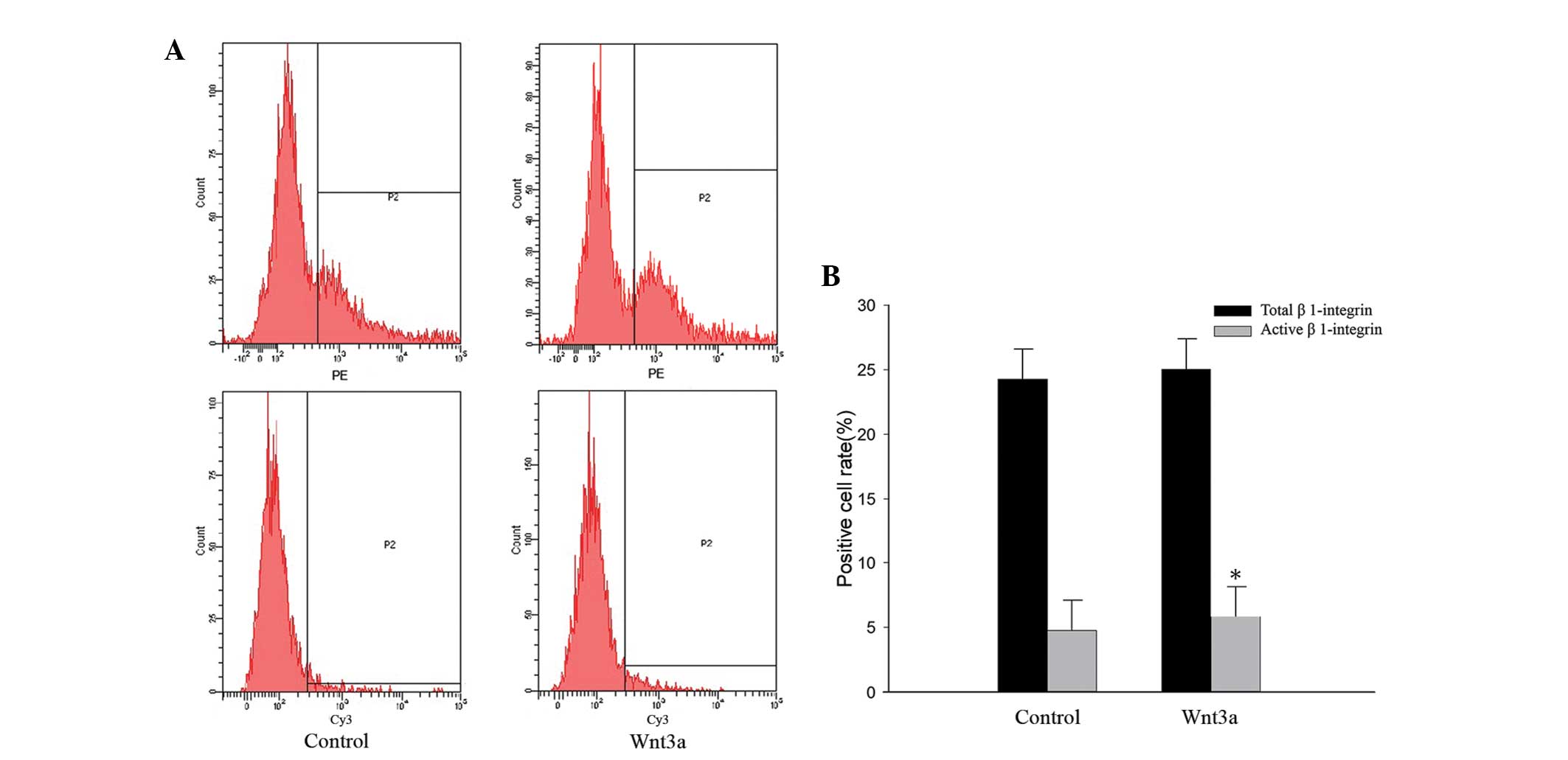
Wnt3a activates β1-integrin
Since treatment with Wnt3a did not alter the
expression of β1-integrin, the expression of active β1-integrin on
the surface of VSMCs was quantified using flow cytometry (Fig. 4A). The anti-active-β1-integrin
antibody is specific for the active conformation of rat
β1-integrin, and it may also discriminate between the activated
states. Therefore, it is useful to investigate how β1-integrin
activation is regulated. VSMCs that were stimulated with Wnt3a for
three days bound more active-β1-integrin antibodies compared with
the unstimulated cells. However, Wnt3a treatment had no effect on
the total quantity of β1-integrin expression on the surface of
VSMCs. These results demonstrate that Wnt3a treatment activates
β1-integrin without changing its expression levels (Fig. 4).
Discussion
It is well known that the Wnt signaling pathways
affect migration and adhesion via downstream effectors (14–18).
The canonical Wnt pathway controls cell migration and adhesion by
regulating the stability of β-catenin, the major downstream target
of Wnt signaling (14–18). Previous studies have determined a
multitude of points of crosstalk between the Wnt pathways and the
mechanisms that control cellular architecture, from the level of
receptors to the level of transcription. Cellular mechanisms that
are responsible for the regulation of migration and adhesion also
function to modulate the activity of several Wnt pathway components
(14,19,20).
Activation of β-catenin signaling may occur by inhibition of GSK-3β
and GSK-3β-mediated phosphorylation of residues in the N-terminal
region of β-catenin may lead to rapid degradation. However,
β-catenin is also activated by phosphorylation of serine 675, in
the C-terminal region of the protein. This phosphorylation
facilitates the nuclear translocation of β-catenin and enhances its
transcriptional activity, as has been shown to occur in VSMCs in
vitro upon activation by v-akt murine thymoma viral oncogene
homolog 1 (Akt) or cAMP-dependent protein kinase, catalytic subunit
C α (PKA) (20–23). Enhanced β-catenin phosphorylation
at Ser675 and enhanced GSK-3β phosphorylation at Ser9 in rat VSMCs
were observed following Wnt3a treatment. Phosphorylation of GSK-3β
at Ser9 may inactivate the protein. Therefore, although a role of
PKA, Akt, or other signaling pathways in β-catenin phosphorylation
may not be excluded, the current data suggest that stimulation of
VSMCs with Wnt3a activates the Wnt/β-catenin pathway.
VSMC migration and proliferation contribute to
arterial wound repair and thickening of the intimal layer in
atherosclerosis and restenosis. These processes are influenced by
cell adhesion to molecules present in the ECM and are regulated by
the integrin family of cell-surface matrix receptors. An important
signaling molecule acting downstream of integrin receptors is ILK.
ILK has been implicated in the control of cancer cell growth and
survival through modulation of downstream targets, notably Akt and
GSK-3β (24–26). Evidence also exists to establish
ILK as a molecular adaptor protein linking integrins to the actin
cytoskeleton and regulating actin polymerization (8,24–27).
In vitro, ILK may phosphorylate and inactivate GSK-3β to
promote the nuclear accumulation of β-catenin and activation of the
canonical Wnt pathway (6–10). However, it remains unclear how
activation of the canonical Wnt pathway influences the activity of
ILK. In the current study, the role of the canonical Wnt pathway in
ILK activation was investigated by culturing rat VSMCs with
recombinant Wnt3a, and assessing ILK protein expression and
β1-integrin activity. The results demonstrate that Wnt3a, not only
activates the canonical Wnt pathway, but also increases ILK protein
expression and β1-integrin activity.
The present study also demonstrates that treatment
with Wnt3a significantly increases VSMC migration and adhesion.
Cell adhesion to the ECM via integrins triggers the assembly of the
actin cytoskeleton and regulatory proteins that form a large
multiprotein complex. Through their association with the actin
cytoskeleton and signaling molecules, adhesion complexes generate
elaborate networks that control a variety of cellular processes in
normal and pathological conditions, including cell migration,
proliferation, survival, and also invasion and metastasis (28–31).
Adhesion of VSMCs to collagen type I has been shown to be
β1-integrin-dependent (11,32).
Although adhesion of VSMCs to collagen type I is predominantly
mediated via β1-integrin receptors, the cell surface expression
levels of β1-integrin were not altered by Wnt3a stimulation
(33). As demonstrated by flow
cytometry, Wnt3a treatment activated β1-integrin in rat VSMCs. The
mechanism by which intracellular signals alter the affinity and
avidity of integrin receptors, termed ‘inside-out’ signaling, has
been characterized by several groups (4,5,34).
ILK is known to bind to the cytoplasmic domain of integrin
subunits. This binding either increases affinity by triggering
conformational changes in the integrin subunits or increases
avidity by stimulating integrin clustering in the cell membrane
(8,24–27).
The results of the current study lead to the conclusion that Wnt3a
activates ILK and β1-integrin, which then regulates VSMC migration
and adhesion.
In conclusion, the Wnt/β-catenin pathway in rat
VSMCs has been observed to activate β1-integrin without leading to
quantitative changes in its expression on the cell surface. ILK
binds to β1-integrin to increase the affinity of β1-integrin for
the ECM by inside-out signaling, which may increase cell migration
and adhesion. Wnt3a has been observed to regulate the migration and
adhesion of VSMCs, and may therefore be a valuable therapeutic
target for the prevention and treatment of vascular disease.
However, vascular disease is a complicated process that is
modulated by multiple signaling pathways. Future studies should
investigate the mechanisms and functions of Wnt3a signaling in
other cell lines and model systems.
Acknowledgements
This study was supported by the National Science
Foundation of China (grant nos. 81170195 and 81200156). The authors
would like to thank all teachers from the Renmin Hospital of Wuhan
University for their technical assistance.
References
|
1
|
van de Schans VA, Smits JF and
Blankesteijn WM: The Wnt/frizzled pathway in cardiovascular
development and disease: friend or foe? Eur J Pharmacol.
585:338–345. 2008.
|
|
2
|
Rao TP and Kühl M: An updated overview on
Wnt signaling pathways: a prelude for more. Circ Res.
106:1798–1806. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsaousi A, Mill C and George SJ: The Wnt
pathways in vascular disease: lessons from vascular development.
Curr Opin Lipidol. 22:350–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Montanez E, Ussar S, Schifferer M, et al:
Kindlin-2 controls bidirectional signaling of integrins. Genes Dev.
22:1325–1330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim C, Ye F and Ginsberg MH: Regulation of
integrin activation. Annu Rev Cell Dev Biol. 27:321–345. 2011.
View Article : Google Scholar
|
|
6
|
Schambony A, Kunz M and Gradl D:
Cross-regulation of Wnt signaling and cell adhesion.
Differentiation. 72:307–318. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oloumi A, McPhee T and Dedhar S:
Regulation of E-cadherin expression and beta-catenin/Tcf
transcriptional activity by the integrin-linked kinase. Biochim
Biophys Acta. 1691:1–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hannigan G, Troussard AA and Dedhar S:
Integrin-linked kinase: a cancer therapeutic target unique among
its ILK. Nat Rev Cancer. 5:51–63. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rallis C, Pinchin SM and Ish-Horowicz D:
Cell-autonomous integrin control of Wnt and Notch signalling during
somitogenesis. Development. 137:3591–3601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oloumi A, Syam S and Dedhar S: Modulation
of Wnt3a-mediated nuclear beta-catenin accumulation and activation
by integrin-linked kinase in mammalian cells. Oncogene.
25:7747–7757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blaschke F, Stawowy P, Goetze S, et al:
Hypoxia activates beta(1)-integrin via ERK 1/2 and p38 MAP kinase
in human vascular smooth muscle cells. Biochem Biophys Res Commun.
296:890–896. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stringa E, Knäuper V, Murphy G and
Gavrilovic J: Collagen degradation and platelet-derived growth
factor stimulate the migration of vascular smooth muscle cells. J
Cell Sci. 113:2055–2064. 2000.PubMed/NCBI
|
|
13
|
Bao XL, Song H, Chen Z and Tang X: Wnt3a
promotes epithelial-mesenchymal transition, migration, and
proliferation of lens epithelial cells. Mol Vis. 18:1983–1990.
2012.PubMed/NCBI
|
|
14
|
Amin N and Vincan E: The Wnt signaling
pathways and cell adhesion. Front Biosci (Landmark Ed). 17:784–804.
2012. View Article : Google Scholar
|
|
15
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan CW, Gardiner BS, Hirokawa Y, Layton
MJ, Smith DW and Burgess AW: Wnt signalling pathway parameters for
mammalian cells. PLoS One. 7:e318822012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parmalee NL and Kitajewski J: Wnt
signaling in angiogenesis. Curr Drug Targets. 9:558–564. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samarzija I, Sini P, Schlange T, Macdonald
G and Hynes NE: Wnt3a regulates proliferation and migration of
HUVEC via canonical and non-canonical Wnt signaling pathways.
Biochem Biophys Res Commun. 386:449–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nusse R: Wnt signaling in disease and in
development. Cell Res. 15:28–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brembeck FH, Rosário M and Birchmeier W:
Balancing cell adhesion and Wnt signaling, the key role of
beta-catenin. Curr Opin Genet Dev. 16:51–59. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marchand A, Atassi F, Gaaya A, et al: The
Wnt/beta-catenin pathway is activated during advanced arterial
aging in humans. Aging Cell. 10:220–232. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taurin S, Sandbo N, Yau DM, Sethakorn N
and Dulin NO: Phosphorylation of beta-catenin by PKA promotes
ATP-induced proliferation of vascular smooth muscle cells. Am J
Physiol Cell Physiol. 294:C1169–C1174. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mill C and George SJ: Wnt signalling in
smooth muscle cells and its role in cardiovascular disorders.
Cardiovasc Res. 95:233–240. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ho B, Hou G, Pickering JG, Hannigan G,
Langille BL and Bendeck MP: Integrin-linked kinase in the vascular
smooth muscle cell response to injury. Am J Pathol. 173:278–288.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ho B and Bendeck MP: Integrin linked
kinase (ILK) expression and function in vascular smooth muscle
cells. Cell Adh Migr. 3:174–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McDonald PC, Fielding AB and Dedhar S:
Integrin-linked kinase - essential roles in physiology and cancer
biology. J Cell Sci. 121:3121–3132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hannigan GE, Coles JG and Dedhar S:
Integrin-linked kinase at the heart of cardiac contractility,
repair, and disease. Circ Res. 100:1408–1414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huttenlocher A and Horwitz AR: Integrins
in cell migration. Cold Spring Harb Perspect Biol. 3:a0050742011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang HQ, Bai L, Shen BR, Yan ZQ and Jiang
ZL: Coculture with endothelial cells enhances vascular smooth
muscle cell adhesion and spreading via activation of beta1-integrin
and phosphatidylinositol 3-kinase/Akt. Eur J Cell Biol. 86:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carlson TR, Hu H, Braren R, Kim YH and
Wang RA: Cell-autonomous requirement for beta1 integrin in
endothelial cell adhesion, migration and survival during
angiogenesis in mice. Development. 135:2193–2202. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parsons JT, Horwitz AR and Schwartz MA:
Cell adhesion: integrating cytoskeletal dynamics and cellular
tension. Nat Rev Mol Cell Biol. 11:633–643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kappert K, Schmidt G, Doerr G,
Wollert-Wulf B, Fleck E and Graf K: Angiotensin II and PDGF-BB
stimulate beta(1)-integrin-mediated adhesion and spreading in human
VSMCs. Hypertension. 35:255–261. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Olivares-Navarrete R, Hyzy SL, Park JH, et
al: Mediation of osteogenic differentiation of human mesenchymal
stem cells on titanium surfaces by a Wnt-integrin feedback loop.
Biomaterials. 32:6399–6411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Régent M, Planus E, Bouin AP, et al:
Specificities of β1 integrin signaling in the control of cell
adhesion and adhesive strength. Eur J Cell Biol. 90:261–269.
2011.
|