Introduction
Bladder cancer is the sixth most common type of
cancer in the US, with 72,570 diagnoses estimated in 2013 (1). Patients with locally advanced or
metastatic bladder cancer exhibit poor 5-year overall survival
rates of 10–15% (2). The
mechanisms of progression from superficial to muscle-invasive, and
then to metastatic bladder cancer remain largely unknown. The
activation of an epithelial-mesenchymal transition (EMT) program
has been proposed as the critical mechanism for the acquisition of
the invasive phenotype and the subsequent systemic spread of cancer
cells. The loss of epithelial markers and the acquisition of
mesenchymal morphological features are characterized by the
disassembly of tight junctions and loss of normal cellular polarity
due to decreasing expression of E-cadherin and increasing
expression of various mesenchymal markers, including vimentin and
N-cadherin (3). It is widely
accepted that EMT is altered in various types of tumor, including
bladder cancer (4).
Transforming growth factor-β1 (TGF-β1) controls cell
proliferation and differentiation during embryonic development
(3), and numerous studies have
indicated that it is also associated with migration and metastases
in multiple types of malignant tumor (5–7).
TGF-β1 has been demonstrated to induce the expression of matrix
metalloproteinases (MMPs), which facilitate tumor cell invasion and
metastasis via the degradation of extracellular matrix. MMP-16 is a
membrane-anchored MMP that is able to activate other MMPs (MMP-2
and 9), growth factors and receptors, and thereby facilitates a
local cellular mechanism for migration (8). TGF-β signaling also regulates
pathological EMT, inducing a number of diseases ranging from
inflammatory disorders to fibrosis and cancer (9).
microRNAs (miRNAs) are short, single-stranded RNA
molecules that interact with the 3′ untranslated region (UTR) of
mRNAs to regulate gene expression (10). An increasing number of studies have
documented a link between the expression of miRNA and cancer
pathogenesis. Several independent studies have indicated that the
miR-200 family (miR-200a, 200b, 200c, 141 and 429) are
downregulated in aggressive human tumors, and are potent inhibitors
of EMT, tumor invasion and metastasis (4,11,12).
Therefore, the present study hypothesized that
TGF-β1 may be involved in the regulation of EMT and the expression
of MMP-16 in bladder cancer. The present study was designed to
investigate whether TGF-β1 is able to induce EMT and upregulation
of MMP-16, and to identify an association between EMT and MMP-16 in
bladder cancer.
Materials and methods
Cell culture and reagents
The human bladder cancer cell lines, HTB9 and T24
(American Type Culture Collection, Manassas, VA, USA) were cultured
in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA)
supplemented with 3.75% fetal bovine serum (Gemini Bio-Products,
Woodland, CA, USA) and 100 U/ml streptomycin-penicillin sulfate
(Life Technologies, Grand Island, NY, USA) at 37°C in a humidified
atmosphere with 5% CO2. Recombinant human TGF-β1 was
obtained from R&D Systems (Minneapolis, MN, USA) and used at
the specified concentrations. TGF-β1 treatment was conducted
without serum deprivation.
Semi-quantitative polymerase chain
reaction (qPCR)
Total RNA from cell samples was extracted using the
RNeasy Mini kit (Qiagen, Alameda, CA, USA). Complementary DNA
(cDNA) was synthesized using the High-Capacity cDNA Archive kit
(Applied Biosystems, Foster City, CA, USA), according to the
manufacturer’s instructions. The cDNA was synthesized from 2 μg
total RNA on a PTC-200 Peltier Thermal Cycler DNA engine (MJ
Research Inc., Waltham, MA, USA). The thermal cycler was used with
a Chromo 4™ Real-Time Detection system and Opticon Monitor
software, version 3.1 (Bio-Rad, Hercules, CA, USA) for qPCR
analysis. Cycle threshold (Ct) values were normalized to the
housekeeper GAPDH gene, and comparative quantification was
performed based on a 2−ΔΔCt calculation method (13). The specific primer sequences were
as follows: GAPDH F, 5′-ACCACAGTCCATGCCATCAC-3′ and R, 5′-TCCACCACC
CTGTTGCTGTA-3′; N-cadherin F, 5′-AACCCTTATTTTGCC CCCAAT-3′ and R,
5′-TCAACATGGTACCGGCATGA-3′; E-cadherin F,
5′-CGGGAATGCAGTTGAGGATC-3′ and R, 5′-AGGATGGTGTAAGCGATGGC-3′;
vimentin F, 5′-GAC CTCTACGAGGAGGAGAT-3′ and R, 5′-TTGTCAACATCCTG
TCTGAA-3′.
For mature miRNA analysis, total RNAs were extracted
from cell cultures with a mirVana™ miRNA Isolation kit according to
the instructions of the manufacturer (Applied Biosystems). For
miRNA detection, qPCR was performed using TaqMan miRNA assays
(Applied Biosystems) with specific commercial primer sets. All
reagents and protocols were from Applied Biosystems, and detection
was performed using U6 as an internal control. miRNA-specific qPCR
was performed in triplicate and repeated three times.
Protein characterization
For western blot assessment, the cells were plated
in culture dishes. Following treatment, the untreated control and
TGF-β1-treated cells were harvested by scraping and then washed
with phosphate-buffered saline (PBS). Cells were collected
following centrifugation at 856 × g and pellets were resuspended in
lysis solution (10 mM Tris, 150 mM NaCl, 1% Triton X-100, 1 mM
EDTA, 1 mM EGTA, 0.2 mM sodium orthovanadate, 0.5% NP-40, 0.3 mM
PMSF, 10 μg/ml Aprotinin). Protein estimation was performed with
crystal violet protein dye (Sigma-Aldrich) using a DU 800
spectrophotometer (Beckman Coulter, Fullerton, CA, USA). Protein
(50 μg) was electrophoresed using NuPAGE 4–12% Bis-Tris gel
(Invitrogen Life Technologies, Carlsbad, CA, USA). The protein was
transferred to a nitrocellulose membrane (Invitrogen Life
Technologies) using a wet method at 100 volts for 1 h. The membrane
was then blocked with 5% (w/v) milk and placed in a rotator for 1 h
at room temperature. The primary antibody was added to the culture
with milk (2.5% w/v) and allowed to incubate overnight at 4°C. The
membrane was then washed with PBS/0.05% Tween-20 three times (15
min each) prior to the addition of the appropriate horseradish
peroxidase-linked secondary antibody and incubation for 1 h at room
temperature. The primary antibodies were anti-E-cadherin,
anti-vimentin (Cell Signaling Technology, Danvers, MA, USA),
anti-MMP-16 (Chemicon, Temecula, CA, USA), anti-N-cadherin (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and anti-GAPDH
(Santa Cruz Biotechnology, Inc.). The membrane was then washed
three times for 15 min each, prior to the addition of SuperSignal
Chemiluminescent substrate (Pierce Biotechnology, Inc., Rockford,
IL, USA) and then immediately visualized using a ChemiDoc Imaging
system (Bio-Rad).
Plasmid construction and dual-luciferase
reporter assay
To validate whether miR-200b was able to suppress
the expression of MMP-16 by directly targeting the 3′UTR region,
luciferase assays were performed. Changsha Yinrun Biotechnologies
Inc. (Changsha, China) constructed pGL3-MMP-16 by amplifying the
3′UTR of the MMP-16 gene harboring the miR-200b binding site, as
predicted by TargetScan (http://www.targetscan.org), and subsequently cloning
it into the pGL3 control vector (Promega, Madison, WI, USA) at the
Xbal site immediately downstream of firefly luciferase
(f-luc). pGL3-MMP-16-mut, which contained deletions of the MMP-16
3′UTR target regions, was generated to be used as a negative
control. For the luciferase assay, HTB9 and T24 cells were cultured
in 12-well plates and each cotransfected with 400 ng of either
pGL3-MMP-16 or pGL3-MMP-16-mut, 50 ng pRL-TK (Promega) and 50
nmol/l 200b mimics or negative control (NC). Cells were lysed after
48 h, and luciferase activity was determined using the
Dual-Luciferase Reporter Assay system (Promega). The results were
expressed as relative luciferase activity (firefly
luciferase/Renilla luciferase).
Transwell assay
Cells were transfected with control or miR-200b
mimic for 48 h and then were treated with or without TGF-β1 (10
ng/ml) for 24 h. 5×104 cells were resuspended and added
to the upper chamber of a Transwell system (BD Biosciences,
Franklin Lakes, NJ, USA). Cells were incubated for 24 h, and those
that did not migrate through the pores were removed by scraping the
upper surface of the membrane with a cotton swab. Cells that had
migrated to the lower surface of the membrane were fixed for 5 min
in 100% methanol (Sigma-Aldrich) and stained with 0.2% crystal
violet. The cells that migrated through the insert were counted in
five random fields and expressed as the average number of
cells/field. All experiments were conducted in triplicate and
performed at least three times.
Statistical analysis
Data were analyzed using SPSS software, version 11.0
(SPSS Inc., Chicago, IL, USA) for Windows. P<0.05 was considered
to indicate a statistically significant difference.
Results
TGF-β1 induces EMT in HTB9 and T24
bladder cancer cell lines
EMT is associated with progression and poor
prognosis in numerous types of cancer, and EMT can be induced by
TGF-β1 (14,15). In the present study, a time-course
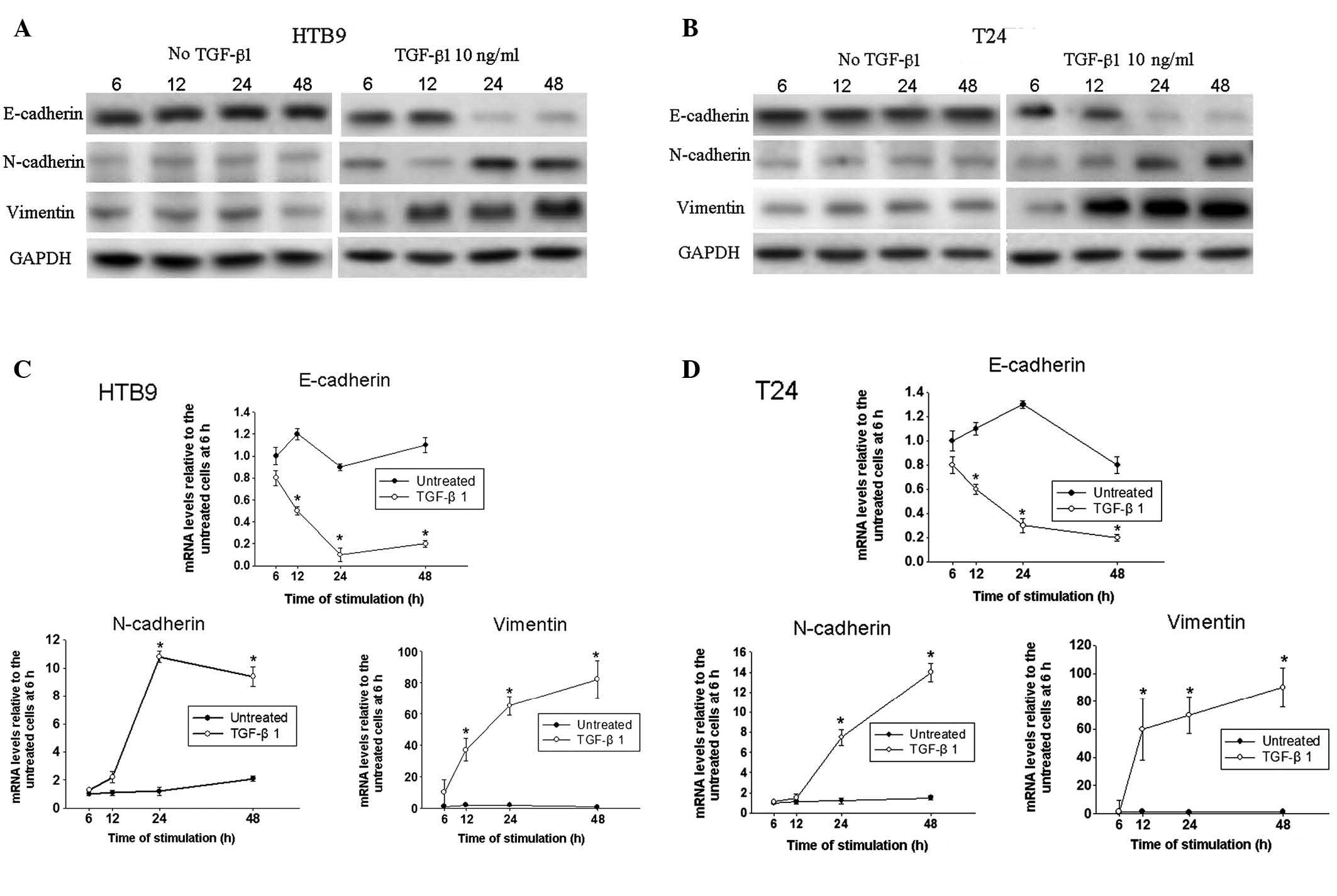
western blot analysis was designed to investigate the expression of
EMT markers, including E-cadherin, N-cadherin and vimentin. The
expression of all these markers remained stable at the protein
level in the untreated HTB9 and T24 cells throughout the 48 h
(Fig. 1A and B). However,
following TGF-β1 treatment of HTB9 and T24 cells, the expression of
E-cadherin was progressively downregulated in a time-dependent
manner, while the expression levels of N-cadherin and vimentin were
increased, particularly after 24 h. qPCR was used to confirm
whether mRNA levels were altered in a manner consistent with the
changes observed at the protein level. A reduction in E-cadherin
mRNA levels in the TGF-β1-treated cells compared with those in
control cells was first observed after 6 h of treatment (Fig. 1C and D). N-cadherin and vimentin
mRNA expression levels were significantly upregulated following
TGF-β1 treatment, which is consistent with the observations from
the western blots.
TGF-β1 reduces the expression of
miR-200b
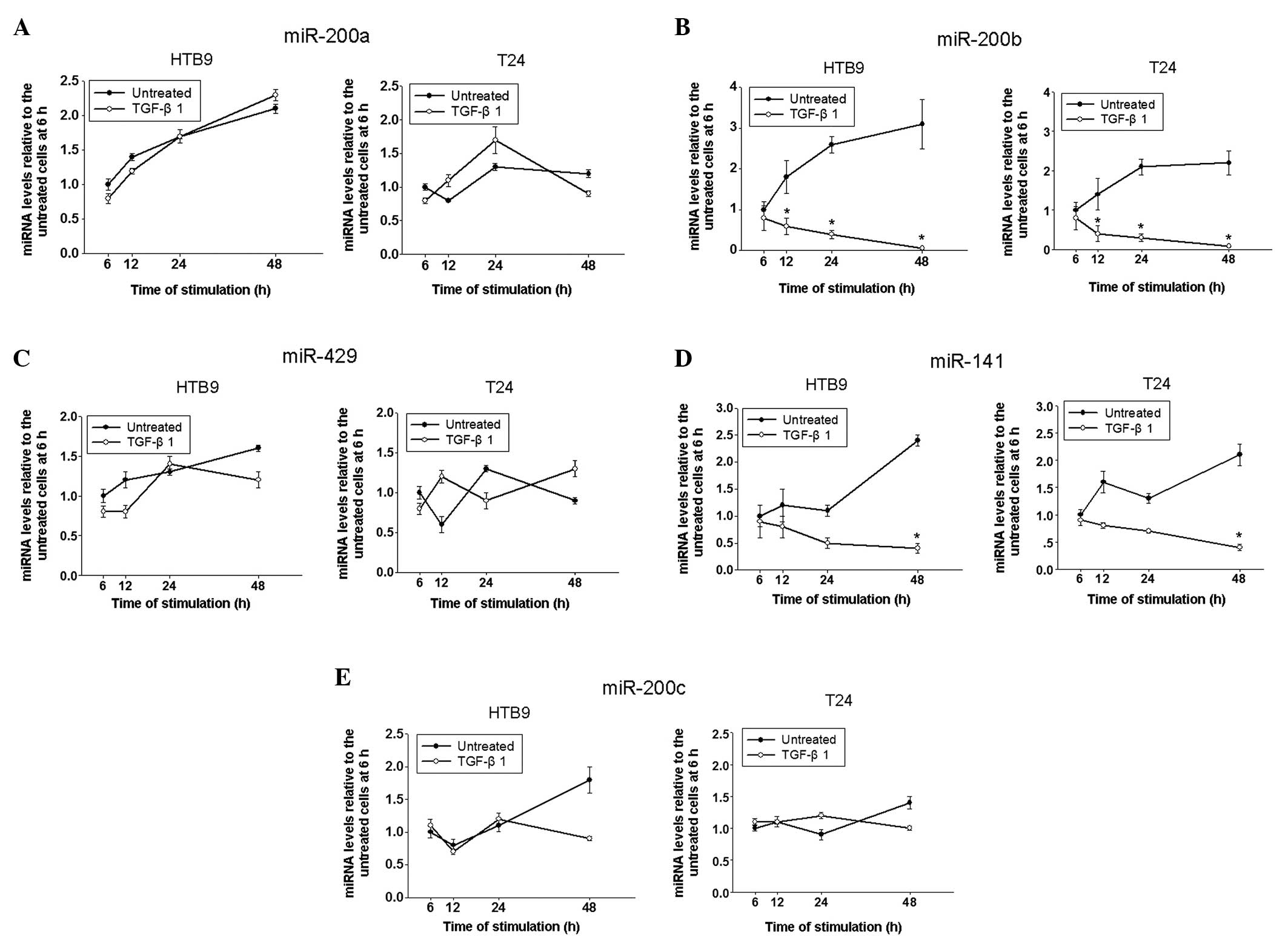
The miR-200 family is considered to be essential in
EMT. Therefore, changes in expression levels of these miRNAs in
untreated and TGF-β1-treated bladder cancer cells were analyzed at
different time points in the current study (Fig. 2). There were no significant
differences in the expression levels of miR-200a, 429 and 200c
between untreated and treated cells (Fig. 2A, C and E). No significant
difference in levels of miR-141 expression between untreated and
treated cells was detected until 48 h after TGF-β1-treatment
(Fig. 2D). However, expression
levels of miR-200b were significantly downregulated from 12 h
post-TGF-β1 treatment (Fig. 2B).
TGF-β1 reduced the expression levels of miR-200b progressively in a
time-dependent manner in HTB9 and T24 cells. This suggests that
miR-200b is a key factor in TGF-β1-induced EMT.
Expression of MMP-16 in HTB9 and T24
cells increases following TGF-β1 treatment
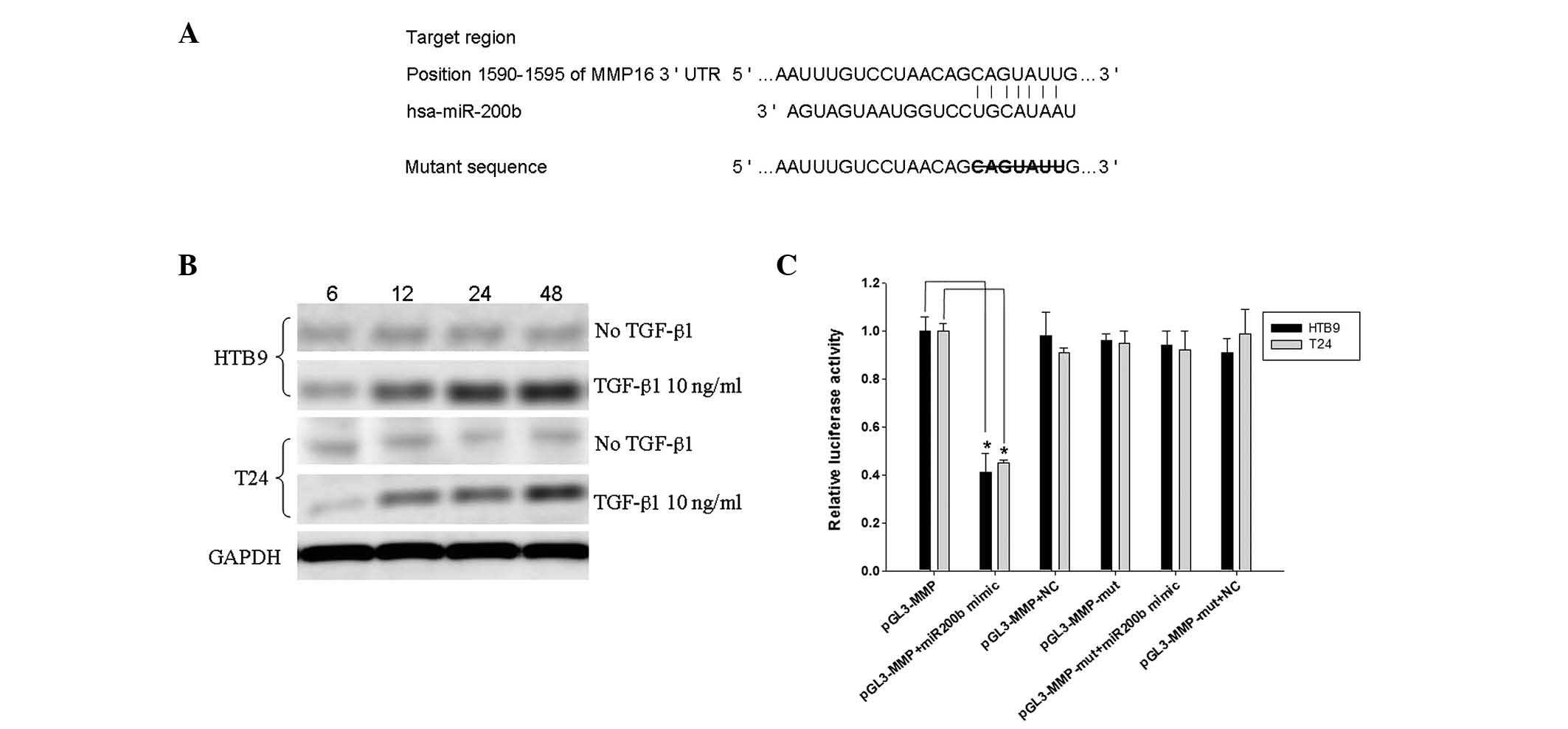
Bioinformatic analysis indicated that MMP-16 may be
the target of miR-200b; positions 1590–1595 of the MMP-16 3′UTR are
the interaction sites of miR-200b, as predicted by TargetScan
(Fig. 3A). Therefore, the
expression of MMP-16 in the two cell lines was examined, and the
western blots implied that its expression was increased by TGF-β1
in a time-dependent manner in the two cell types (Fig. 3B).
MMP-16 is a direct downstream functional
target of miR-200b
To validate the direct binding of miR-200b to the
MMP-16 3′ UTR region, an miR target reporter luciferase assay was
performed in HTB9 and T24 cells. Levels of luciferase activity in
HTB9 or T24 cells cotransfected with miR-200b mimics and the
pGL3-MMP-16 vector were reduced compared with levels in controls
(Fig. 3C), indicating that MMP-16
is one of the functional downstream targets of miR-200b.
miR-200b overexpression inhibits
expression of MMP-16 and the migration of bladder cancer cells
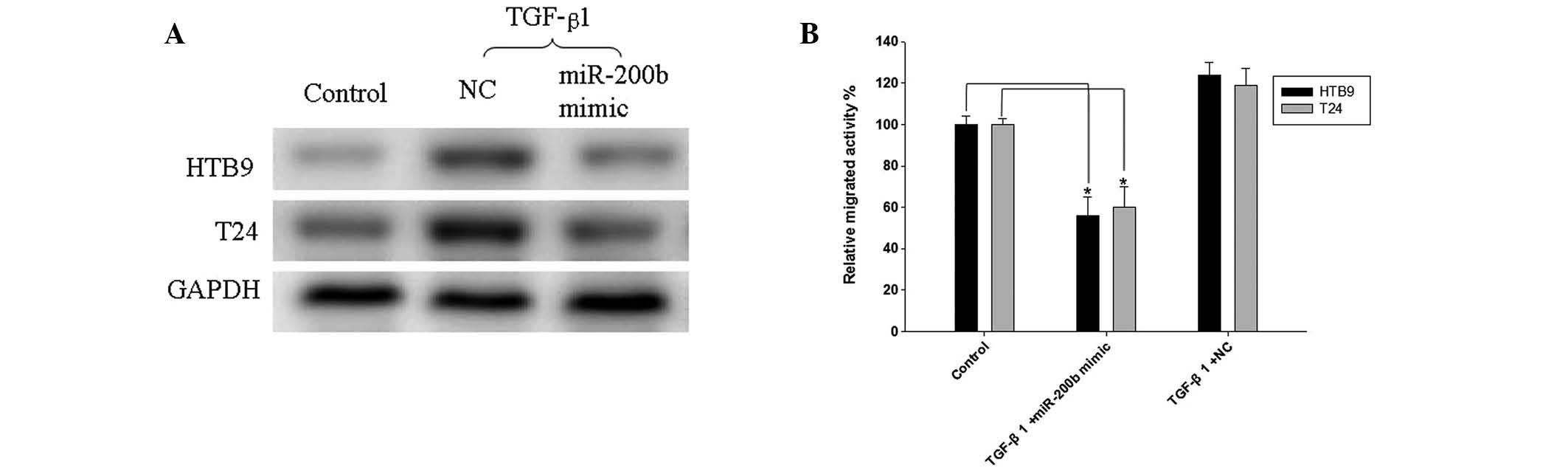
The expression levels of MMP-16 were measured, in
view of the changes of miR-200b expression levels in HTB9 and T24
cells induced by miR-200b mimic or NC transfection (Fig. 4A). The expression of MMP-16 was
downregulated following miR-200b mimic transfection compared with
that following NC transfection. A Matrigel migration assay
demonstrated that TGF-β1 slightly enhanced the migration of HTB9
and T24 cells transfected with NC compared with that of non-treated
control cells (Fig. 4B). However,
miR-200b mimic transfection significantly inhibited the migration
in the two cell lines when treated with TGF-β1 (Fig. 4B).
Discussion
In the present study, the results indicated that
exogenous TGF-β1 leads to reduced expression of E-cadherin and
downregulated levels of miR-200b in the bladder cancer cell lines,
HTB9 and T24. Bioinformatic analysis demonstrated that MMP-16 may
be a novel downstream target of miR-200b, and this was confirmed in
two cell lines. Further experiments indicated that miR-200b
overexpression inhibited TGF-β1-induced MMP-16 upregulation and the
migration of bladder cancer cells. The results of the present study
indicated that TGF-β1 downregulated the levels of miR-200b and
increased those of MMP-16. This may be a novel mechanism of
invasion and metastasis in bladder cancer.
TGF-β1 is a cytokine that enhances cell
characteristics associated with invasiveness, metastasis and
motility (14,16). EMT is associated with progression
and poor prognosis in various types of cancer, and can be induced
by TGF-β1 (14). In a study by Koo
et al (15), AY-27 rat
bladder cancer cells were incubated with TGF-β1 and were observed
to have reduced E-cadherin and increased vimentin immunoreactivity.
Their results were consistent with results observed in the present
study.
Gregory et al (17) treated Madin-Darby canine kidney
epithelial cells with TGF-β1 and noted that all the members of the
miR-200 family were selectively downregulated, indicating that the
miR-200 family is involved in TGF-β1-induced EMT. Slabáková et
al (14) and Starsíchová et
al (18) described that BPH-1
benign prostate hyperplasia cells underwent EMT following TGF-β1
treatment, but also that there were no apparent changes in the
expression levels of the miR-200 family members following TGF-β1
treatment. They hypothesized that the expression of the miR-200
family members was not regulated by TGF-β1 at the onset of the EMT
process, but may oppose the reversal of the EMT phenotype. In the
present study, only miR-200b was significantly downregulated
following TGF-β1 treatment in two bladder cancer cell lines. It was
hypothesized that miR-200b may be a key member in the
TGF-β1-induced EMT process of bladder cancer. The discrepancy
between results of the current study and those of previous studies
may be due to the use of different cell lines in the experiments.
The previous cell lines were derived from benign tissue, while the
cells used in the current study were malignant.
The miR-200 family is a cluster of miRNAs that are
positioned at two different loci in the genome (19). Two studies by Hurteau et al
(20,21) revealed that one individual member
of the miR-200 family, miR-200c, was able to simulate the effects
of the function of the cluster and be sufficient to regulate ZEB1
and restore E-cadherin in breast cancer cells. Loss of expression
of the miR-200 family members may be critical in the repression of
E-cadherin during EMT, thereby enhancing migration and invasion
during cancer progression (22).
In the present study, the results suggested that TGF-β1 treatment
led to reduced expression levels of E-cadherin and one individual
member, miR-200b.
The enzymatic activity of MMPs is tightly controlled
at multiple levels. MMP-16 can not only directly degrade certain
matrix molecules, but can also activate other MMPs (23). miR-15 and miR-146-5p have been
demonstrated to induce MMP-16 to inhibit migration and invasion of
glioma and cardiomyocyte progenitor cells (8,23,24).
To the best of our knowledge, the current study is the first to
provide evidence that miR-200b targeted the MMP-16 3′ UTR regions
via reporter luciferase assay in two bladder cancer cell lines.
Additionally, the upregulation approach was employed to investigate
the role of miR-200b in the invasive ability of bladder cancer
cells. miR-200b inhibited cell migration, establishing that
miR-200b serves as a metastasis-inhibiting miRNA in bladder
cancer.
A limitation of the present study was that the
mechanism of the TGF-β1-induced repression of miR-200b was not
assessed. DNA hypermethylation has previously been associated with
the silencing of miR-200 family members in muscle-invasive bladder
tumors and poorly differentiated bladder cell lines (4). It was therefore hypothesized that
TGF-β1 mediates the epigenetic silencing of miR-200 family members,
which requires further investigation.
In summary, exogenous TGF-β1 led to the induction of
EMT and the downregulation of miR-200b in bladder cancer cells. To
the best of our knowledge, the current study provided evidence that
MMP-16 is a direct target of miR-200b for the first time.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (81001137), and by the China
Hunan Provincial Science and Technology Department
(2010sk3102).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
von der Maase H, Sengelov L, Roberts JT,
et al: Long-term survival results of a randomized trial comparing
gemcitabine plus cisplatin, with methotrexate, vinblastine,
doxorubicin, plus cisplatin in patients with bladder cancer. J Clin
Oncol. 23:4602–4608. 2005.
|
|
3
|
Mongroo PS and Rustgi AK: The role of the
miR-200 family in epithelial-mesenchymal transition. Cancer Biol
Ther. 10:219–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiklund ED, Bramsen JB, Hulf T, et al:
Coordinated epigenetic repression of the miR-200 family and miR-205
in invasive bladder cancer. Int J Cancer. 128:1327–1334. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Yang K, Mao Q, et al: Inhibition of
TGF-beta receptor I by siRNA suppresses the motility and
invasiveness of T24 bladder cancer cells via modulation of
integrins and matrix metalloproteinase. Int Urol Nephrol.
42:315–323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Michl P, Ramjaun AR, Pardo OE, et al:
CUTL1 is a target of TGF(beta) signaling that enhances cancer cell
motility and invasiveness. Cancer Cell. 7:521–532. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Subramanian G, Schwarz RE, Higgins L, et
al: Targeting endogenous transforming growth factor beta receptor
signaling in SMAD4-deficient human pancreatic carcinoma cells
inhibits their invasive phenotype1. Cancer Res.
64:5200–5211. 2004. View Article : Google Scholar
|
|
8
|
Liu J, van Mil A, Aguor EN, et al: MiR-155
inhibits cell migration of human cardiomyocyte progenitor cells
(hCMPCs) via targeting of MMP-16. J Cell Mol Med. 16:2379–2386.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mamuya FA and Duncan MK: aV integrins and
TGF-β-induced EMT: a circle of regulation. J Cell Mol Med.
16:445–455. 2012.
|
|
10
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gottardo F, Liu CG, Ferracin M, et al:
Micro-RNA profiling in kidney and bladder cancers. Urol Oncol.
25:387–392. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dyrskjøt L, Ostenfeld MS, Bramsen JB, et
al: Genomic profiling of microRNAs in bladder cancer: miR-129 is
associated with poor outcome and promotes cell death in vitro.
Cancer Res. 69:4851–4860. 2009.PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔCT) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Slabáková E, Pernicová Z, Slavíčková E, et
al: TGF-β1-induced EMT of non-transformed prostate hyperplasia
cells is characterized by early induction of SNAI2/Slug. Prostate.
71:1332–1343. 2011.
|
|
15
|
Koo V, El Mekabaty A, Hamilton P, et al:
Novel in vitro assays for the characterization of EMT in
tumourigenesis. Cell Oncol. 32:67–76. 2010.PubMed/NCBI
|
|
16
|
Zheng Q, Safina A and Bakin AV: Role of
high-molecular weight tropomyosins in TGF-beta-mediated control of
cell motility. Int J Cancer. 122:78–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Starsíchová A, Lincová E, Pernicová Z, et
al: TGF-beta1 suppresses IL-6-induced STAT3 activation through
regulation of Jak2 expression in prostate epithelial cells. Cell
Signal. 22:1734–1744. 2010.PubMed/NCBI
|
|
19
|
Chan YC, Khanna S, Roy S and Sen CK:
miR-200b targets Ets-1 and is down-regulated by hypoxia to induce
angiogenic response of endothelial cells. J Biol Chem.
286:2047–2056. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hurteau GJ, Carlson JA, Spivack SD and
Brock GJ: Overexpression of the microRNA hsa-miR-200c leads to
reduced expression of transcription factor 8 and increased
expression of E-cadherin. Cancer Res. 67:7972–7976. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hurteau GJ, Carlson JA, Roos E and Brock
GJ: Stable expression of miR-200c alone is sufficient to regulate
TCF8 (ZEB1) and restore E-cadherin expression. Cell Cycle.
8:2064–2069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar
|
|
23
|
Li Y, Wang Y, Yu L, et al: miR-146b-5p
inhibits glioma migration and invasion by targeting MMP16. Cancer
Lett. 339:260–269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xia H, Qi Y, Ng SS, et al: microRNA-146b
inhibits glioma cell migration and invasion by targeting MMPs.
Brain Res. 1269:158–165. 2009. View Article : Google Scholar : PubMed/NCBI
|