Introduction
Osteoarthritis (OA) is the most widespread
degenerative joint disease affecting articular cartilage and
subchondral bone. Individuals suffering from OA often experience
chronic pain, tenderness, restriction of movement, crepitus and
limited intraarticular inflammation (1). The etiology of OA is a complex
process centered on the disruption of anabolic-catabolic pathways
in the bodily tissues, induced by a number of biochemical,
biomechanical, and genetic factors (2). One key event in the development of OA
that has garnered much attention, is the induction of apoptosis
within chondrocytes. A previous study by Hashimoto et al
linked chondrocyte apoptosis in human OA to cartilage degradation
following the examination of the apoptotic rates via in situ
and in vitro methods (3).
Furthermore, Heraud et al observed that 18–21% of
chondrocytes from OA cartilage exhibited apoptotic features
compared with 2–5% in normal cartilage (4). Based on this evidence and other
studies, investigations are now focusing on the chondrocyte, as
well as the expression of cytokines, cell signaling molecules and
pro-apoptotic proteins, as mediators of OA pathogenesis.
Interleukin-1β (IL-1β) is a pro-inflammatory
cytokine secreted by chondrocytes that has been linked to cartilage
degradation in OA (5). In 2000,
Heraud et al demonstrated that IL-1β increased the
percentage of apoptotic cells in both normal and OA cartilage in a
dose-dependent manner (4). IL-1β
was also demonstrated to promote mitochondrial dysfunction and
energy depletion in mouse chondrocyte-like ATDC5 cells following 48
h of treatment (6). Furthermore,
Lopez-Armada et al described an upregulation of
pro-apoptotic Bcl-2 family proteins in human articular chondrocytes
in response to IL-1β (7). In the
present study, due to its apoptosis-inducing effect on
chondrocytes, the IL-1β-induced model of apoptosis was selected to
investigate the mechanisms of cell death in cultured cells.
PUMA, a pro-apoptotic protein belonging to the Bcl-2
protein family, is closely associated with mitochondrial-dependent
apoptosis and serves as a major effector of p53-mediated cell
death. Upregulation of PUMA has been observed in a variety of
apoptosis models, including oligodendroglial cell death in toxic
demyelination (8),
microglia-derived TNFα induces apoptosis in neural precursor cells
(9) and crizotinib induces
apoptosis in colon cancer cells (10). Knockdown or knockout of PUMA in
SH-SY5Y neuroblastoma and PC-12 cells induces a significant delay
in cellular apoptosis (11,12),
demonstrating that PUMA-mediated apoptosis is a widespread and
conserved mechanism. However, until now, there have been no studies
illustrating the role of PUMA in OA.
The involvement of c-Jun N-terminal kinase (JNK) in
signaling transduction pathways has been well-characterized in
mammalian cells (13), including
human articular chondrocytes (14). In particular, immunostaining
studies by Fan et al demonstrated the activation of the JNK
signaling cascade in cultured, normal articular chondrocytes
following treatment with IL-1β (15). c-Jun, a member of the AP-1
transcriptional complex, is preferentially phosphorylated and
activated by JNK and is central in the regulation of several matrix
metalloproteinases that promote the destruction of OA chondrocytes
(16–18). Of note, the JNK/c-Jun pathway has
also been reported to mediate the gene expression of PUMA in
neuronal and tumor apoptosis (19,20).
Based on these results and others, the JNK/c-Jun pathway posed as
an attractive target in our investigations to elucidate the
mechanism of apoptosis in chondrocytes.
In the present study, we investigated whether the
JNK/c-Jun pathway was involved in the induction of PUMA and thus
contributes to the pathological process of OA. The results
demonstrate that PUMA levels are upregulated and mediated by
JNK/c-Jun pathway during the IL-1β-induced apoptosis of
chondrocytes. These data suggest a role for PUMA and the JNK/c-Jun
pathway in the regulation of chondrocyte apoptosis during OA.
Materials and methods
Isolation and culture of human articular
chondrocytes
Human articular cartilage samples from 11 patients
with OA (range, 61–72 years) were obtained at the time of total
knee replacement. Normal cartilage was obtained from eight
post-mortem donors (range, 61–69 years) with no previous history of
joint pain. Informed consent was obtained in accordance with the
local ethics commission and all studies were conducted under the
approval of the Research Ethics Board of The Third Affiliated
Hospital of Sun Yat-sen University (Guangzhou, China). Cartilage
samples were harvested and fragmented into small pieces. Following
the digestion of the cartilage samples with collagenase D, the
resulting cells were incubated at 37°C in a 5% CO2
humidified atmosphere in DMEM containing 10% fetal bovine serum
(FBS) and 1% penicillin/streptomycin.
Isolation and culture of mouse articular
chondrocytes
Articular chondrocytes were isolated from the
femoral heads, femoral condyles and tibial plateau of 5- to
6-day-old C57BL/6 mice. Briefly, articular cartilage tissues were
minced (<1 mm3), and digested with 0.3 and 0.05%
collagenase D at 37°C for 45 min and overnight, respectively. The
cell suspension was filtered through a sterile 40 μM nylon mesh
cell strainer and the released cells were cultured in DMEM medium
supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in
a humidified 5% CO2 atmosphere. Cell viability of
isolated chondrocytes was determined by trypan blue exclusion
assay. Primary cells were maintained as a monolayer culture
throughout this study.
Apoptosis assays
Apoptotic cells were detected by fluorescence
microscopy with cell permeable Hoechst 33258 dye (Invitrogen Life
Technologies, Carlsbad, CA, USA). Following 48 h of transfection
with siRNA fragments (Shanghai Genepharma, Co., Ltd, Shanghai,
China), a subconfluent monolayer of mouse chondrocytes was
subjected to stress with 10 ng/ml IL-1β for 48 h and stained with
Hoechst 33258 dye (5 μg/ml). The nuclear morphology of cells was
examined for the presence of condensed nuclei (apoptotic cells) and
images were captured on an inverted fluorescence microscope (Axio
Observer Z1; Carl Zeiss AD, Germany). The percentage of apoptotic
cells was calculated from a minimum of 8 fields with >100
cells/field in an unbiased manner and the cells were scored in a
blinded manner without previous knowledge of their treatments.
Immunohistochemical analysis
Immunohistochemistry for PUMA was performed using a
polymer-based technology (Envision; Dako, Glostrup, Denmark).
Briefly, human articular cartilage samples were dissected and
post-fixed overnight in freshly prepared 4% paraformaldehyde in PBS
(pH 7.4) at 4°C. Subsequently, the tissue samples were embedded in
paraffin, cut into 5 μm thick sections and mounted onto
gelatin-coated slides. Paraffin sections were incubated for 1 h at
60°C, deparaffinized in xylenes and rehydrated in a series of
graded alcohols. Following three washes in phosphate-buffered
saline (PBS; pH 7.4), endogenous peroxidase was inactivated by
incubation in 3% H2O2 in methanol for 10 min.
Antigen retrieval was then performed by microwaving slides in 10 mM
sodium citrate buffer (pH 6.0) for 15 min followed by cooling to
room temperature. Sections were incubated with an anti-PUMA
antibody (1:100) (ab33906; Abcam, Cambridge, MA, USA) at 4°C
overnight. Following washing in PBS, a polymer reagent was applied
for 30 min and the expression of PUMA was visualized by incubation
with diaminobenzidine tetrahydrochloride (DAB). Sections were then
washed with PBS, counterstained with hematoxylin for 1 min,
dehydrated for 15 min and mounted in neutral gum. The expression of
PUMA was evaluated by IPP (version 6.0; Media Cybernetics, Silver
Spring, MD, USA) following the collection of ten digital images at
1360×1024 pixel resolution at a magnification of ×400 using a
BX51WI microscope (Olympus, Tokyo, Japan). The measurement
parameters included density mean, area sum and inter ocular
distance (IOD). Following calibration of the optical density, the
image was converted to gray scale and the values were counted.
Western blotting and antibodies
Whole cell lysates were prepared from chondrocytes
lysed in ice-cold RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS,
0.5% sodium deoxycholate, 1% Triton X-100, 10 mM NaF, 0.1 mM
Na3VO4 and 1 mM phenylmethylsulfonyl
fluoride). Lysates were cleared by centrifugation at 30,000 × g for
10 min at 4°C and protein concentrations were determined by an
Bradford assay. A 30 μg aliquot of each cell lysate was subjected
to SDS-polyacrylamide gel electrophoresis (PAGE) as described by
22506026. Proteins were then transferred to polyvinylidene
difluoride membranes (Millipore Corporation, Billerica, MA, USA)
and membranes were probed with polyclonal antibodies specific for
PUMA (diluted 1:1,000; Abcam), Bax, c-Jun and GAPDH (diluted
1:1,000; Cell Signaling Technology, Inc) overnight at 4°C.
Following washing, membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (dilution, 1:1,000;
Jackson ImmunoResearch, West Grove, PA, USA) for 60 min and the
proteins were visualized using the ECL chemiluminescence system
(Forevergen Bioscience, China).
Reverse transcription (RT)-PCR
Total RNA was extracted from isolated chondrocytes
using TRIzol reagent (Invitrogen, USA) according to the
manufacturer’s recommendations. First strand cDNA was synthesized
from 1 μg of total RNA using M-MLV Reverse Transcriptase (Promega,
Southampton, UK) and oligo(dB) primers. Reverse transcription was
performed using SuperScript reverse transcriptase (Invitrogen Life
Technologies) and oligo-dT primers. The following sense and
anti-sense primers for Puma, c-jun, and
β-actin were used: Puma: forward,
5′-AGCGGCGGAGACAAGAA-3′ and reverse, 5′-CAAGTCCGTATCTCCATCAGTG-3′;
c-jun: forward, 5′-CCTTCTACGACGATGCCCTC-3′ and reverse,
5′-GGTTCAAGGTCATGCTCTGTTT-3′; β-actin: forward,
5′-GGCTGTATTCCCCTCCATCG-3′ and reverse
5′-CCAGTTGGTAACAATGCCATGT-3′.
siRNA knockdown
Mouse chondrocytes were transfected with 100 nM of
siRNA against Puma using Lipofectamine reagent (Invitrogen Life
Technologies) according to the manufacturer’s instructions. siRNA
sequences were as follows: si-PUMA-1,
5′-CCTGGAGGGTCATGTACAATCTCTT-3′; si-PUMA-2,
5′-GGAGGGTCATGTACAATCTCTTCAT-3. One scrambled siRNA was designed to
serve as a siRNA transfection control with the following sequence;
5′-UGGUUUACAUGUCGACUAA-3′. The cells were lysed 72 h following
siRNA transfection and the degree of knockdown was assessed by
RT-PCR and western blot analysis.
Results
PUMA mRNA and protein levels are
upregulated in IL-1β treated mouse chondrocytes
The apoptosis of chondrocytes has a key role in the
development of OA, however, the mechanisms responsible for the
induction of apoptosis remain unclear. In order to study the
apoptotic signaling pathway, mouse chondrocytes were cultured and
treated with 10 ng/ml of IL-1β to mimic the pathogenesis of OA.
Since it has been demonstrated that PUMA regulates extracellular
apoptotic pathways, we investigated the effects of IL-1β treatment
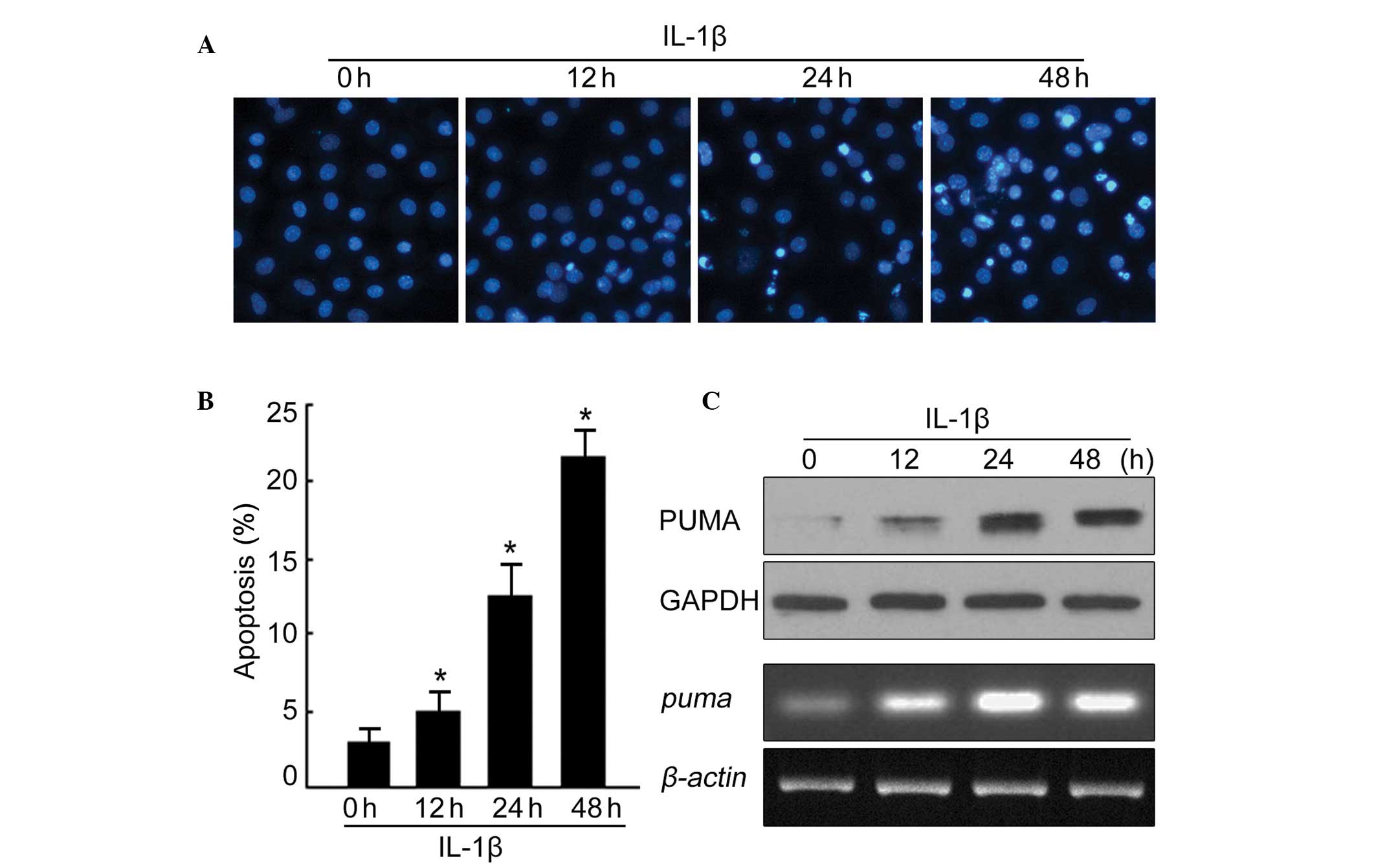
on PUMA levels during chondrocyte apoptosis. As illustrated in
Fig. 1A, chondrocytes underwent
apoptosis after 48 h of treatment with IL-1β. The apoptotic rate
was quantified following Hoechst staining and demonstrated that to
be ~23% in treated chondrocytes, however only 3% in untreated cells
(Fig. 1B). To determine PUMA
protein and mRNA levels, IL-1β-treated chondrocytes were then
subjected to western blotting and RT-PCR. As illustrated in
Fig. 1C, the PUMA protein and mRNA
levels were significantly increased throughout the time course of
IL-1β treatment. Together, these data indicate that IL-1β treatment
not only has the ability to induce chondrocyte apoptosis, but also
to alter PUMA protein and mRNA levels during the apoptotic
process.
PUMA upregulation is critical for IL-1β
induced chondrocyte apoptosis
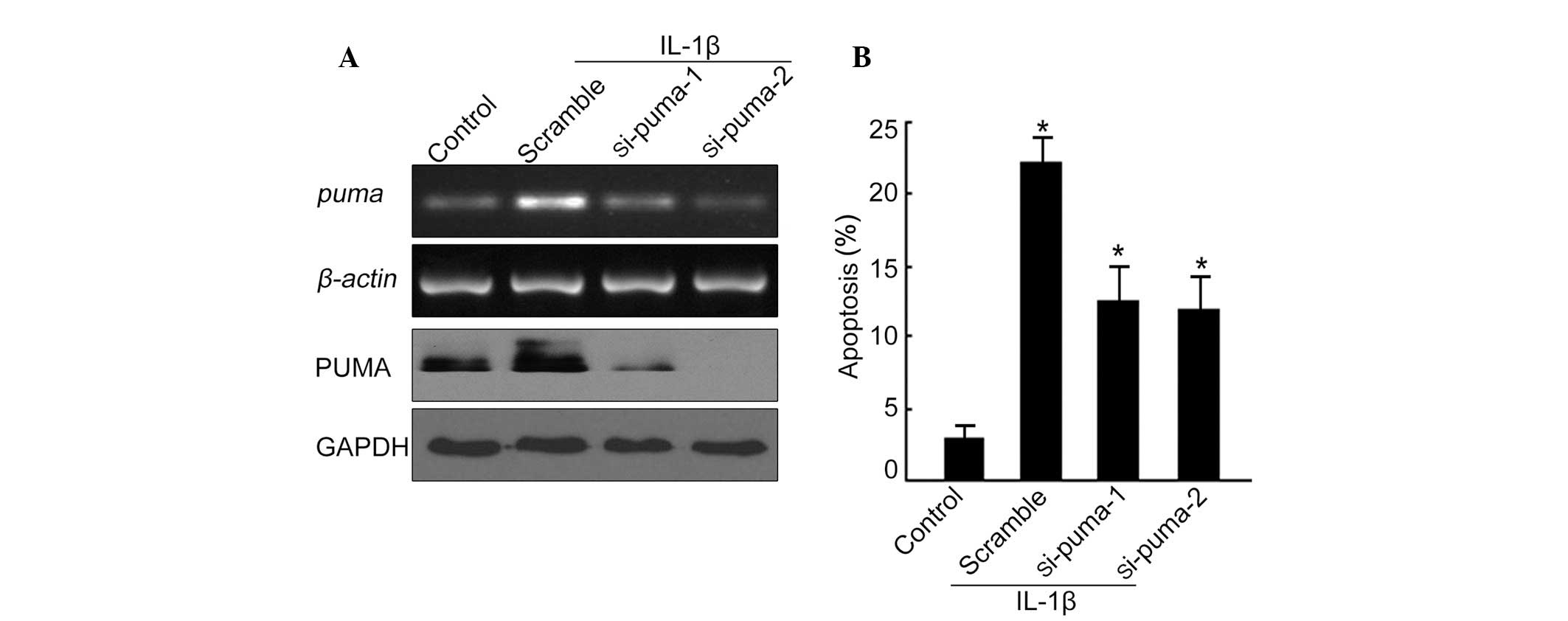
Since PUMA mRNA and protein levels were upregulated
in cultured chondrocytes following IL-1β stimulation, we then
investigated whether PUMA upregulation was essential for
chondrocyte apoptosis. Two different siRNA fragments specifically
targeting PUMA were designed. The knockdown efficiency of each
siRNA was determined by RT-PCR and western blot analysis following
72 h of transfection. Of the two siRNAs tested, si-PUMA-2 gave an
enhanced knockdown efficiency at a concentration of 100 nM. As
demonstrated in Fig. 2A (upper
panel), the mRNA of PUMA was decreased by si-PUMA-1 and si-PUMA-2,
respectively, as compared with the scrambled siRNA. PUMA protein
levels were also significantly reduced upon treatment with
si-PUMA-1 and si-PUMA-2 (Fig. 2A,
lower panel), indicating a successful knockdown effect with these
siRNA fragments. In addition to this, knockdown of PUMA triggered a
marked decreased in the apoptotic rate in the presence of IL-1β
(Fig. 2B). These data suggest that
PUMA is critical for IL-1β induced chondrocyte apoptosis.
JNK/c-Jun pathway mediates upregulation
of PUMA in IL-1β treated chondrocytes
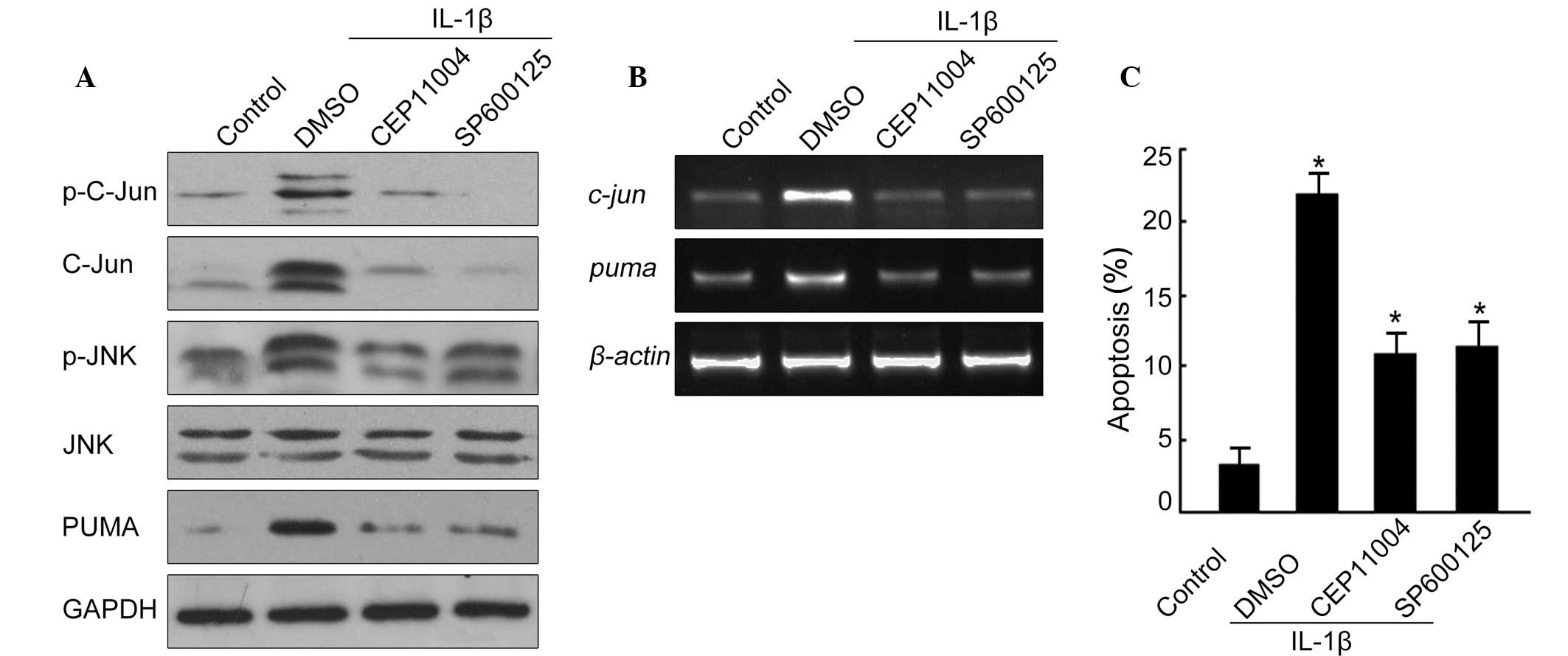
Since the JNK/c-Jun pathway has been reported to
regulate PUMA induction, it was considered that this pathway may
also be involved in the induction of PUMA in an IL-1β-induced model
of apoptosis in mouse chondrocytes. Two potent and specific
inhibitors of the JNK/c-Jun pathway were used; SP600125 to inhibit
JNK and CEP11004 to inhibit MLK, an upstream kinase of JNK. The
results identified an increased phosphorylation of JNK and c-Jun
following IL-1β treatment, indicating activation of the JNK/c-Jun
pathway (Fig. 3A). Upon the
addition of 10 μM SP600125 or 2 μM CEP11004, there was a decrease
in the phosphorylation levels of c-Jun and JNK, as well as a
decrease in the protein levels of PUMA (Fig. 3A). PUMA protein levels were
decreased with the two inhibitors following IL-1β treatment
(Fig. 3A). Simultaneously, PUMA
mRNA was also suppressed upon treatment with SP600125 or CEP11004
in the presence of IL-1β (Fig.
3B).
Following this, we determined whether inhibition of
the JNK/c-Jun pathway would impact the rate of apoptosis induced by
IL-1β. As demonstrated in Fig. 3C,
apoptosis induced by IL-1β was significantly suppressed following
the incubation of cells with SP600125 and CEP11004, respectively.
This accumulative evidence suggests that the JNK/c-Jun pathway may
be activated by IL-1β treatment and that it acts upstream of PUMA
to mediate chondrocyte apoptosis.
PUMA and c-Jun are activated in
chondrocyte tissues of OA patients
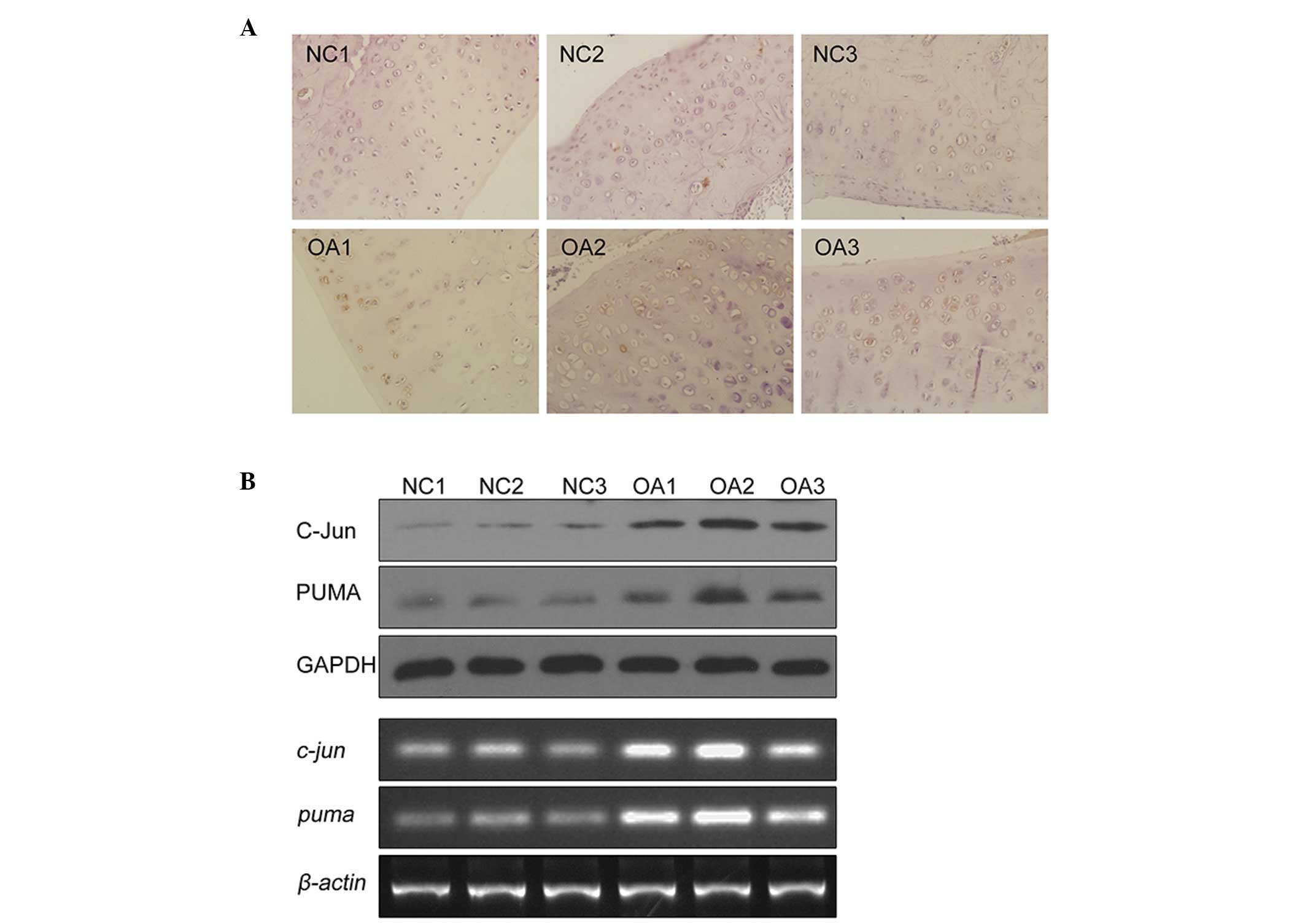
To determine whether c-Jun and PUMA are upregulated
in chondrocyte tissues isolated from OA patients, the expression
level of c-Jun and PUMA was examined in human articular cartilage
and in primary cultured chondrocytes from OA patients. Using
immunohistochemistry, an increased staining of PUMA was observed in
the articular cartilage of OA patients as compared with the normal
tissues (Fig. 4A). Positive
staining for PUMA was identified in 56% of OA tissues tested (data
not presented). Then, chondrocytes from the tissues of patients
with or without a history of OA were isolated and cultured. Cell
lysates were prepared and subject to western blot analysis. As
compared with chondrocytes derived from healthy tissue (Fig. 4; NC1,2,3), the protein levels of
c-Jun and PUMA in chondrocytes from OA patients were elevated
(Fig. 4; OA1, 2, 3). Similarly,
the mRNA levels for c-Jun and PUMA were also elevated in OA
patients (Fig. 4, lower panel).
These data demonstrated that PUMA and c-Jun are significantly
upregulated in OA patients, suggesting they have a role in the
apoptosis observed in the tissues of OA patients.
Discussion
Numerous different types of cell death have been
reported in cartilage-apoptosis, necrosis and chondroptosis.
However, much controversy remains regarding which of these types
predominate during OA (21).
Numerous investigators have demonstrated correlations between
increased rates of chondrocyte apoptosis and the severity of OA in
animals (22–26) and in humans (27–32).
These studies have used a wide range of analytical tools to
demonstrate the correlation between apoptosis and OA, including
histology, TUNEL staining, caspase-3 activation, ELISA and
fluorescence-activated cell sorter analysis (FACS). Despite this,
the relative importance of apoptosis in OA remains controversial
because other studies have failed to confirm the presence of large
numbers of apoptotic cells in OA cartilage (33). Therefore, elucidating the mechanism
of cell death in cartilage may be vital for identifying new
therapeutic agents for OA.
In the present study, it was demonstrated for the
first time, to the best of our knowledge, that upregulated PUMA is
critical for OA apoptosis in cultured chondrocytes. The results
reveal that PUMA protein levels are significantly increased in
human OA cartilage tissues and in chondrocytes cultured from these
tissues. Additionally, chondrocytes isolated from human OA tissues
also exhibit elevated PUMA mRNA levels as compared with
chondrocytes derived from normal tissue. Similar to human
chondrocytes, mouse chondrocytes also exhibited increased PUMA
protein and mRNA levels following IL-1β treatment. This
upregulation of PUMA implicates mitochondrial apoptosis in the
progression of OA. However, we identified that Bax levels do not
change in chondrocytes cultured human OA cartilage or in
chondrocytes exposed to IL-1β (data not presented). These results
may indicate the involvement of other Bcl-2 pro-apoptotic
mediators, such as Bid, Bak and/or Bad. Future studies are
necessary to determine the role(s) of other pro-apoptotic mediators
in the pathogenesis of OA.
Activation of the JNK/c-Jun pathway has been
reported in human OA cartilage and chondrocytes treated with IL-1β
(14–16). Although PUMA is reported to be a
downstream target of the JNK/c-Jun pathway in neurons, the
correlation between the JNK/c-Jun pathway and PUMA in OA has not
yet been determined. In the present study, it was demonstrated that
c-Jun is largely upregulated in cultured human and mouse
chondrocytes upon IL-1β treatment, which supports previous evidence
in the literature. Furthermore, we identified that PUMA functions
as a downstream effector of the JNK/c-Jun pathway because
pharmacological inhibition of this pathway attenuated the
expression of PUMA at transcriptional and post-transcriptional
levels. Together, these results indicate that the JNK/c-Jun pathway
may alter PUMA induction and may have a central role in the
pathological process of OA.
In summary, the present study has demonstrated that
PUMA is significantly upregulated in OA and the upregulation of
PUMA is induced by the JNK/c-Jun pathway. This JNK/c-Jun regulation
of PUMA expression is associated with cartilage damage and may
provide a new therapeutic target for the development of treatment
strategies for OA in the future.
Acknowledgements
The authors are grateful for financial support from
the National Natural Science Foundation of China (no. 81272040 and
30600632) and the Natural Science Foundation of Guangdong Province,
China (no. S2011010004808).
References
|
1
|
Goldring MB and Goldring SR:
Osteoarthritis. J Cell Physiol. 213:626–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arden N and Nevitt MC: Osteoarthritis:
epidemiology. Best Pract Res Clin Rheumatol. 20:3–25. 2006.
View Article : Google Scholar
|
|
3
|
Hashimoto S, Ochs RL, Komiya S and Lotz M:
Linkage of chondrocyte apoptosis and cartilage degradation in human
osteoarthritis. Arthritis Rheum. 41:1632–1638. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heraud F, Heraud A and Harmand MF:
Apoptosis in normal and osteoarthritic human articular cartilage.
Ann Rheum Dis. 59:959–965. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi M, Squires GR, Mousa A, et al:
Role of interleukin-1 and tumor necrosis factor alpha in matrix
degradation of human osteoarthritic cartilage. Arthritis Rheum.
52:128–135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yasuhara R, Miyamoto Y, Akaike T, et al:
Interleukin-1beta induces death in chondrocyte-like ATDC5 cells
through mitochondrial dysfunction and energy depletion in a
reactive nitrogen and oxygen species-dependent manner. Biochem J.
389:315–323. 2005. View Article : Google Scholar
|
|
7
|
Lopez-Armada MJ, Carames B, Lires-Dean M,
et al: Cytokines, tumor necrosis factor-alpha and
interleukin-1beta, differentially regulate apoptosis in
osteoarthritis cultured human chondrocytes. Osteoarthritis
Cartilage. 14:660–669. 2006. View Article : Google Scholar
|
|
8
|
Hagemeier K, Lurbke A, Hucke S, et al:
Puma, but not noxa is essential for oligodendroglial cell death.
Glia. 61:1712–1723. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guadagno J, Xu X, Karajgikar M, Brown A
and Cregan SP: Microglia-derived TNFalpha induces apoptosis in
neural precursor cells via transcriptional activation of the Bcl-2
family member Puma. Cell Death Dis. 4:e5382013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng X, He K, Zhang L and Yu J:
Crizotinib induces PUMA-dependent apoptosis in colon cancer cells.
Mol Cancer Ther. 12:777–786. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reimertz C, Kogel D, Rami A, Chittenden T
and Prehn JH: Gene expression during ER stress-induced apoptosis in
neurons: induction of the BH3-only protein Bbc3/PUMA and activation
of the mitochondrial apoptosis pathway. J Cell Biol. 162:587–597.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zou CG, Cao XZ, Zhao YS, et al: The
molecular mechanism of endoplasmic reticulum stress-induced
apoptosis in PC-12 neuronal cells: the protective effect of
insulin-like growth factor I. Endocrinology. 150:277–285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Islam S, Kermode T, Sultana D, et al:
Expression profile of protein tyrosine kinase genes in human
osteoarthritis chondrocytes. Osteoarthritis Cartilage. 9:684–693.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan Z, Soder S, Oehler S, Fundel K and
Aigner T: Activation of interleukin-1 signaling cascades in normal
and osteoarthritic articular cartilage. Am J Pathol. 171:938–946.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mengshol JA, Vincenti MP, Coon CI,
Barchowsky A and Brinckerhoff CE: Interleukin-1 induction of
collagenase 3 (matrix metalloproteinase 13) gene expression in
chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear
factor kappaB: differential regulation of collagenase 1 and
collagenase 3. Arthritis Rheum. 43:801–811. 2000. View Article : Google Scholar
|
|
17
|
Newton R, Stevens DA, Hart LA, Lindsay M,
Adcock IM and Barnes PJ: Superinduction of COX-2 mRNA by
cycloheximide and interleukin-1beta involves increased
transcription and correlates with increased NF-kappaB and JNK
activation. FEBS Lett. 418:135–138. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ray A, Shakya A and Ray BK:
Inflammation-responsive transcription factors SAF-1 and c-Jun/c-Fos
promote canine MMP-1 gene expression. Biochim Biophys Acta.
1732:53–61. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ambacher KK, Pitzul KB, Karajgikar M,
Hamilton A, Ferguson SS and Cregan SP: The JNK- and AKT/GSK3beta-
signaling pathways converge to regulate Puma induction and neuronal
apoptosis induced by trophic factor deprivation. PloS One.
7:e468852012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao Z, Wang J, Tang J, et al: JNK- and
Akt-mediated Puma expression in the apoptosis of
cisplatin-resistant ovarian cancer cells. Biochem J. 444:291–301.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zamli Z and Sharif M: Chondrocyte
apoptosis: a cause or consequence of osteoarthritis? Int J Rheum
Dis. 14:159–166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carames B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
D’Lima D, Hermida J, Hashimoto S, Colwell
C and Lotz M: Caspase inhibitors reduce severity of cartilage
lesions in experimental osteoarthritis. Arthritis Rheum.
54:1814–1821. 2006.PubMed/NCBI
|
|
24
|
Zemmyo M, Meharra EJ, Kuhn K,
Creighton-Achermann L and Lotz M: Accelerated, aging-dependent
development of osteoarthritis in alpha1 integrin-deficient mice.
Arthritis Rheum. 48:2873–2880. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mistry D, Oue Y, Chambers MG, Kayser MV
and Mason RM: Chondrocyte death during murine osteoarthritis.
Osteoarthritis Cartilage. 12:131–141. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thomas CM, Fuller CJ, Whittles CE and
Sharif M: Chondrocyte death by apoptosis is associated with
cartilage matrix degradation. Osteoarthritis Cartilage. 15:27–34.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blanco FJ, Guitian R, Vazquez-Martul E, de
Toro FJ and Galdo F: Osteoarthritis chondrocytes die by apoptosis.
A possible pathway for osteoarthritis pathology. Arthritis Rheum.
41:284–289. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sharif M, Whitehouse A, Sharman P, Perry M
and Adams M: Increased apoptosis in human osteoarthritic cartilage
corresponds to reduced cell density and expression of caspase-3.
Arthritis Rheum. 50:507–515. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsuo M, Nishida K, Yoshida A, Murakami T
and Inoue H: Expression of caspase-3 and -9 relevant to cartilage
destruction and chondrocyte apoptosis in human osteoarthritic
cartilage. Acta Med Okayama. 55:333–340. 2001.PubMed/NCBI
|
|
30
|
Kim HA, Lee YJ, Seong SC, Choe KW and Song
YW: Apoptotic chondrocyte death in human osteoarthritis. J
Rheumatol. 27:455–462. 2000.
|
|
31
|
Hashimoto S, Takahashi K, Amiel D, Coutts
RD and Lotz M: Chondrocyte apoptosis and nitric oxide production
during experimentally induced osteoarthritis. Arthritis Rheum.
41:1266–1274. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hashimoto S, Nishiyama T, Hayashi S, et
al: Role of p53 in human chondrocyte apoptosis in response to shear
strain. Arthritis Rheum. 60:2340–2349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aigner T, Hemmel M, Neureiter D, et al:
Apoptotic cell death is not a widespread phenomenon in normal aging
and osteoarthritis human articular knee cartilage: a study of
proliferation, programmed cell death (apoptosis), and viability of
chondrocytes in normal and osteoarthritic human knee cartilage.
Arthritis Rheum. 44:1304–1312. 2001.
|