Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common type of cancer worldwide and the third most common cause of
cancer-related mortality (1). HCC
usually occurs in the presence of continuous inflammation and
hepatocyte regeneration during chronic hepatitis and cirrhosis;
thus, mediators of inflammation are involved in the development of
hepatic carcinogenesis (2). Drug
treatment is the main therapy for patients in the advanced stages
of disease. However, the response rate to traditional chemotherapy
for HCC patients is far from satisfactory (3). Therefore, novel and effective
pharmacological strategies are urgently required for the treatment
of advanced HCC.
The non-steroidal anti-inflammatory drug celecoxib
is a member of the cyclooxygenase-2 (COX-2) inhibitor drug family
and is a potent and specific inhibitor of human COX-2 (4,5). The
anticancer effects of celecoxib have been demonstrated in various
different tumor types, including colorectal, lung, breast, prostate
and head/neck cancer (6–8). The mechanism underlying the effects
of celecoxib may be associated with COX-2-dependent or -independent
pathways (9,10). A number of studies have
investigated the mechanisms of the anticancer action of celecoxib.
Celecoxib and derived compounds have been suggested to induce cell
cycle arrest or apoptosis, inhibit tumor growth and suppress tumor
neoangiogenesis (11). However,
the mechanisms underlying celecoxib treatment of HCC are not yet
completely understood.
In recent years, numerous advances in the
understanding of HCC have been reported (12). The failure of transformed cells to
undergo apoptosis markedly disrupts tissue homeostasis and allows
proliferation of the resistant clone, a phenomenon frequently
observed in HCC (13). Apoptosis
is an evolutionarily conserved programmed mode of cell death and is
critical for the sustained tissue homeostasis. Apoptosis signaling
is tightly regulated by two main apoptotic pathways, termed the
‘extrinsic pathway’ and the ‘intrinsic pathway’. These pathways
involve either cell surface death receptors, or the mitochondria
and the endoplasmic reticulum, respectively (14,15).
We have previously focused on the effects of celecoxib on the
arachidonic acid (AA) signaling pathway in H22 mouse hepatoma
cells. The imbalance between AA and prostaglandin (PG)E2,
characterized by increased AA at a low dosage of celecoxib and
reduced PGE2 at a high dosage of celecoxib, was demonstrated to be
a significant indicator of celecoxib-mediated apoptosis in H22
cells (16). The present study was
designed to clarify the targeting of the apoptotic pathway by
celecoxib and possible interconnections between COX-2 and the
peroxisome proliferator-activated receptor (PPAR)γ/nuclear factor
(NF)-κB signaling pathway.
Materials and methods
Reagents and antibodies
Celecoxib (purity, >98%) was purchased from
Sunheat Chemicals (Shanghai, China). High-glucose Dulbecco’s
modified Eagle’s medium (DMEM) was obtained from Gibco-BRL
(Carlsbad, CA, USA). Dimethyl sulfoxide, methanol, ethanol,
chloroform, phosphoric acid and acetic acid were obtained from
Beijing Chemistry Company (Beijing, China). Sulforhodamine B and
proteinase inhibitors were bought from Sigma-Aldrich (St. Louis,
MO, USA). Fetal bovine serum (FBS), SDS,
tetramethylethylenediamine, glycine, ammonium, persulfate,
acrylamide, Tris, Trizol, agarose and Tween-20 were purchased from
Beijing Dingguo Changsheng Biological Technology Co., Ltd (Beijing,
China). The protein assay kit, radioimmunoprecipitation assay
(RIPA) lysis buffer, Rhodamine 123 and Cell Apoptosis Assay kit
were obtained from Beyotime Institute of Biotechnology (Jiangsu,
China). Antibodies against β-actin (mouse, monoclonal, sc-47778),
Bax (mouse, monoclonal, sc-7480), B-cell CLL/lymphoma 2 (Bcl-2;
mouse, monoclonal, sc-7382), cytochrome c (mouse,
monoclonal, sc-13561), caspase-3 (rabbit, polyclonal, sc-98785),
caspase-9 (mouse, monoclonal, sc-133109), apoptosis-inducing factor
(AIF; mouse, monoclonal, sc-55519), PPARγ (rabbit, polyclonal,
sc-7196) and NF-κB (rabbit, polyclonal, sc-298) were purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture
The H22 mouse hepatoma cell line was originally
obtained from the American Type Culture Collection (Rockville, MD,
USA) and stored at −80°C. The H22 cells were cultured in DMEM
supplemented with 10% FBS (v/v), 100 μM penicillin G (Sigma
Aldrich, St. Louis, MO, USA) and 0.1 mg/ml streptomycin (Sigma
Aldrich). The cultures were maintained in a humidified incubator at
37°C in 5% CO2.
Determination of cell viability by a
sulforhodamine B (SRB) assay
The cells were seeded in 96-well plates in medium
with 10% FBS. Following incubation with celecoxib at concentrations
ranging between 25 and 400 μM for 48 h, the cells were fixed with
10% trichloroacetic acid (Sigma Aldrich), and 0.4% (w/v) SRB in 1%
acetic acid was added to stain the cells. Unbound SRB was washed
away with 1% acetic acid and SRB-bound cells were rendered soluble
with 10 mM Trizma base (pH 10.5; Sigma Aldrich). The absorbance was
read at a wavelength of 570 nm with a 680 microplate enzyme-linked
immunosorbent assay (ELISA) reader (Bio-Rad Laboratories, Hemel
Hempstead, UK). Using the following absorbance measurements: Time
zero (T0), control growth (C) and cell growth in the
presence of the compound (Tx), the percentage growth was
calculated at each compound concentration level. Percentage growth
inhibition was calculated as:
100-[(Tx-T0)/(C-T0)] × 100. The
50% growth inhibition (IC50) value was determined to be
the compound concentration that resulted in a 50% reduction of the
total protein increase in the control cells during the compound
incubation.
Determination of apoptosis by Annexin
V/propidium iodide (PI) staining
Apoptosis was measured using a fluorescein
isothiocyanate-Annexin V apoptosis detection kit (BD Pharmingen,
San Diego, CA, USA). Following treatment, the cells were harvested
by trypsinization, washed with ice-cold PBS and suspended in
binding buffer at a density of 1×106 cells/ml. The cell
suspension was stained with 5 μl Annexin V and PI, and analyzed by
a FACSort flow cytometer (Becton-Dickinson Biosciences, Franklin
Lakes, NJ, USA).
Measurement of mitochondrial membrane
potential (ΔΨm)
The cells were treated with celecoxib. At 30 min
prior to incubation termination, Rhodamine 123 solution (final
concentration of 10 μg/ml) was added to the cells and incubated for
the final 30 min at 37°C. The cells were harvested and the
accumulation of Rhodamine 123 was determined using FACScan flow
cytometric analysis.
Preparation of cell extracts and western
blot analysis
Subsequent to treatment, the cells were harvested
with trypsinization, centrifuged and lysed in 0.1 ml lysis buffer
containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EGTA, 1%
Triton X-100, 1mM phenylmethanesulfonyl fluoride, 10 mg/ml
leupeptin, 10 mg/ml aprotinin, 50 mM NaF and 100 mM sodium
orthovanadate. Total protein was quantified, mixed with sample
buffer and boiled at 90°C for 5 min. Equal quantities of protein
(30 mg) were separated by gel electrophoresis in 8 or 12% SDS-PAGE,
and transferred to polyvinylidene difluoride membranes. The blots
were blocked in Tris-buffered saline containing 0.05% Tween-20
(TBST) and 5% non-fat dry milk for 1 h at room temperature. The
membrane was incubated overnight at 4°C with the primary
antibodies. Following repeated washings with TBST, the membranes
were incubated with the HRP-conjugated mouse anti-rabbit secondary
antibody or HRP-conjugated rabbit anti-mouse secondary antibody
(Santa Cruz Biotechnology, Inc.) for 1 h at room temperature prior
to additional washes with TBST. Detection of antibody binding was
performed using the enhanced chemiluminescence kit (Amersham
Pharmacia Biotech, Amersham, UK). Equal loading was verified using
the antibodies against β-actin. All western blot analyses were
repeated three times.
Statistical analysis
The data are reported as the mean ± standard error
of the mean (n=3 per group). Statistical analysis was performed
using the unpaired Student’s t-test and P<0.05 was considered to
indicate a statistically significant difference.
Results
Celecoxib inhibits cell
proliferation
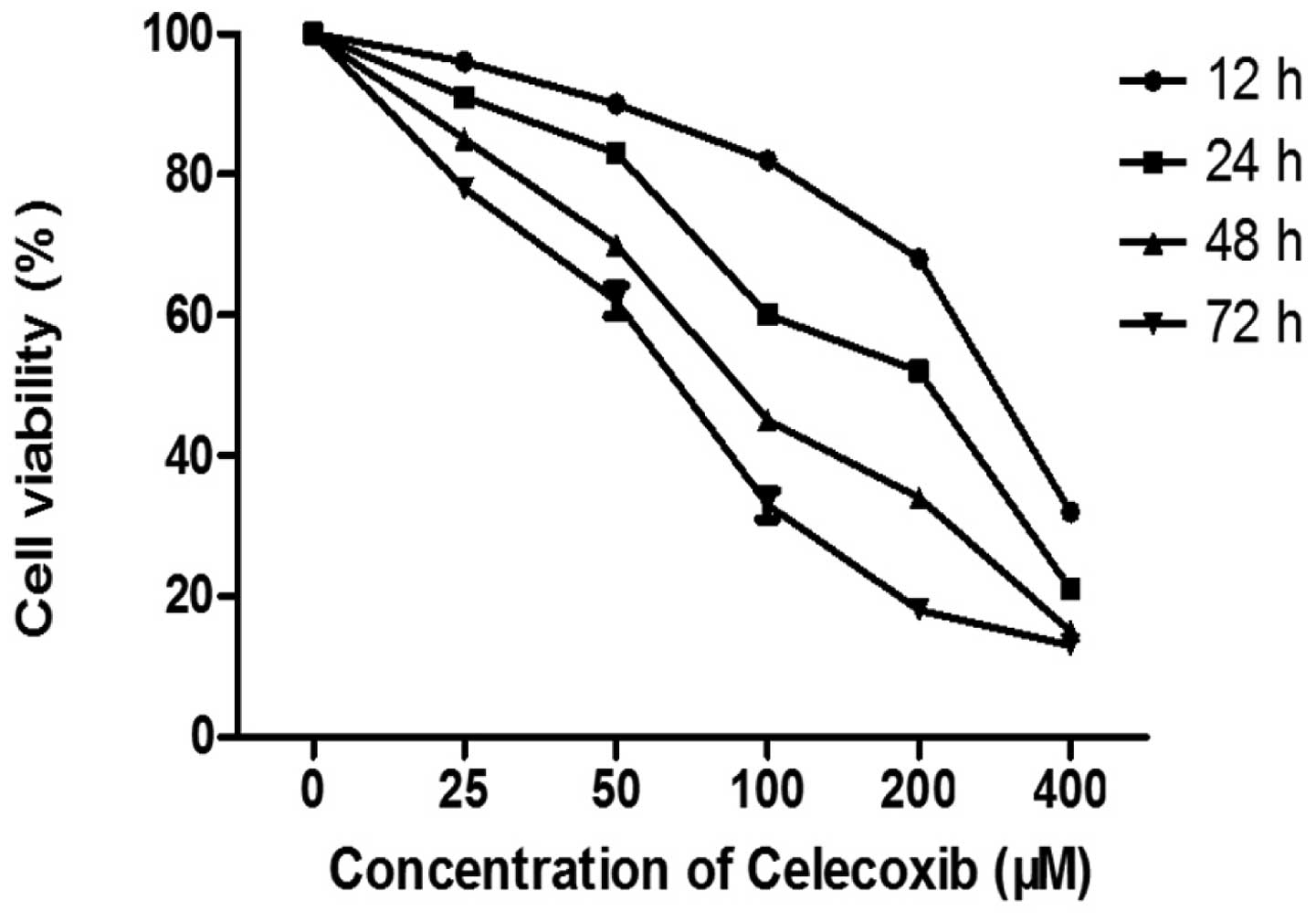
The effect of celecoxib on cell viability was
determined by an SRB assay. The H22 cells were treated with
celecoxib at concentrations ranging between 25 and 400 μM for 12,
24, 48 and 72 h. As shown in Fig.
1, celecoxib exposure significantly reduced the viability of
the H22 cells. The celecoxib IC50 values were 256.6±1.5
μM, 155.4±0.6 μM, 99.2±0.4 μM and 70.6±0.6 μM at 12, 24, 48 and 72
h, respectively. These results suggest that celecoxib induces cell
death and inhibits cell proliferation in a dose- and time-dependent
manner.
Celecoxib induces apoptosis in H22
cells
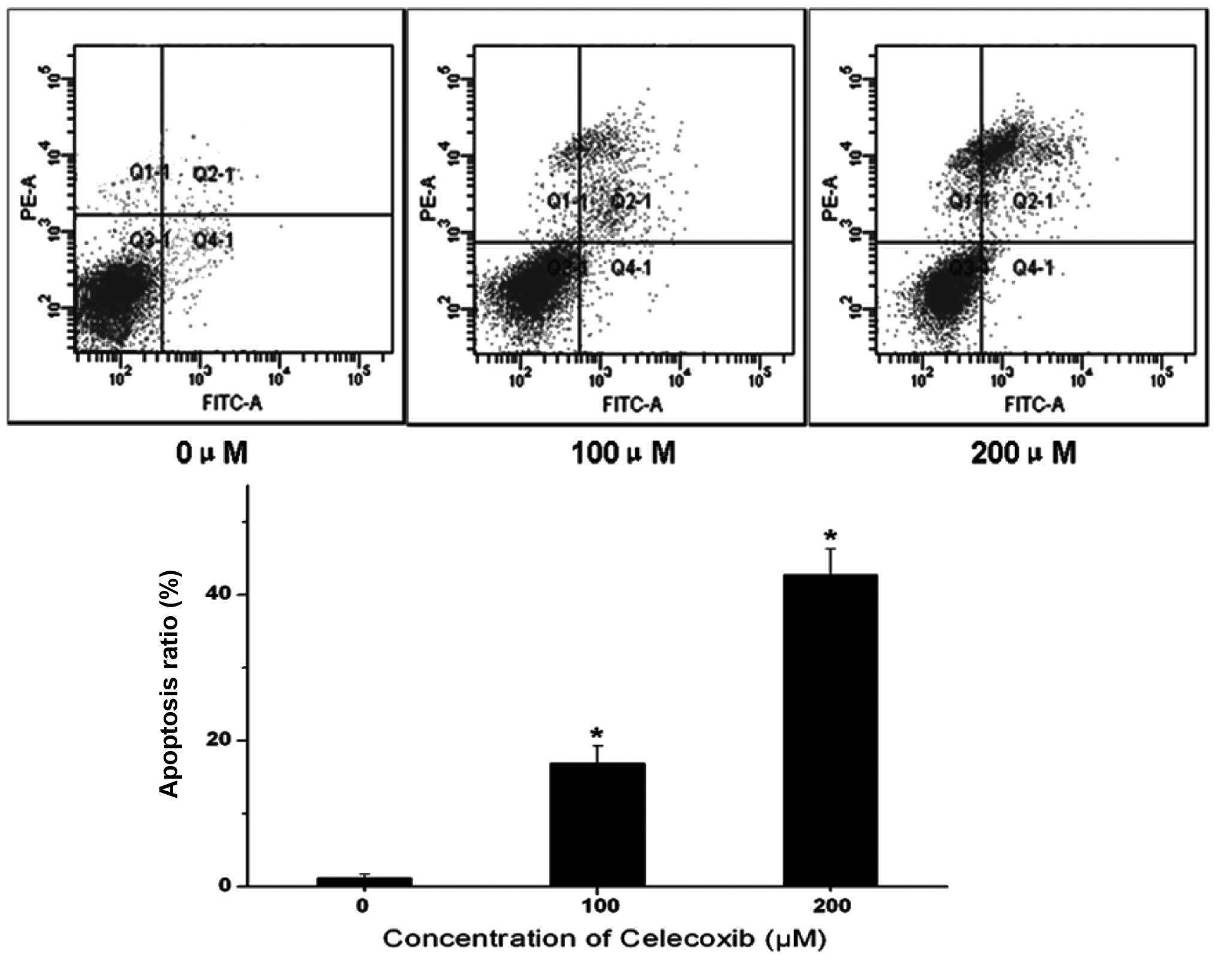
To further understand whether celecoxib-induced cell
death is mediated by apoptosis or necrosis, apoptotic cell death
was evaluated using Annexin V/PI double staining, which
specifically labels apoptotic cells. Fig. 2 shows that H22 cells without
celecoxib treatment were mostly detected in the Q3 quadrant,
indicating that these cells were viable. Following celecoxib
treatment, however, an increased number of late apoptotic cells
were detected in the Q2 quadrant. Therefore, celecoxib markedly
increased the proportion of apoptotic cells in a dose-dependent
manner. These data suggest that apoptotic cell death events
contribute to the growth inhibitory effect of celecoxib.
Celecoxib alters mitochondrial function
and ΔΨm
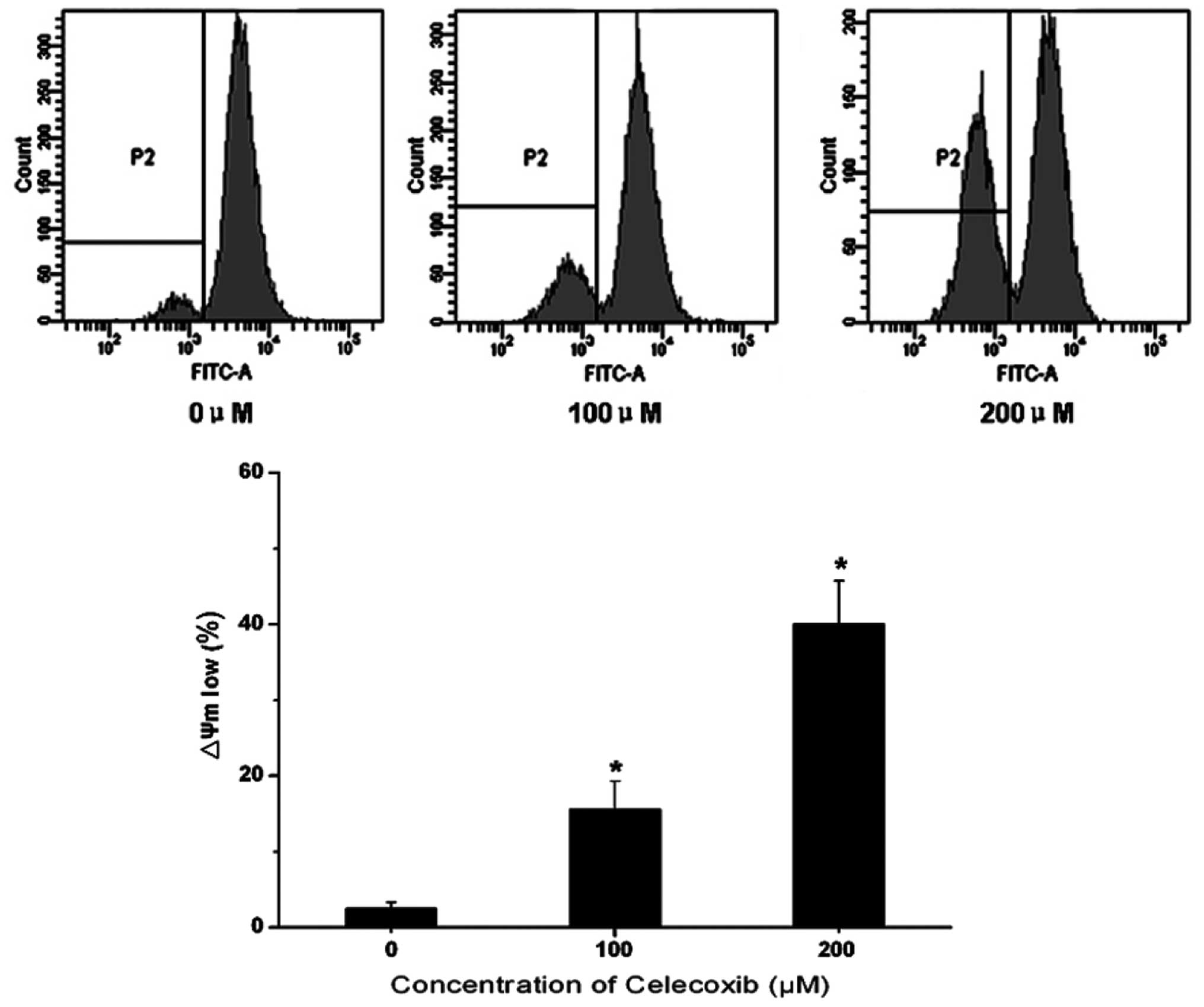
Mitochondrial function is critical to cell
viability. Disruption of the mitochondrial membrane potential has
been reported to irreversibly result in cell apoptosis, the release
of cytochrome c and a reduction in adenosine triphosphate
(ATP) generation. In order to gain an improved understand of the
mechanism of celecoxib-induced H22 cell apoptosis, Rhodamine 123
was used to ascertain the mitochondrial membrane potential through
examining the fluorescent intensity. As shown in Fig. 3, the fluorescence intensity was
reduced with increases in celecoxib concentration. A
concentration-dependent reduction in Rhodamine 123 fluorescence was
detected following celecoxib treatment, compared with the control
group. This indicates that celecoxib was able to induce
mitochondrial membrane potential disruption in H22 cells.
Celecoxib induces apoptosis via the
mitochondria-dependent pathway
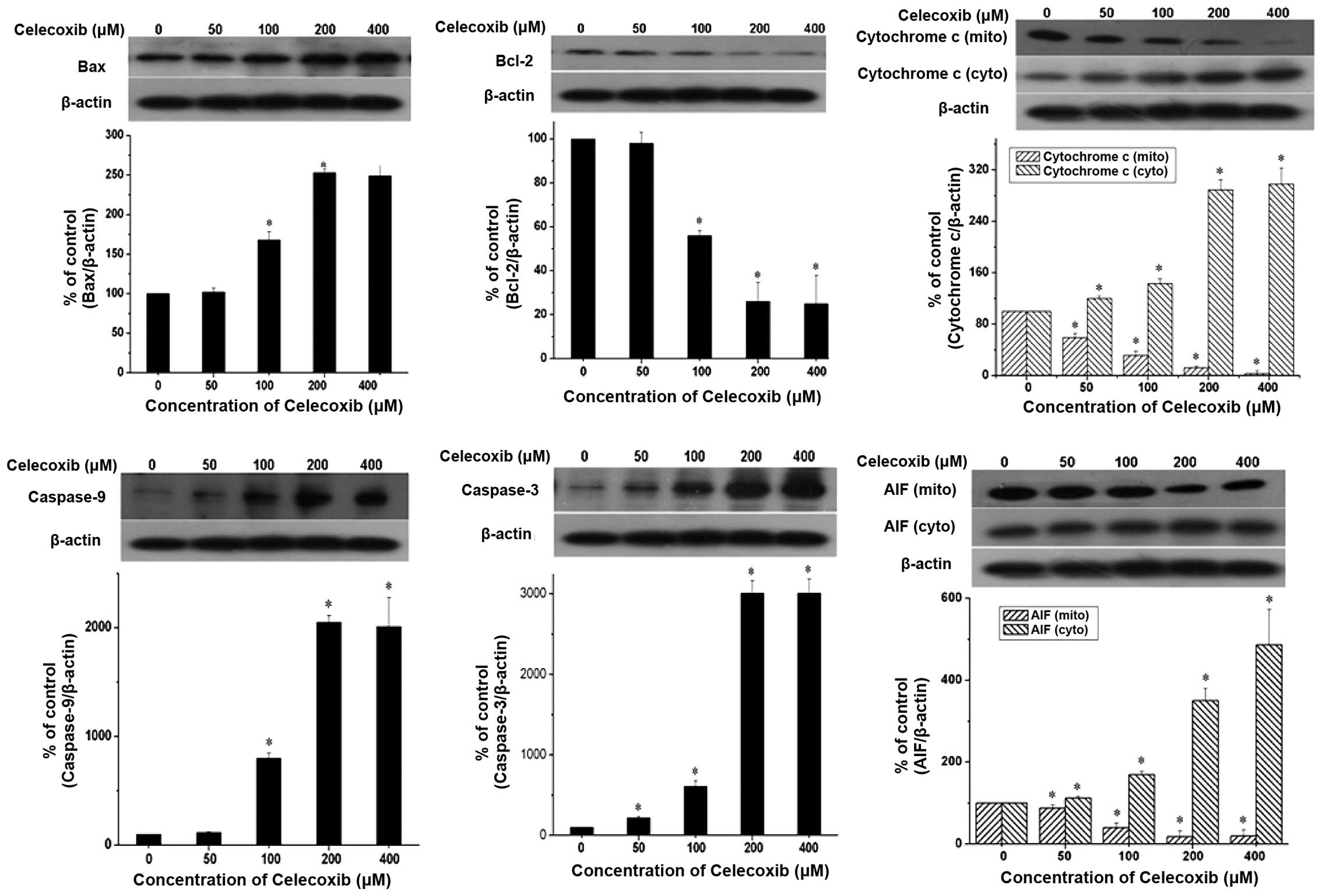
In chemically-induced apoptosis, mitochondria are
central in the cellular commitment to apoptosis through cytochrome
c-dependent or -independent pathways. To elucidate the
molecular mechanism of celecoxib-induced apoptosis in H22 cells,
the expression levels of proteins associated with apoptosis were
examined. H22 cells were exposed to the indicated celecoxib
concentrations for 24 h (Fig. 4).
The data revealed that celecoxib induced the release of cytochrome
c from the mitochondria to the cytosol and AIF from the
cytosol to the nucleus in a concentration-dependent manner.
Caspase-3 and caspase-9, well-known to be activated downstream of
cytochrome c, were also cleaved in a concentration-dependent
manner. By contrast, the expression levels of Bcl-2 protein, an
anti-apoptotic molecule, were reduced in a concentration-dependent
manner. However, celecoxib induced an increase in Bax expression
levels; the complementary pro-apoptotic Bax proteins of the
Bcl-2-family are essential for the activation of the intrinsic
death pathway. These data suggest that celecoxib induced
mitochondria-dependent apoptosis through the mitochondria-dependent
pathway.
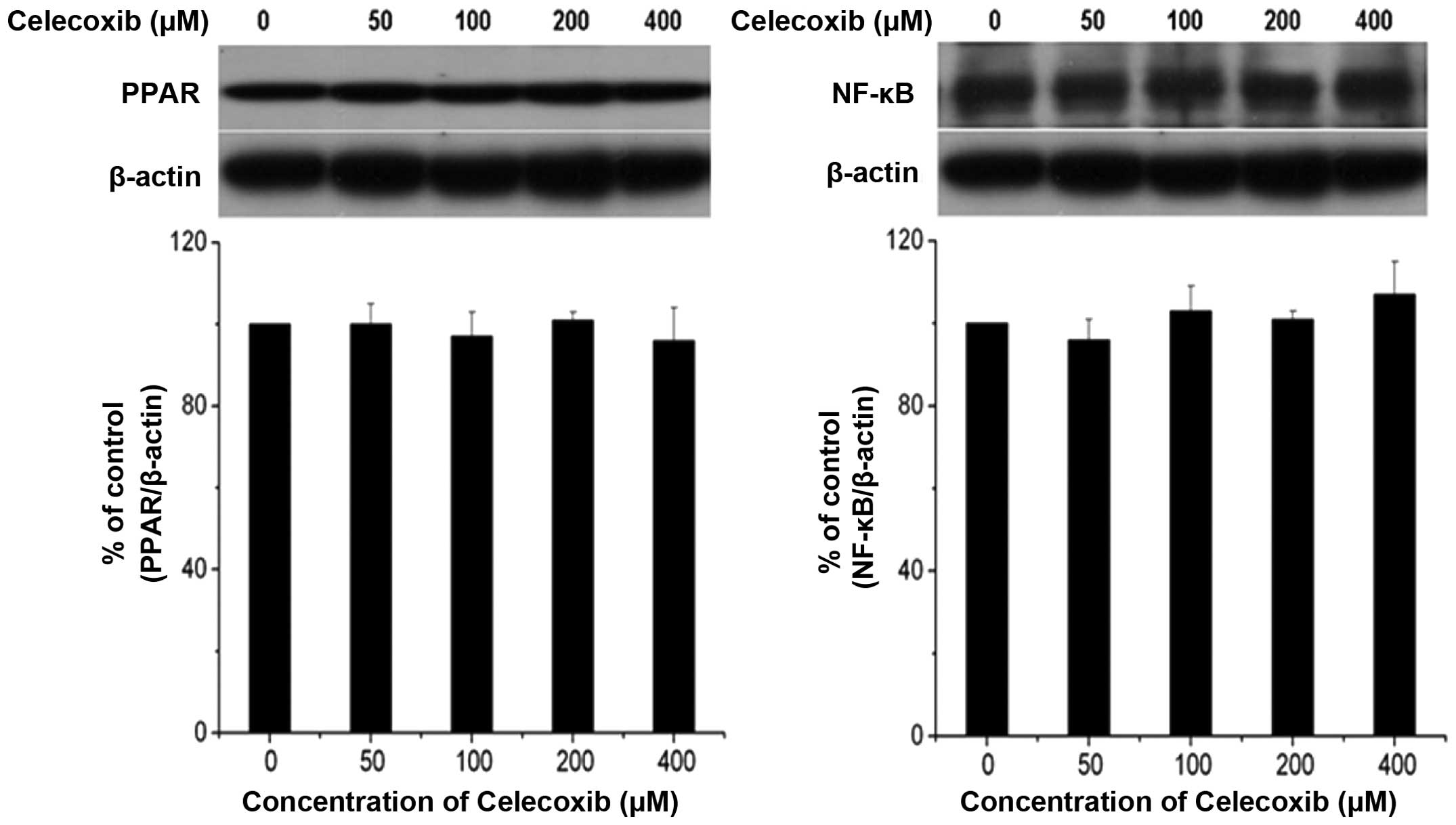
Celecoxib does not induce apoptosis
through the PPARγ/NF-κB signaling pathway
Since PPAR-γ agonists are known to exhibit
growth-inhibitory effects in tumor cells, including HCC cell lines,
the effect of celecoxib on PPAR-γ expression levels in H22 cells
was investigated. In Fig. 5,
western blot analysis of PPARγ and NF-κB did not reveal a
differential effect on PPARγ and NF-κB expression levels during
celecoxib-induced apoptosis, indicating that celecoxib induced
apoptosis via the mitochondria-dependent pathway rather than the
PPARγ/NF-κB signaling pathway.
Discussion
Celecoxib may reduce the risk of cancer formation by
altering AA metabolism, which has been implicated in the
development of cancer. Although the antitumor mechanism of
celecoxib is not completely understood, a large number of studies
have demonstrated that celecoxib prevents carcinogenesis through
inhibition of COX-2 activity and the resulting reduction in PGE2
expression levels (17). In
previous studies, the inhibitory effects of low celecoxib doses on
hepatoma H22 cells were indicated to be induced by AA released
through cPLA2 activity (16).
Although celecoxib at low doses induced different changes in the AA
metabolism pathway compared with high doses, celecoxib was, to the
best of our knowledge, found for the first time to increase the
ratio of AA to PGE2 in a dose-dependent manner, correlating with
the repressing of cell viability on H22 cells. Therefore, the
increased ratio of AA to PGE2 may be an important indicator of
celecoxib cytotoxicity and may be used to evaluate antitumor
activity (16).
Recently, data have indicated that the effects of
celecoxib, a selective COX-2 inhibitor, are COX-2-dependent and
-independent in HCC (18,19). The pro-apoptotic effects of
celecoxib were first attributed to the inhibitory action of the
drug on COX-2. By inhibiting COX-2, celecoxib has been suggested to
interfere with prostaglandin-mediated upregulation of
anti-apoptotic proteins (20,21).
However, a study has demonstrated that celecoxib is able to
suppress tumor growth without an apparent involvement of the target
protein, COX-2 (22). Important
molecular targets of the COX-2-independent celecoxib activity
include protein kinase B and its upstream kinase
3-phosphoinositide-dependent kinase-1 (23), cyclin-dependent kinase inhibitors
and cyclins, the anti-apoptotic proteins survivin, Bcl-2 and Mcl-1,
as well as sarcoplasmic/endoplasmic reticulum calcium ATPase
(24,25). Thus, celecoxib acts as a
multifunctional drug. Although celecoxib constitutes a prototype of
drugs that induce cell death independently from COX-2, inhibition
of COX-2 in COX-2-expressing cells may also contribute to the
cytotoxic effects of the drug. COX-2 inhibition may be particularly
important for the in vivo effects of celecoxib in
COX-2-expressing tumors, as COX-inhibition affects
prostaglandin-mediated angiogenesis in xenografts and newly formed
tumors (26).
Intrinsic apoptotic signaling occurs in response to
stimuli such as DNA damage, growth-factor withdrawal and exposure
to certain chemotherapeutic agents, which result in the release of
cytochrome c and other pro-death factors from the
intermembrane spaces of the mitochondria, and subsequent downstream
signaling through the initiator caspase-9. The mitochondria are
important for apoptosis. With external or internal apoptotic signal
stimulation in tumor cells, the mitochondrial membrane permeability
changes, and cytochrome c, AIF and other apoptotic factors
are released into the cytoplasm. Cytochrome c may result in
cell apoptosis following the formation of the apoptosome with
Apaf-1 and caspase-9, activating caspase-3, caspase-6 or caspase-7
downstream. The Bcl-2 family proteins, including the anti-apoptotic
protein Bcl-2 and pro-apoptotic protein Bax, are important factors
that regulate the changes of the mitochondrial membrane
permeability, and are important in the mitochondrial apoptotic
pathway (27).
PPARγ is a ligand-activated transcription factor
with a DNA-binding domain that recognizes response elements in the
promoter regions of specific target genes associated with
inflammation, cell proliferation, apoptosis and differentiation
(28,29). NF-κB is aberrantly activated in
tumor cells, contributing an advantage in cellular survival and
proliferation (30). The NF-κB
activation mechanism in tumor cells is not well-elucidated, but it
appears complex and varies in different tumor types. In a previous
study, celecoxib upregulated expression of PPARγ and inhibited
growth of Lewis cancer cells in a dose-dependent manner (31). Whether increased PPARγ expression
levels occurred directly or indirectly due to elevated AA levels
remains unclear. However, our recent data has not demonstrated a
differential effect on PPARγ and NF-κB expression levels during
celecoxib-induced H22 cell apoptosis. The mechanism of celecoxib
action appears to vary according to the capacity of the cell type
being treated. Celecoxib effects depend not only on the conditions
under which it is administered to the cells and the cell type, but
also on key pathways, such as COX-2, PPARγ and NF-κB. This
highlights the requirement to closely analyze the mechanisms
underlying celecoxib action within and among different tumor
types.
In conclusion, the results of the present study
demonstrate that celecoxib reduced the percentage of viable H22
cells in a dose- and time-dependent manner, which was associated
with cell apoptosis. Celecoxib induced apoptosis via the
mitochondria-dependent pathway, including through mitochondrial
dysfunction, release of AIF and cytochrome c from the
mitochondria, and the activation of caspase-9 and caspase-3.
Celecoxib also increased the abundance of the pro-apoptotic protein
Bax and reduced the levels of the anti-apoptotic protein Bcl-2.
Therefore, the data indicate that celecoxib may be an effective
therapy in HCC.
Acknowledgements
The authors would like to thank Dr Zhigang Xu, Dr
Qiongshu Li, Miss Huilin Zhen and Mr. Xin Zhang for their help.
Funding for this study was provided by the Jilin Science &
Technology Development Plan (grant nos. 20090441, 201205006 and
2012747), the Graduate Innovation Fund of Jilin University (grant
no. 20121118) and the Opening Project of State Key Laboratory of
Supramolecular Structure and Materials of Jilin University (grant
nos. SKLSSM 200912 and SKLSSM 201317).
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar
|
|
3
|
Raoul JL, Sangro B, Forner A, et al:
Evolving strategies for the management of intermediate-stage
hepatocellular carcinoma: available evidence and expert opinion on
the use of transarterial chemoembolization. Cancer Treat Rev.
37:212–220. 2011. View Article : Google Scholar
|
|
4
|
Fort J: Celecoxib, a COX-2-specific
inhibitor: the clinical data. Am J Orthop (Belle Mead NJ). 28(3
Suppl): 13–18. 1999.PubMed/NCBI
|
|
5
|
Chakraborti AK, Garg SK, Kumar R, et al:
Progress in COX-2 inhibitors: a journey so far. Curr Med Chem.
17:1563–1593. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie SQ, Zhang YH, Li Q, et al:
COX-2-independent induction of apoptosis by celecoxib and polyamine
naphthalimide conjugate mediated by polyamine depression in
colorectal cancer cell lines. Int J Colorectal Dis. 27:861–868.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang ZL, Fan ZQ, Jiang HD, et al:
Selective Cox-2 inhibitor celecoxib induces epithelial-mesenchymal
transition in human lung cancer cells via activating MEK-ERK
signaling. Carcinogenesis. 34:638–646. 2013. View Article : Google Scholar
|
|
8
|
Bocca C, Bozzo F, Bassignana A and
Miglietta A: Antiproliferative effects of COX-2 inhibitor celecoxib
on human breast cancer cell lines. Mol Cell Biochem. 350:59–70.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bastos-Pereira AL, Lugarini D, de
Oliveira-Christoff A, et al: Celecoxib prevents tumor growth in an
animal model by a COX-2 independent mechanism. Cancer Chemother
Pharmacol. 65:267–276. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang AH, Tian XY, Yu JJ, et al: Celecoxib
radiosensitizes the human cervical cancer HeLa cell line via a
mechanism dependent on reduced cyclo-oxygenase-2 and vascular
endothelial growth factor C expression. J Int Med Res. 40:56–66.
2012. View Article : Google Scholar
|
|
11
|
Jendrossek V: Targeting apoptosis pathways
by Celecoxib in cancer. Cancer Lett. 332:313–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bruix J and Sherman M: American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: an update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schattenberg JM, Schuchmann M and Galle
PR: Cell death and hepatocarcinogenesis: Dysregulation of apoptosis
signaling pathways. J Gastroenterol Hepatol. 26(Suppl 1): 213–219.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan TT and White E: Therapeutic targeting
of death pathways in cancer: mechanisms for activating cell death
in cancer cells. Adv Exp Med Biol. 615:81–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu Z, Zhang M, Lv X, et al: The inhibitory
effect of celecoxib on mouse hepatoma H22 cell line on the
arachidonic acid metabolic pathway. Biochem Cell Biol. 88:603–609.
2010.PubMed/NCBI
|
|
17
|
Dai ZJ, Ma XB, Kang HF, et al: Antitumor
activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on
breast cancer in vitro and in vivo. Cancer Cell Int. 12:532012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maier TJ, Schilling K, Schmidt R, et al:
Cyclooxygenase-2 (COX-2)-dependent and -independent
anticarcinogenic effects of celecoxib in human colon carcinoma
cells. Biochem Pharmacol. 67:1469–1478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cervello M, Bachvarov D, Cusimano A, et
al: COX-2-dependent and COX-2-independent mode of action of
celecoxib in human liver cancer cells. OMICS. 15:383–392. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin MT, Lee RC, Yang PC, et al:
Cyclooxygenase-2 inducing Mcl-1-dependent survival mechanism in
human lung adenocarcinoma CL1.0 cells. Involvement of
phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem.
276:48997–49002. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan YX, Li WZ, Huang YQ, et al: The COX-2
inhibitor Celecoxib enhances the sensitivity of KB/VCR oral cancer
cell lines to Vincristine by down-regulating P-glycoprotein
expression and function. Prostaglandins Other Lipid Mediat.
97:29–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grosch S, Maier TJ, Schiffmann S and
Geisslinger G: Cyclooxygenase-2 (COX-2)-independent
anticarcinogenic effects of selective COX-2 inhibitors. J Natl
Cancer Inst. 98:736–747. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pyrko P, Soriano N, Kardosh A, et al:
Downregulation of survivin expression and concomitant induction of
apoptosis by celecoxib and its non-cyclooxygenase-2-inhibitory
analog, dimethyl-celecoxib (DMC), in tumor cells in vitro
and in vivo. Mol Cancer. 5:192006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rudner J, Elsaesser SJ, Müller AC, et al:
Differential effects of anti-apoptotic Bcl-2 family members Mcl-1,
Bcl-2, and Bcl-xL on celecoxib-induced apoptosis. Biochem
Pharmacol. 79:10–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson AJ, Hsu AL, Lin HP, et al: The
cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular
calcium by inhibiting endoplasmic reticulum
Ca2+-ATPases: a plausible link with its anti-tumour
effect and cardiovascular risks. Biochem J. 366:831–837.
2002.PubMed/NCBI
|
|
26
|
Rahman M, Selvarajan K, Hasan MR, et al:
Inhibition of COX-2 in colon cancer modulates tumor growth and
MDR-1 expression to enhance tumor regression in therapy-refractory
cancers in vivo. Neoplasia. 14:624–633. 2012.
|
|
27
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ricote M, Li AC, Willson TM, et al: The
peroxisome proliferator-activated receptor-gamma is a negative
regulator of macrophage activation. Nature. 391:79–82. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Birnbaum Y, Long B, Qian J, et al:
Pioglitazone limits myocardial infarct size, activates Akt, and
upregulates cPLA2 and COX-2 in a PPAR-gamma-independent manner.
Basic Res Cardiol. 106:431–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin Y, Bai L, Chen W and Xu S: The
NF-kappaB activation pathways, emerging molecular targets for
cancer prevention and therapy. Expert Opin Ther Targets. 14:45–55.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang M, Xu ZG, Shi Z, et al: Inhibitory
effect of celecoxib in lung carcinoma by regulation of
cyclooxygenase-2/cytosolic phospholipase A2 and
peroxisome proliferator-activated receptor gamma. Mol Cell Biochem.
355:233–240. 2011. View Article : Google Scholar : PubMed/NCBI
|