Introduction
Nemaline myopathy (NM) is a rare congenital muscle
disease with an incidence of 0.02 per 1,000 live births (1), which is characterized by the presence
of rod-like structures in skeletal muscle fibers. In 1963, Conen
et al (2) and Shy et
al (3) were the first to
describe NM. The common clinical manifestation of NM is its onset,
characterized by hypotonia or general weakness predominantly
affecting facial, axial and proximal limb muscles. An important
clinical feature of NM is that weakness is more predominant in the
respiratory muscles compared to other muscle groups, and this is
the main cause of mortality of NM patients. According to the degree
of muscle weakness, its severity and the age at onset, six types
have been clinically defined by the European Neuromuscular Center
(ENMC) International Consortium on NM (4): severe congenital, intermediate
congenital, typical congenital, mild childhood, adult onset and
other unusual types. Cases are often sporadic, but other cases with
family history exhibit either autosomal recessive or dominant
patterns of inheritance; mutations associated with the disease have
been discovered in seven genes to date (5): TPM3, NEB, ACTA1,
TPM2, TNNT1, CFL2 and KBTBD13. Although
NM is a disease of high clinical and genetic heterogeneity, its
diagnosis is based on standard criteria, namely, the presence of
characteristic purple subsarcolemmal or cytoplasmic rods,
visualized with the modified Gömöri trichrome dye and observed
under a light microscope, or the presence of high electron-dense
nemaline bodies of Z-line origin, observed by electron
microscopy.
Since the first NM case reported by Cao et al
(6) from the Beijing Friendship
Hospital (Beijing, China), a total of 16 cases have been reported
in China (7–16), but no study has systematically
focused on the clinical and pathological features of NM. In our
study, we present clinical and pathological data from a total of 28
NM patients from China, which we used to investigate the clinical
diversity and pathological features of NM.
Patients and methods
Patients and ethics
A total of 28 patients were included in this study.
These patients were divided in two groups. Clinical and
pathological data from a total of 28 NM patients from China is
presented and the clinical diversity and pathological features of
Chinese patients with NM were investigated. Their enrollment was
based on the references of these studies, available from the China
Academic Journal Electronic Publishing House (http://www.cnki.net) and from the Medline database.
This group comprised a total of 16 patients from 11 studies. The
second group comprised 12 patients, admitted in the Department of
Neurology at the Chinese People’s Liberation Army General Hospital
from 1986 until 2011. The 12 patients underwent muscle biopsy and
were diagnosed on the presence of rods in the muscle fibers.
Consent to analyze the muscle biopsies was obtained in the
out-patient clinic or during hospital admission for diagnostic
investigation. This study was approved by the hospital’s
Institutional Review Board.
Clinical information and laboratory
examination
Study protocol data collected from each patient
included: i) detailed personal history (age at onset, initial
symptom or sign, motor milestone delay, clinical course and family
history); ii) clinical features (distribution of muscle weakness
and atrophy, progression pattern, cardiac or respiratory issues,
dysphagia and mental retardation); iii) dysmorphic features
(pseudohypertrophy, high arched palate and foot, elongated face and
spinal deformity); iv) laboratory data [level of serum creatine
kinase (CK), results from electromyography and nerve conduction
velocity]; and v) muscle power (from manual muscle testing).
Electromyography
Electromyography and nerve conduction velocity tests
were performed at the same time with the electromyography equipment
(keypoint 2000; Dantec Dynamics, Skovlunde, Denmark). After
informed consent was signed by the patients, a needle containing
two fine-wire electrodes was inserted through the skin into the
muscle tissue. A trained neurologist observed and recorded the
electrical activity while inserting the electrode. The bilateral
deltoid muscle, biceps brachii muscle, quadriceps femoris muscle
and anterior tibial muscle were assessed. Spontaneous signals from
a muscle at rest were measured. Then, the patients were asked to
contract slightly. The shape, size and frequency of the motor-unit
action potentials were recorded. In addition, the recruitment was
recorded when the patients were asked to contract vigorously. In
the nerve conduction velocity test, small electrical shocks were
delivered to a nerve at set locations. Electrical leads attached
elsewhere on the body picked up the electrical signal, and the
motor nerve velocity and sensory nerve velocity were recorded.
Muscle biopsy
Open muscle biopsy was performed on the biceps
brachii or the quadriceps femoris under local anesthesia. Following
removal, muscle samples were immediately frozen in isopentane
cooled with liquid nitrogen and were stored at -80°C. Transversal
serial 5-μm frozen muscle sections were stained with hematoxylin
and eosin, modified Gömöri trichrome stain, nicotinamide
dehydrogenase tetrazolium reductase (NADH-TR) and adenosine
triphosphatase (ATPase) after incubation at pH 4.3, 4.5 and 10.6.
Morphometric evaluation of the muscle specimens was performed under
a light microscope (BX51; Olympus, Tokyo, Japan). A 0.5×0.5×1.0
cm-size muscle sample was stored in glutaraldehyde and observed
under an electron microscope (JEM1230; JEOL, Ltd., Tokyo, Japan).
All reagents were purchased from Sinopharm Chemical Reagent Co.,
Ltd. (Beijing, China).
Results
Patient information
From April 1986 to December 2011, 4,127 muscle
biopsies were performed on suspected myopathy cases in our
hospital. The proportion of NM cases in this group was 0.29%
(12/4,127). In the 28 patients evaluated in this study, there were
18 male (M) and 10 female (F), with a ratio M:F=1.8:1. The
patients’ age ranged from 4 to 74 years, with a mean age 22.5±16.7
years. The disease course ranged from 1 month to 35 years, with a
mean disease course of 107.9±107.2 months. A total of 26 patients
with no family history were considered as sporadic cases, with 14
of these patients recruited from other studies and 12 from the
Neurology Department in our hospital. The remaining 2 patients
belong to a family with positive family history and were considered
as family cases.
Clinical information
Details on each patient are shown in Table I. Among the 28 patients, 15
presented mild hypotonia and chronic limb weakness during the
neonatal period, with delayed motor milestone in a few cases.
Symptoms such as lower limb weakness, dyspnea and myalgia developed
rapidly in 7 cases during adulthood. Three of these patients
displayed rapid clinical progression and acute respiratory
insufficiency, while their prognosis was poor. Lower limb weakness
at childhood or adolescence was the most common initial symptom
observed in the remaining 6 patients. Each of these patients leaded
an independent life with mild muscle weakness. According to the age
at onset, severity of muscle weakness, development of clinical
progression and respiratory involvement, we classified the three
groups of patients as typical congenital, adult onset and childhood
onset on the basis of NM classification criteria defined by the
ENMC International Consortium.
 | Table IClinical and pathological
characteristics of the 28 cases with nemaline myopathy. |
Table I
Clinical and pathological
characteristics of the 28 cases with nemaline myopathy.
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 |
|---|
| Subtype | Adult onset | Typical
congenial | Mild Childhood | Typical
congenital | Typical
congenital | Mild Childhood | Typical
congenital | Typical
congenital | Mild Childhood | Typical
congenital | Typical
congenital | Adult onset | Typical
congenital | Adult onset | Typical
congenital | Adult onset | Adult onset | Adult onset | Typical
congenital | Typical
congenital | Typical
congenital | Typical
congenital | Adult onset | Childhood onset | Childhood onset | Childhood onset | Typical
congenital | Typical
congenital |
| Gender/agea | M/49 | F/10 | F/21 | M/6 | F/35 | M/22 | M/27 | M/31 | M/21 | M/6 | M/17 | M/32 | M/7 | M/74 | M/4 | M/34 | F/52 | F/36 | F/10 | F/6 | F/5 | M/13 | M/35 | F/20 | M/14 | M/24 | M/9 | F/11 |
| Family history | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | Younger
brother | Elder sister |
| Age at
onseta | 45 | Birth | 17 | Birth | Birth | 16 | Birth | Birth | 18 | Birth | Birth | 25 | Birth | 74 | 4 | 34 | 47 | 36 | Birth | Birth | Birth | Birth | 35 | 17 | 6 | 7 | Birth | Birth |
| Mode of onset | Subacute | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Chronic | Subacute | Chronic | Acute | Subacute | Subacute | Chronic | Chronic | Chronic | Chronic | Acute | Chronic | Chronic | Chronic | Chronic | Chronic |
| Course of
diseasea | 4 | 10 | 4 | 6 | 35 | 6 | 27 | 31 | 4 | 6 | 17 | 7 | 7 | 1 month | 4 | 10 months | 5 | 8 months | 10 | 6 | 5 | 13 | 1 month | 3 | 8 | 17 | 9 | 11 |
| Initial
symptoms | L/EX weakness,
dyspnea | L/EX weakness | L/EX weakness | L/EX weakness | Infantile
hypotonia, EX weakness | L/EX weakness | L/EX weakness | L/EX weakness | L/EX weakness | EX weakness | EX weakness | EX weakness, muscle
atrophy | EX weakness,
delayed motor milestone | EX myalgia,
dyspnea | L/EX weakness | EX weakness, muscle
atrophy | L/EX weakness | L/EX neck weakness,
dyspnea | L/EX weakness | L/EX weakness | EX weakness | L/EX weakness | L/EX weakness | Mild muscle atrophy
of L/EX | L/EX weakness | L/EX weakness | Infantile
hypotonia, EX weakness | Infantile
hypotonia, EX weakness |
| Muscle weakness
distribution |
| Neck | 2 | 2+ | 3+ | 5− | 3 | 3+ | 3+ | 3+ | 4 | 3 | 3+ | 3 | 2 | 5 | 5 | 2 | 3 | 3 | 5 | 5 | 4 | 4 | 5 | 5 | 4 | 4 | 3 | 3 |
| Upper limb |
| Proximal | 1 | 4 | 5 | 5 | 3+ | 5 | 4 | 3+ | 4 | 3 | 4 | 3 | 1 | 5− | 5 | 1 | 4 | 1 | 4 | 4 | 4 | 4 | 5− | 5 | 5 | 3 | 3 | 4 |
| Distal | 5− | 5 | 5 | 5 | 4+ | 5 | 4 | 5− | 5 | 4+ | 5 | 4+ | 2 | 5 | 5 | 5 | 5− | 3 | 5 | 4 | 5 | 5− | 5− | 5 | 5 | 5 | 4 | 4+ |
| Lower limb |
| Proximal | 2 | 4 | 4+ | 5 | 3+ | 3+ | 4 | 3+ | 3 | 3 | 3 | 3− | 1 | 5− | 4 | 1 | 4 | 4 | 4 | 4 | 4 | 4 | 5− | 4 | 4 | 3 | 3 | 4 |
| Distal | 5− | 5 | 5 | 5 | 4+ | 5 | 2+ | 5− | 4+ | 4− | 5− | 4− | 2 | 5 | 5 | 5 | 5 | 5 | 5 | 4 | 5 | 5 | 5− | 3 | 5− | 4 | 4 | 4+ |
| Muscle atrophy | Trunk, proximal
EX | No | Gastroc-nemius
muscle | No | No | Proximal EX | L/EX, severe in
distal | Four EX | No | Proximal EX | No | Proximal EX | Proximal L/EX | Trunk | No | Trunk, proximal
EX | Proximal EX | U/EX | No | No | No | No | No | Distal L/EX | No | Mild muscle atrophy
of EX | Proximal EX | Proximal EX |
| Dysmorphic
features | No | No | No | High arched
feet | No | No | High arched
feet | No | High arched
feet | No | High arched
feet | No | No | No | No | No | No | No | No | No | No | No | No | No | High arched
feet | No | High arched palate
and feet, elongated face | High arched palate
and feet, elongated face |
| Clinical
progression | Rapid | No | Slow | No | No | Slow | Slow | Slow | Slow | Slow | Slow | Progressive | Progressive | Rapid | No | Rapid | Progressive | Progressive | No | No | No | Slow | Rapid | Slow | Slow | Slow | Slow | Slow |
| Deep tendon
reflex | ↓↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓↓ | ↓ | ↓ | No | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | No | No | No |
| CK (IU/l) | 107.6 | 32 | 56.5 | 195.1 | 194.1 | 235 | 110.4 | 69 | 113.2 | 111 | 133 | 132 | 80 | 133 | 216 | 78 | 106 | 91 | 61 | 76 | 93 | 261 | 133 | 86 | 70 | 93 | 28 | 35 |
| NCV/EMG | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M | N/M |
| Muscle biopsy
findings |
| Nemaline body in
fibers | Type I and II | Type I | Type I | Type I and II | Type I and II | Type I | Type I | Type I and II | Type I | Type I and II | Type I | Type I | Type I and II | Type I | Type I and II | Type I | Type I | Type I | Type I and II | Type I | Type I | Type I | Type I | Type I | Type I and II | Type I and II | Type I | Type I |
| Fiber typeI | PD | PD/Atrophy | PD | PD | PD/Atrophy | PD/Atrophy | PD | PD | PD/Atrophy | PD | PD | PD/Atrophy | PD | PD | PD | PD/HT | PD/HT | PD/HT | PD | PD | PD | PD | PD/HT | PD | PD/HT | PD | PD/HT | PD/HT |
Most patients walked slowly or easily lost balance
due to the lower limb muscle weakness, especially in the proximal
parts. However, two patients (nos. 7 and 24) showed a different
muscle weakness distribution, with higher severity of weakness
observed in the distal rather than the proximal parts. Muscle
weakness would then develop in the upper limb muscle, neck flexor
and facial muscle groups, but cardiomyopathy and ophthalmoplegia
were not detected in our study. Respiratory muscle involvement,
causing respiratory insufficiency and associated with poor
prognosis, was only found in adult onset type patients (42.9%,
3/7).
Similar to the muscle weakness distribution, the
muscle atrophy mostly occurred in the four limbs and markedly
affected the proximal parts. Three out of the 7 adult onset type
patients showed trunk muscle atrophy, and 9 out of the 15 typical
congenital type patients showed no muscle atrophy. In 15 typical
congenital NM cases, 7 showed high arched palates, 2 showed
elongated faces and no dysmorphic features in other cases were
identified. All patients exhibited reduced to absent tendon
reflexes. Needle electromyography revealed normal nerve conduction
and a combination of small, brief, motor-unit action potentials
with reduced recruitment in all patients; these are myopathic
electromyography presentations. The serum CK level ranged from 28
to 261 IU/l (normal range 2–200 IU/l) with a mean value 111.7±60.1
IU/l. The CK level was within the normal range for 89.3% of the
patients (25/28), while it was mildly to moderately elevated in
10.7% of the patients (3/28).
Pathological information
Histological analysis of the muscles biopsies was
performed in all cases, and similar abnormalities were found among
the samples. Hematoxylin and eosin staining revealed variations in
the fiber size, with areas of small groups of atrophic muscle
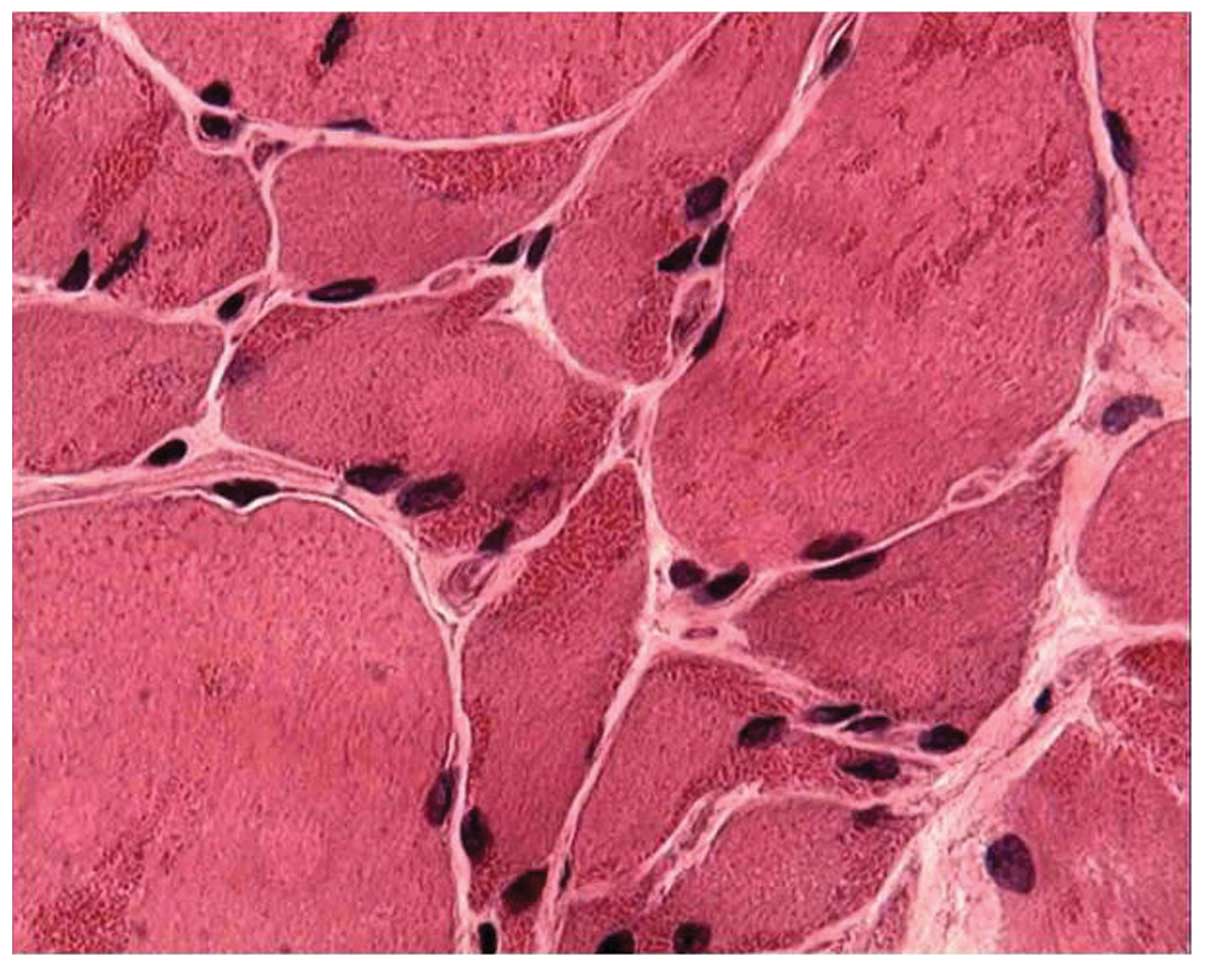
fibers. A frequency of red-stained rods >90% was found in the
cytoplasm and the subsarcolemmal region of muscle fibers in 19
patients (67.9%) (Fig. 1), while a
frequency of 70% was found in 7 (25%) and of 50% in 2 (7.1%)
patients. Some of the rods were detectable with hematoxylin and
eosin, but most of them became evident upon modified Gömöri
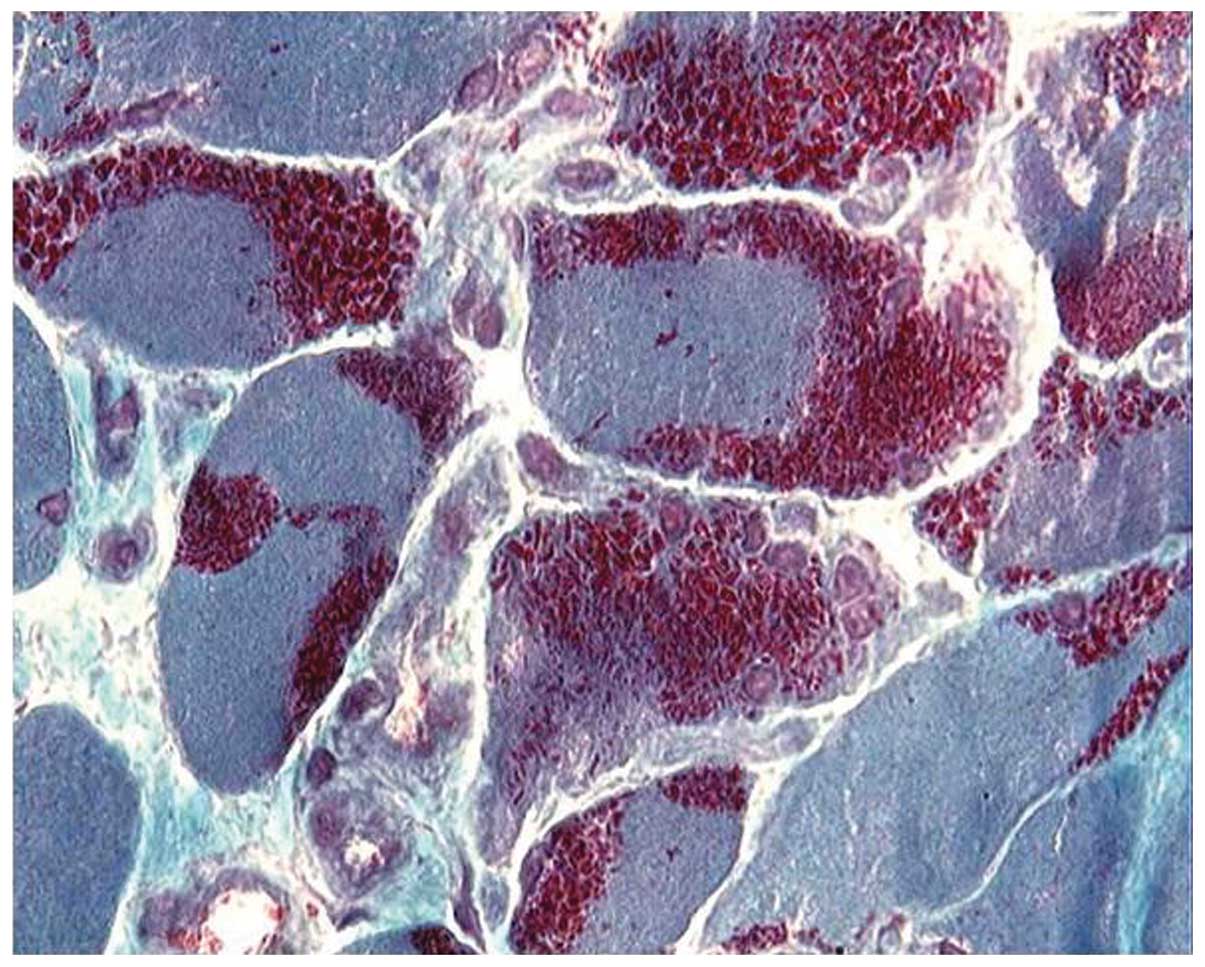
trichrome staining (Fig. 2).
Characteristic purple-colored rods were observed in the cytoplasmic
and subsarcolemmal region of muscle fibers stained with modified
Gömöri trichrome, which were more prominent in type I fibers.
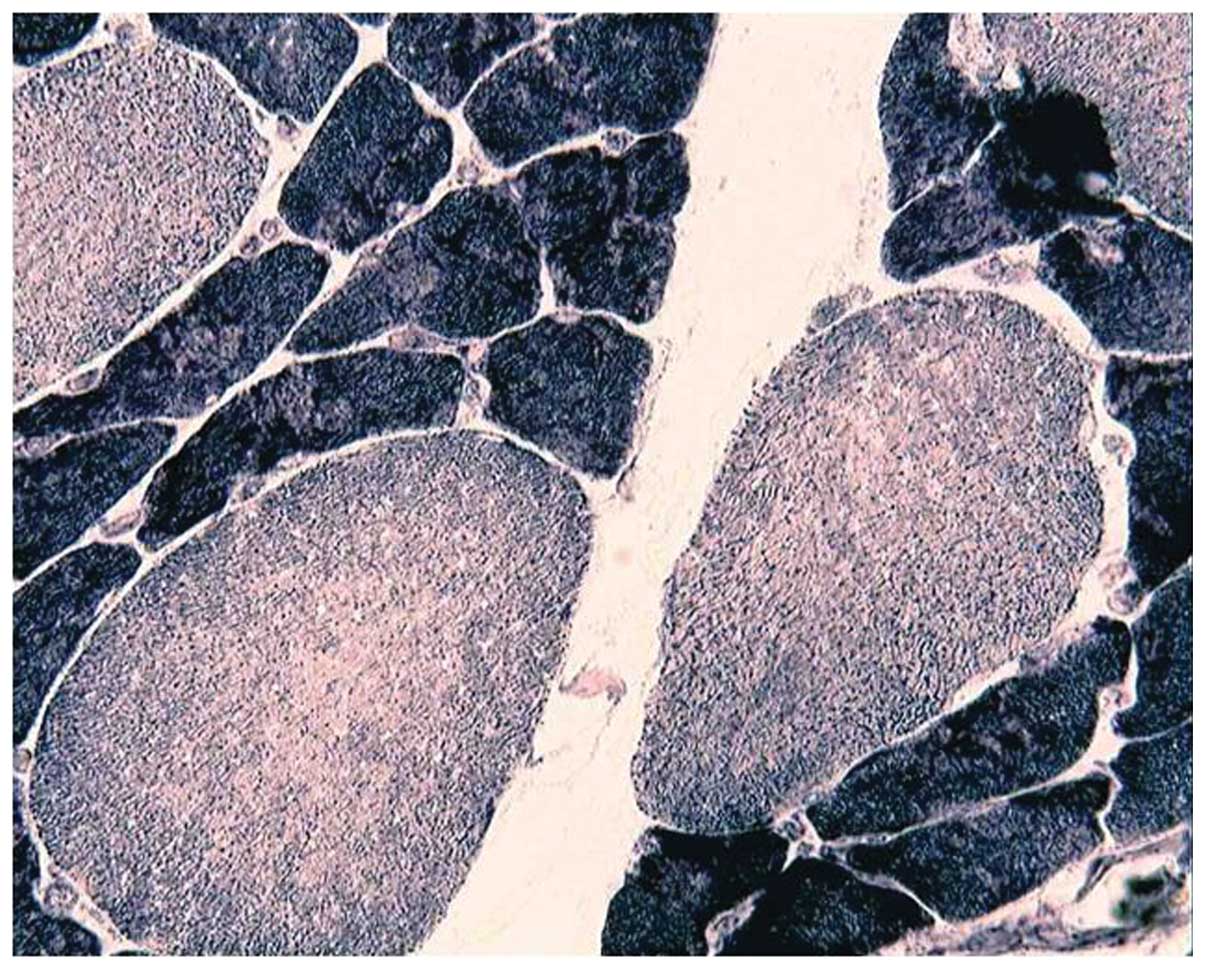
ATPase stain showed the predominance of type I fibers in all
patients (Fig. 3) and the type I
fiber atrophy in 12 patients. Nemaline rods were confined in type I
fibers in 18 patients, while rods were never observed to be
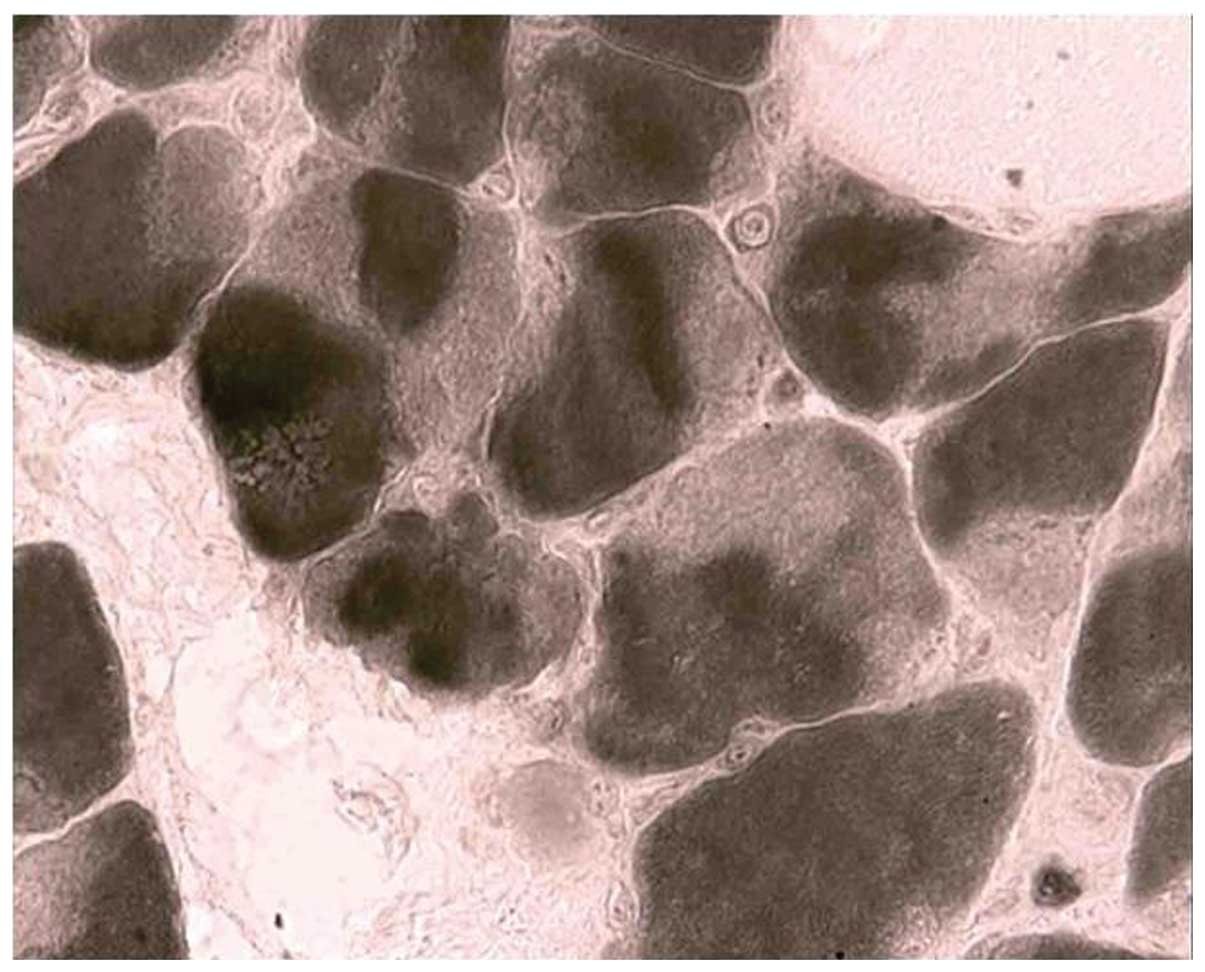
confined in type II fibers. A histochemical reaction for NADH-TR
showed that the darker, type I fibers are smaller in diameter than
the pale, type II fibers, and revealed the presence of core-like
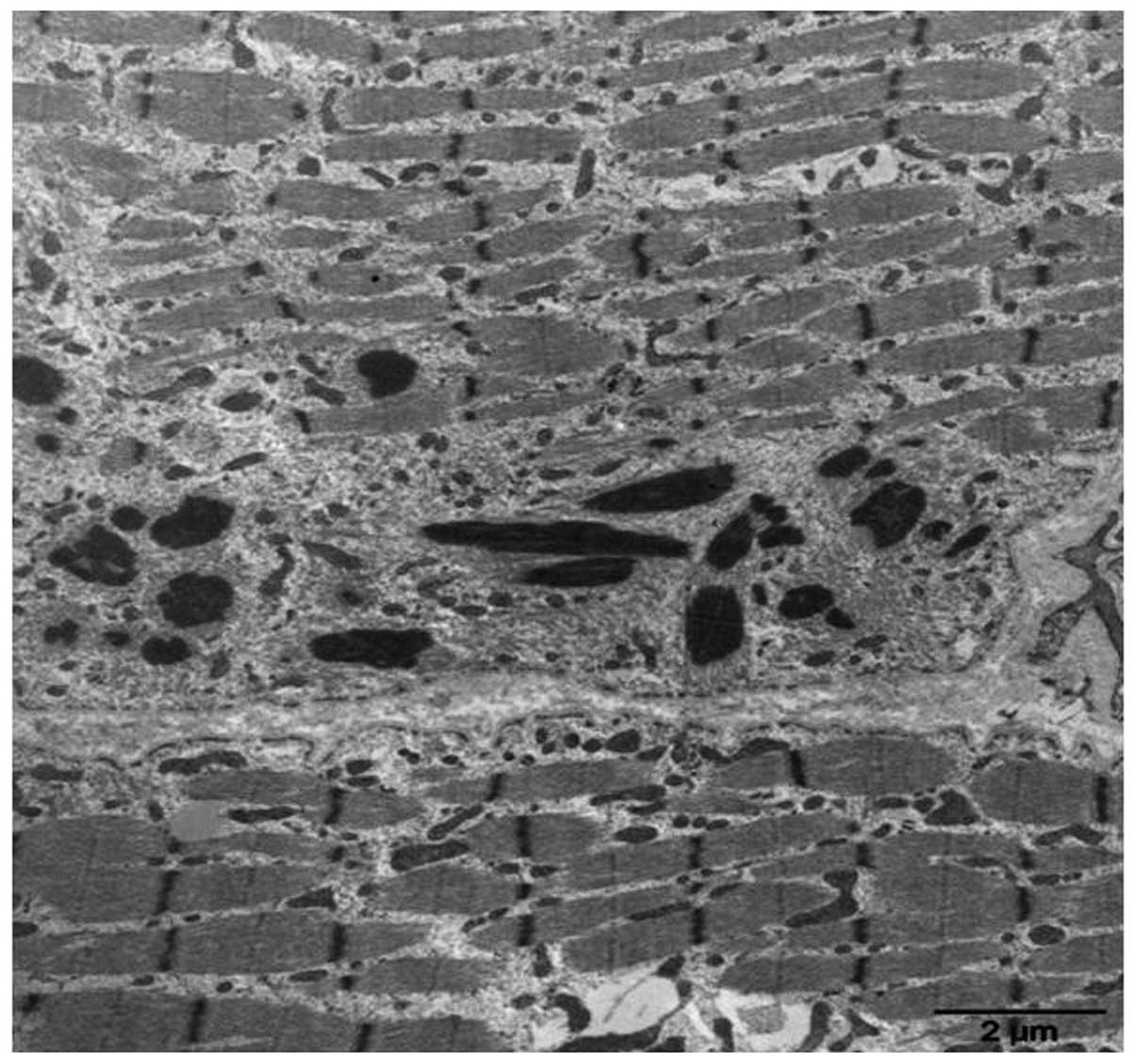
areas devoid of enzyme activity in some larger fibers (Fig. 4). Electron microscopy allowed
detection of nemaline rods as high electron-dense rod-shaped or
ovoid structures; these structures located in the middle of the
muscle fibers or were accompanied by a disorganized myofibrillar
apparatus, with broken myofilaments, and irregular myofibrils and Z
lines (Fig. 5).
Discussion
NM is a clinically heterogeneous congenital
myopathy, characterized by the presence of rod-like structures in
the cytoplasm of muscle fibers. The rods, or nemaline bodies, are
stained purple with the modified Gömöri trichrome staining method
or can be visualized by electron microscopy as high electron-dense
bodies similar to Z bands. Similar to other congenital myopathies,
NM was named after the Greek word ‘nema’ for the thread observed in
muscle biopsies. Although a relatively rare disease, NM is the most
common non-dystrophic congenital myopathy, with an estimated
incidence of 0.02 per 1,000 live births worldwide. In China, only a
few NM cases were reported, and related epidemiological data have
not yet become available. In our study, we screened, using muscle
biopsy, a total of 4,127 patients all over China from 1986 to 2011,
and identified 12 patients with histopathological signs of NM. The
proportion of NM in this pool of suspected myopathy cases is
~0.29%.
The clinical features of NM patients are highly
variable, ranging from neonatal death to mild limb weakness. The
onset age also ranges from neonates to several decade-old
individuals. In 2000, according to the age of patients at onset,
severity of muscle weakness, development of clinical progression
and respiratory involvement, the ENMC International Consortium
classified NM into six types (4).
The severe congenital type is the most severe type of NM. The first
symptoms manifest at birth, with hypotonia and profound muscle
weakness, particularly affecting the diaphragm and intercostal
muscles, and eventually leading to severe respiratory
insufficiency. Patients can perform few spontaneous movements and
have difficulties in swallowing and sucking. Most cases die within
one year. Patients of the intermediate congenital type can perform
spontaneous movements or respiration at birth, but as the disease
develops, most cases become unable to achieve ambulation or
respiratory independence at early childhood. Characteristic of
patients of this type is that walking is performed with the help of
a wheelchair before the age of 11 years. Most NM cases belong to
the typical congenital type. Although muscle weakness and hypotonia
occur during the neonatal period, these patients often improve
strength with age and lead an independent life. The clinical
features of patients of the mild childhood type are similar to
those of patients of the typical congenital type, except for the
onset age. The NM adult onset type comprises a heterogeneous group
of patients, with the disease onset reported between the third and
the sixth decade of life. Respiratory muscle involvement along the
rapidly progressive disease course is characteristic of this type,
and often causes respiratory dysfunction, while its is also
associated with poor prognosis. The other unusual types of NM
include rare features such as cardiomyopathy, ophthalmoplegia and
the detection of intranuclear nemaline bodies.
Three groups of patients were included in our study:
typical congenital, adult onset and mild childhood, accounting for
53.6, 25 and 21.4% of the studied population, respectively, This
distribution is similar to that reported by Ryan et al
(17). Our study highlighted the
clinical heterogeneity of NM. On the one hand, patients of the same
type shared a number of clinical features: limb weakness and
delayed motor milestone at birth in the typical congenital,
proximal limb weakness at early childhood in the mild childhood and
rapidly progressive disease course in the adult onset type. On the
other hand, there were also slight differences among patients of
the same type regarding aspects such as initial symptoms, muscle
weakness distribution and clinical progression. One finding from
the present study that is different from previous studies conducted
on patients from other countries is the high proportion of adult
onset type patients (17). The
reason for missing severe and intermediate type patients may relate
to the small number of patients enrolled in our study, and to a
bias for exclusion of neonatal and infantile patients: Patients in
neurological departments are often adolescents and adults, since NM
children can seldom survive long. A future study will include a
higher number of NM patients to confirm the validity of our
findings.
Because of the great heterogeneity of NM patients,
muscle biopsy analysis used for diagnosis of NM was mainly based on
the presence of rod-like structures or nemaline bodies in the
muscle fibers. Haematoxylin and eosin-stained sections of skeletal
muscle from patients with NM appeared normal, or exhibited a
certain variation in fiber size, but staining of frozen sections
with the modified Gömöri trichrome dye revealed the presence of
rods, the hallmark of this disorder, which were stained purple in
contrast to the pale blue-green myofibrils. Our study also showed a
tendency for rods to be confined in type I fibers, in clusters
under the sarcolemmal membrane, or rarely in the nuclei, which is
often associated with poor prognosis (18). Similar to a previous report
(19), muscle type disproportion
was common, and fiber type I predominance and muscle atrophy were
frequently observed in NM patients. The distribution and number of
rods was variable between different muscles or different patients,
and sometimes a second biopsy was needed. There was no obvious
correlation between the number of rods and the clinical severity of
the disease. A correlation between the severity of clinical
features and alterations in the size and proportion of type I
myofibers was previously reported (20).
When observed under an electron microscope, rods
appeared as high electron-dense ovoid structures that were often
parallel to the long axis of the sarcomere or located near the Z
disc, measuring 1–7 μm in length and 0.3–2 μm in width. The rods
altered the normal structure of the sarcomere, while in the area
without rods, the sarcomere was well organized. Although the exact
source of rods is still unclear, these structures may derive from
the Z disc. Luther and Squire (21) demonstrated with
electron microscopy experiments that rods have a lattice structure
similar to that of Z disks, while the major constituent of rods is
α-actinin, which is also the component of the Z disc.
Genetic studies have identified mutations in seven
distinct genes that are associated with the onset of NM:
slow-muscle α-tropomyosin (TPM3) (22), nebulin (NEB) (23), α-actin (ACTA1) (24), β-tropomyosin (TPM2)
(25), troponin T1 (TNNT1)
(26), cofilin-2 (CFL2) and
kelch repeat and BTB domain-containing 13 (KBTBD13)
(27). Except for KBTBD13,
the other six genes all encode protein components of the muscle
thin filaments (28). Importantly,
this molecular variability in NM-causing mutations extends to
inheritance patterns, as mutations in at least a few of these genes
have either autosomal dominant or autosomal recessive patterns of
inheritance. The gene NEB was localized on the human
chromosome 2q21.2-q22 based on a linkage study by
Wallgren-Pettersson et al (23); the gene encodes the protein
nebulin, which is the main component of the axis of the thin
filament. Mutations in NEB are only seen in autosomal
recessive cases (29) and are
associated with 50% of NM cases related to gene mutation. Moreover,
most clinical cases related to NEB mutations are of typical
congenital type (30). Besides
mutations in NEB, the second most common mutation associated
with NM patients was discovered in the ACTA1 gene,
accounting for 10–20% of NM cases caused by mutations (31). About half of the cases related to
mutations in ACTA1 are associated with the severe congenital
type and display autosomal recessive, as well as autosomal dominant
inheritance patterns (32). A
mutation in TNNT1 was identified in Old Order Amish families
with NM history, inherited in an autosomal recessive pattern
(26). A mutation in the
CFL2 gene, locating on 14q12, was reported in four cases
displaying facial weakness and foot drop (33,34).
TPM3 mutations have been discovered in rare dominant and
recessive cases of NM, and rarely are TPM2 in autosomal dominant
mode (25). It is notable that
nemaline bodies are only present in type I fibers of patients with
TPM3 mutations, in accordance with the fact that TPM3
encodes the slow-muscle α-tropomyosin in type I fibers. Recently,
mutations in KBTBD13 have been reported to cause dominantly
inherited NM (26). In contrast to
the other 5 genes mutations in which cause NM, KBTB13
encodes a protein with a kelch repeat and a BTB domain, which is
not related to the thin filament of the myofiber; the function of
this protein is still unclear.
In conclusion, NM is a rare myopathy in China, with
only 0.29% of cases detected among the screened myopathy cases in
this study. The 28 Chinese patients with NM examined herein showed
great heterogeneity in clinical features, and their types ranged
from typical congenital and mildly affected at childhood to adult
onset type with respiratory failure. The diagnosis of NM was mainly
based on the presence of characteristic rods in >50% of the
muscle fibers. Genetic analyses are necessary to further
investigate the pathophysiology of the observed clinical and
pathological features of these patients.
Acknowledgements
This study was supported by a grant from the
Characteristic Clinical Application Program of the Beijing
Municipal Science and Technology Commission (no.
Z111107058811108).
References
|
1
|
Agrawal PB, Strickland CD, Midgett C, et
al: Heterogeneity of nemaline myopathy cases with skeletal muscle
alpha-actin gene mutations. Ann Neurol. 56:86–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Conen PE, Murphy EG and Donohue WL: Light
and electron microscopic studies of ‘myogranules’ in a child with
hypotonia and muscle weakness. Can Med Assoc J. 89:983–986.
1963.
|
|
3
|
Shy GM, Engel WK, Somers JE and Wanko T:
Nemaline myopathy. A new congenital myopathy. Brain. 86:793–810.
1963. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wallgren-Pettersson C and Laing NG: Report
of the 70th ENMC International Workshop: nemaline myopathy, 11–13
June 1999, Naarden, The Netherlands. Neuromuscul Disord.
10:299–306. 2000.PubMed/NCBI
|
|
5
|
Wallgren-Pettersson C, Sewry CA, Nowak KJ
and Laing NG: Nemaline myopathies. Semin Pediatr Neurol.
18:230–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao P, Lu D and Feng LF: A case report of
nemaline myopathy. Hered Dis. 7:243–244. 1990.
|
|
7
|
Li YX, Wu LJ, Chen QT, et al: Adult
nemaline myopathy. J Clin Neurol. 11:369–370. 1998.
|
|
8
|
Li YX, Wu LJ and Chen QT: Clinical and
pathological discussion: the 42nd cases. Chin J Neurol. 34:122–124.
2001.
|
|
9
|
Han Y, Zheng HM, Zhou S, Deng BQ, Wu T, et
al: A discussion of clinical and pathological features and
pathogenesis of nemaline myopathy (NM). J Apoplexy Nerv Dis.
19:90–92. 2002.
|
|
10
|
Yan CZ, Liu SP, Wu JL, et al: Adult form
nemaline myopathy: two cases report with clinicopathological and
ultrastructural study. Chin J Neurol. 36:173–175. 2002.(In
Chinese).
|
|
11
|
Lai HW, Yang CH, Wang FH and Tang GY:
Electron microscopic observation of nemaline myopathy (report of a
case). J Chin Electr Microsc Soc. 23:77–80. 2004.
|
|
12
|
Yuan JH, Hu J, Chen L, Mei L, Kang ZJ, et
al: The clinical and pathological analysis of the nemaline myopathy
complicated with the uniform type I fiber myopathy. J Apoplexy Nerv
Dis. 22:432–433. 2005.
|
|
13
|
Chen L and Hu J: A case report of nemaline
myopathy. Chin J Pract Pediatr. 21:1542006.
|
|
14
|
Lu Y, Da Y, Wang M, Liu L and Jia JP:
Nemaline myopathy - A 2 cases report and review of literatures.
Chin J Neuroimmunol Neurol. 15:236–238. 2008.
|
|
15
|
Lu HD, Li ZF, Qin DX, Zhang SJ, Qian Q, et
al: Clinical and pathological analysis of 3 nemaline myopathy
cases. Chin J Neurol. 41:465–467. 2008.
|
|
16
|
Jiang H1, Xiao B, Jia DD, Zhang N, Xu XP,
et al: Clinical and pathologic analysis of an autosomal recessive
kindred with nemaline myopathy. Zhonghua Yi Xue Za Zhi.
89:3316–3319. 2009.(In Chinese).
|
|
17
|
Ryan MM, Schnell C, Strickland CD, Shield
LK, Morgan G, et al: Nemaline myopathy: a clinical study of 143
cases. Ann Neurol. 50:312–320. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goebel HH, Piirsoo A, Warlo I, Schofer O,
Kehr S, et al: Infantile intranuclear rod myopathy. J Child Neurol.
12:22–30. 1997. View Article : Google Scholar
|
|
19
|
Gurgel-Giannetti J, Reed UC, Marie SK,
Zanoteli E, Fireman MA, et al: Rod distribution and muscle fiber
type modification in the progression of nemaline myopathy. J Child
Neurol. 18:235–240. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bhatt JR and Pascuzzi RM: Neuromuscular
disorders in clinical practice: case studies. Neurol Clin.
24:233–265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luther PK and Squire JM: Muscle Z-band
ultrastructure: Titin Z-repeats and Z-band periodicities do not
match. J Mol Biol. 319:1157–1164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Laing NG, Wilton SD, Akkari PA, Dorosz S,
Boundy K, et al: A mutation in the alpha tropomyosin gene TPM3
associated with autosomal dominant nemaline myopathy NEM1. Nat
Genet. 10:2491995. View Article : Google Scholar
|
|
23
|
Wallgren-Pettersson C, Avela K, Marchand
S, et al: A gene for autosomal recessive nemaline myopathy assigned
to chromosome 2q by linkage analysis. Neuromuscul Disord.
5:441–443. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nowak KJ, Wattanasirichaigoon D, Goebel
HH, Wilce M, Pelin K, et al: Mutations in the skeletal muscle
alpha-actin gene in patients with actin myopathy and nemaline
myopathy. Nat Genet. 23:208–212. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wattanasirichaigoon D, Swoboda KJ, Takada
F, Tong HQ, Lip V, et al: Mutations of the slow muscle
alpha-tropomyosin gene, TPM3, are a rare cause of nemaline
myopathy. Neurology. 59:613–617. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johnston JJ, Kelley RI, Crawford TO,
Morton DH, Agarwala R, et al: A novel nemaline myopathy in the
Amish caused by a mutation in troponin T1. Am J Hum Genet.
67:814–821. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sambuughin N, Yau KS, Olivé M, Duff RM,
Bayarsaikhan M, et al: Dominant mutations in KBTBD13, a member of
the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum
Genet. 87:842–847. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanoudou D and Beggs AH: Clinical and
genetic heterogeneity in nemaline myopathy - a disease of skeletal
muscle thin filaments. Trends Mol Med. 7:362–368. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ottenheijm CA, Hooijman P, DeChene ET,
Stienen GJ, Beggs AH, et al: Altered myofilament function depresses
force generation in patients with nebulin-based nemaline myopathy
(NEM2). J Struct Biol. 170:334–343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wallgren-Pettersson C, Pelin K, Nowak KJ,
Muntoni F, Romero NB, et al: Genotype-phenotype correlations in
nemaline myopathy caused by mutations in the genes for nebulin and
skeletal muscle alpha-actin. Neuromuscul Disord. 14:461–470. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Visegrády B and Machesky LM:
Myopathy-causing actin mutations promote defects in serum response
factor signaling. Biochem J. 427:41–48. 2010.PubMed/NCBI
|
|
32
|
Laing NG, Dye DE, Wallgren-Pettersson C,
Richard G, Monnier N, et al: Mutations and polymorphisms of the
skeletal muscle alpha-actin gene (ACTA1). Hum Mutat. 30:1267–1277.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Agrawal PB, Greenleaf RS, Tomczak KK, et
al: Nemaline myopathy with minicores caused by mutation of the CFL2
gene encoding the skeletal muscle actin-binding protein, cofilin-2.
Am J Hum Genet. 80:162–167. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ockeloen CW, Gilhuis HJ, Pfundt R, et al:
Congenital myopathy caused by a novel missense mutation in the CFL2
gene. Neuromuscul Disord. 22:632–639. 2012. View Article : Google Scholar : PubMed/NCBI
|