Introduction
Non-alcoholic fatty liver disease (NAFLD) is
currently the most common form of chronic liver disease affecting
both adults and children. It is strongly associated with obesity,
diabetes and hyperlipidemia. Thus, NAFLD is often considered as the
hepatic manifestation of metabolic syndrome. NAFLD comprises a
pathological spectrum ranging from simple hepatic steatosis and
non-alcoholic steatohepatitis (NASH) to liver fibrosis, cirrhosis
and hepatocellular carcinoma (1).
Although previously thought to be benign, simple hepatic steatosis
can evolve into more severe liver diseases (2). In addition, individuals with simple
hepatic steatosis are at an increased risk of cardiovascular
disease, diabetes and obesity-related and overall mortality
(3). Aside from lifestyle
modification, there is currently no effective therapy for simple
hepatic steatosis (2).
Glucagon-like peptide-1 (GLP-1) is an incretin
secreted by L-cells in the small intestine in response to food
intake, and is rapidly degraded by dipeptidyl peptidase 4 present
on the endothelial cells lining capillaries of the lamina propria.
The main roles of GLP-1 in the pancreas are the stimulation of
glucose-dependent insulin secretion, inhibition of postprandial
glucagon release and induction of β-cell proliferation (4). Apart from the pancreatic islets,
GLP-1 receptors (GLP-1Rs) are also present in several other tissues
including the nervous system, gastrointestinal tract, lung and
heart. As a result, GLP-1 exerts further beneficial functions
including delaying stomach emptying, increasing satiety, reducing
food intake and triggering weight loss (5). Recently, two studies have suggested
that GLP-1Rs exist in the liver (6,7).
Accumulating evidence reveals that GLP-1-related drugs reduce
hepatic steatosis both in vivo and in vitro (6–10). A
few studies in humans have also proven that utilization of GLP-1R
agonists can improve hepatic steatosis, particularly in type 2
diabetes patients with NAFLD (as reviewed in 11). However, until now, there has been
little research into the effect of GLP-1 on simple hepatic
steatosis.
Autophagy is a highly evolutionarily conserved
process involved in the turnover of long-lived proteins, cytosolic
components or damaged organelles. Three types of autophagy are
known: Macroautophagy, chaperone-mediated autophagy and
microautophagy. Furthermore, macroautophagy (hereafter referred to
as autophagy) is the main regulated catabolic mechanism used by
eukaryotic cells to degrade long-lived proteins and organelles. It
is well known that autophagy is widely involved in the pathogenesis
of numerous diseases and processes including infections, cancer,
neurodegeneration, aging and heart disease (12). Previous studies have discovered new
functions for autophagy in the regulation of lipid metabolism. The
inhibition of autophagy by genetic knockdown of the autophagy gene
ATG5, or treatment with 3-methyladenine (3-MA) in cultured
hepatocytes that were challenged with a lipid load from fatty acid
supplementation, can significantly increase intracellular
triglyceride (TG) content (13).
Another study reported that increased steatosis in models of
alcohol-induced hepatic injury can be induced by inhibition of
autophagy (14). These studies
indicate that modulating autophagy may be a new therapeutic
strategy for NAFLD.
Until now, relatively few studies have investigated
whether GLP-1 reduces lipid accumulation through modulating
autophagy. With this background, we hypothesize that GLP-1 is able
to improve simple hepatic steatosis by enhancing autophagy.
Therefore, in the present study, we selected the L-02 cell line,
which is an immortalized normal human hepatocyte-derived cell line,
to construct a cell model of simple hepatic steatosis.
Subsequently, we used liraglutide (LG), a long-acting GLP-1 analog,
to treat the cell model and measure the change of intracellular
lipid accumulation. Furthermore, we detected the intracellular
level of autophagy by utilizing autophagic tool drugs, which
modulate the activity of autophagy, including autophagy inhibitors
and activators, and LG.
Materials and methods
Cell culture
L-02 cells (Institute of Biochemistry and Cell
Biology, Shanghai Institute for Biological Sciences, Shanghai,
China) were incubated in Dulbecco’s modified Eagle’s medium (DMEM;
Invitrogoen Life Technologies, Carslbad, CA, USA) supplemented with
10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogoen
Life Technologies) at 37°C in an atmosphere containing 5%
CO2. Cells were used for experiments at 75%
confluency.
Steatotic hepatocyte model
construction
Stock solution of oleic acid (OA; Sigma-Aldrich, St.
Louis, MO, USA) and palmitic acid (PA; Sigma-Aldrich) were prepared
as described previously (15). In
brief, a 15-mmol/l solution of OA or PA in 0.01 mmol/l NaOH was
incubated at 70°C for 30 min, and then fatty acid soaps were mixed
with 5% bovine serum albumin (BSA; Millipore, Billerica, MA, USA)
in phosphate-buffered saline (PBS) (the molar ratio of fatty acid
to BSA was 7:1). To induce steatosis, L-02 cells were exposed to 1
mmol/l free fatty acid (FFA) mixture (OA:PA ratio, 2:1). After
incubation for another 24 h, cells were used for later analysis or
experiments.
Experimental grouping
To investigate the role of autophagy in the
treatment of simple hepatic steatosis by LG (donated by Novo
Nordisk, Bagsvaerd, Denmark), L-02 cells were firstly exposed to 1
mmol/l FFA mixture for 24 h and then divided into six groups to be
exposed to the following stimulation: i) saline (model group), ii)
10 nmol/l LG (LG10 group), iii) 100 nmol/l LG (LG100 group), iv)
100 nmol/l LG + 3-MA (autophagic inhibitor, 5 mmol/l;
Sigma-Aldrich) (negative control, LG100M group), v) rapamycin
(RAPA, autophagic enhancer, 1 μmol/l; Sigma-Aldrich) (positive
control, RAPA group), vi) incubated in DMEM for 24 h and then given
saline (normal group). After another 24 h, cells were collected for
further analyses including oil red O staining, intracellular TG
quantification, quantitative polymerase chain reaction (qPCR),
western blot analysis and electronic microscopy. Furthermore, to
assess the autophagic flux, L-02 cells were exposed to 100 nmol/l
bafilomycin (Sigma-Aldrich) following the aforementioned treatments
for 24 h. After 24 h, cell lysates were quantified by western blot
analysis. The differences in the amount of microtubule-associated
protein 1 light chain 3B (LC3B)-II between groups in the presence
and absence of bafilomycin represent the amount of LC3B-II that is
delivered to lysosomes for degradation (16).
Oil red O staining
To observe the lipid droplets in the L-02 cells that
were cultivated in the six-well plates, each group was processed
with the relevant treatment then rinsed three times with PBS, fixed
in 4% paraformaldehyde for 30 min, stained for 15 min at room
temperature in freshly diluted oil red O, decolorized for 15 sec in
70% ethanol solution, redyed for 30 sec in hematoxylin staining
solution and rinsed with PBS twice. Finally, intracellular lipid
droplets were observed and captured with an inverted microscope
(Mode DIMRE2; Leica, Wetzlar, Germany) connected to a digital
camera (Mode DP70; Olympus, Tokyo, Japan_.
Intracellular TG quantification
Intracellular TG quantification assay was performed
according to the manufacturer’s instructions (K622-100; Biovision,
Milpitas, CA, USA).
Cell viability assay
Cell viability was assessed by the Cell Counting
Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). L-02 cells were treated with 1 mmol/l FFA in
96-well plates. Following incubation for 24 h, 10 μl CCK-8 solution
was added to each well and the plates were further incubated at
37°C for 1–2 h. Subsequently, the optical densities of the plates
were read on a microplate reader (model 3550; Bio-Rad Laboratories,
Inc., Hercules, CA, USA) using a test wavelength of 450 nm. The
absorbance was directly proportionate to the number of living
cells.
Evaluation of intracellular oxidative
stress
Malondialdehyde (MDA) is a marker of oxidative
stress. MDA content was evaluated using an intracellular MDA
quantification kit (A003-4; NanJing JianCheng Bioengineering
Institute, Nanjing, China).
Apoptosis analysis
Early- and late-phase apoptotic cells were monitored
with an Annexin V-fluorescein isothiocyanate (FITC) apoptosis
detection kit (KGA108; Nanjing KeyGen Biotech Co., Nanjing, China).
Following treatment with 1 mmol/l FFA, L-02 cells were harvested,
washed twice with cold PBS, resuspended in binding buffer and
incubated with Annexin V-FITC and propidium iodide (PI) staining
solution following the manufacturer’s instructions. Samples of
10,000 stained cells were analyzed using a flow cytometer (BD
Biosciences, San Jose, CA, USA).
qPCR
Total RNA was extracted from L-02 cells and
converted to cDNA using the PrimeScript RT reagent kit (Takara Bio,
Inc., Shiga, Japan). LC3B mRNA levels were measured by qPCR with
the ABI Prism 7900 sequence detection system (Life Technologies,
Carlsbad, CA, USA), with β-actin as an internal reference gene. The
reaction mixture contained 0.1 mol/l of each primer, 2X SYBR-Green
PCR Master mix (Takara Bio., Inc.) and 1 μl cDNA (1:10 dilution).
Each reaction was performed in triplicate. The primers were
designed as follows: Sense, 5′-CAACTGGGACGACATGGAGAAAAT-3′ and
anti-sense, 5′-CCAGAGGCGTACAGGGATAGCAC-3′ for β-actin; sense,
5′-GAGCAGCATCCAACCAAAAT-3′ and anti-sense,
5′-CTGTGTCCGTTCACCAACAG-3′ for LC3B. The relative mRNA expression
levels were quantified using the 2−ΔΔCt method with
β-actin as an endogenous control (16).
Western blot analysis
L-02 cells in each 25-cm2 culture flask
were collected and proteins were extracted with 1 ml mixture of
RIPA lysis buffer (P0013B; Beyotime Institute of Biotechnology,
Haimen, China) and phenylmethylsulfonyl fluoride (final
concentration, 1 mmol/l). Protein concentration was determined
using a bicinchoninic acid protein assay kit (P0012; Beyotime
Institute of Biotechnology). Thirty micrograms of protein samples
were separated by SDS-PAGE and then electro-transferred onto the
polyvinylidene difluoride membranes (Millipore Corporation,
Billerica, MA, USA). Membranes were blocked for 1 h with 5% skimmed
milk in TBST buffer [50 mmol/l Tris (pH 7.6), 150 mmol/l NaCl and
0.1% Tween-20] and incubated with rabbit monoclonal antibodies
against human LC3B (Cell Signaling Technology, Inc., Beverly, MA,
USA) overnight at 4°C. β-actin was used as an internal control and
was detected using a mouse monoclonal antibody against β-actin
(Sigma, St. Louis, MO, USA). Following incubation with secondary
antibodies, enhanced chemiluminescence detection reagents (Beyotime
Institute of Biotechnology) were used to detect the signals. The
intensity of the signals was quantified with Image Lab 3.0 software
(Bio-Rad Laboratories, Inc.).
Electron microscopy
Following administration of the relevant treatment,
L-02 cells were fixed with 2% glutaraldehyde in 0.1 mol/l phosphate
buffer (pH 7.4) followed by 1% OsO4. After dehydration, thin
sections were cut and stained with uranylacetate and lead citrate.
Digital images were obtained using a JEM1200EX transmission
electron microscope.
Statistical analysis
All data points are expressed as the mean ± standard
error. For comparison of two groups, the unpaired Student’s t-test
was used. In instances of multiple means comparisons, one-way
analysis of variance followed by Bonferroni post hoc tests were
used. P<0.05 was considered to indicate a statistically
significant difference. Graphs and statistical analyses were
generated using Graphpad Prism 5 for Windows (Graphpad Software,
Inc., La Jolla, CA, USA).
Results
FFA treatment induced lipid accumulation
in L-02 cells
L-02 cells were incubated in DMEM containing 1
mmol/l FFA mixture (OA:PA ratio, 2:1) for 24 h and then stained
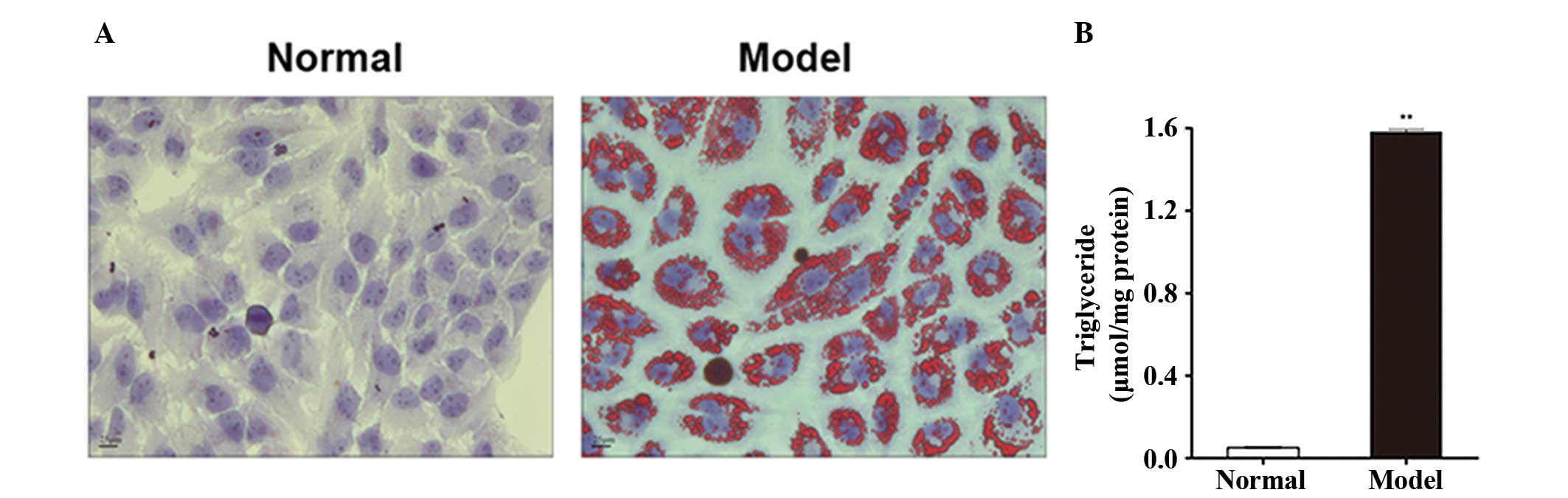
with oil red O. We observed a large quantity of red lipid droplets
in L-02 cells in the model group (Fig.
1A). However, no lipid droplets were detected in L-02 cells in
the normal group (Fig. 1A). As
shown in Fig. 1B, the
intracellular TG level was significantly higher in the model group
than in the normal group (P=3.28×10−8).
FFA treatment did not induce significant
cytotoxicity, oxidative stress or apoptosis
L-02 cells were treated with 1 mmol/l FFA mixture
for 24 h and the cytotoxicity of FFA to L-02 cells was analyzed by
CCK-8 assay. According to the manufacturer’s instructions,
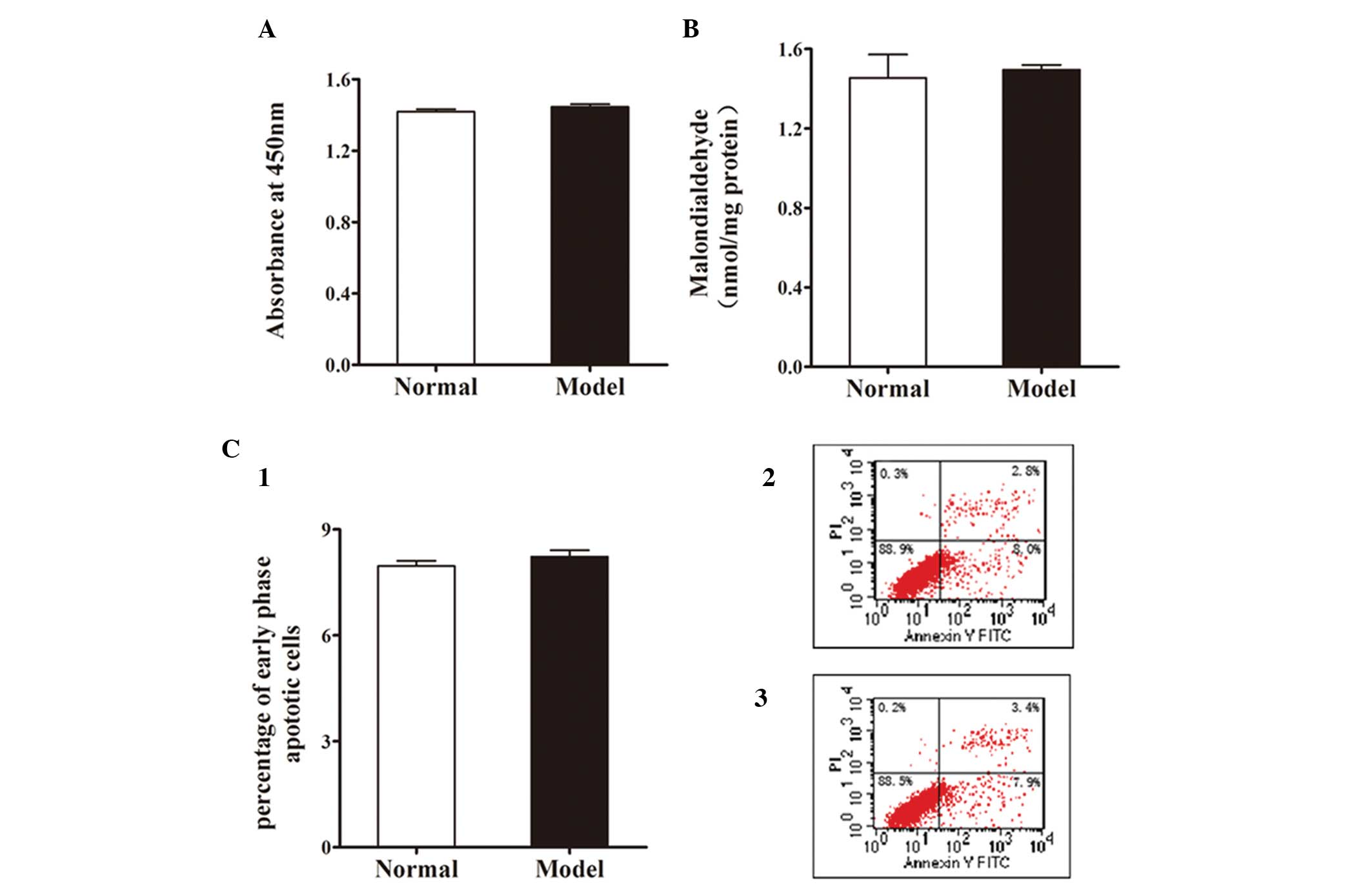
absorbance at 450 nm was positively correlated with cell viability.
Fig. 2A reveals no significant
difference between the normal group and the model group in cell
viability.
There was no significant increase in MDA content, a
widely used marker of intracellular oxidative stress, following FFA
treatment (Fig. 2B).
To evaluate the apoptotic effect of FFA treatment on
L-02 cells, the cells were incubated with 1 mmol/l FFA mixture for
24 h and then stained with Annexin V-FITC/PI. Apoptosis was
measured by flow cytometry. As shown in Fig. 2C, FFA treatment had no apparent
effect on early phase apoptosis in L-02 cells.
These data demonstrate that it is possible to
construct a steatotic hepatocyte model by incubating L-02 cells
with 1 mmol/l FFA mixture (OA:PA=2:1) for 24 h. Notable lipid
accumulation without apparent cytotoxicity, oxidative stress or
apoptosis was detected in this cell model, which was similar to
that observed in simple hepatic steatosis in humans by Farrell and
Larter (17).
LG reduced lipid accumulation in
steatotic L-02 cells
To determine the effect of LG on lipid accumulation
in FFA-treated L-02 cells and the role of autophagy in this
process, steatotic L-02 cells were given various treatments and the
lipid accumulation was quantified, the results of which were
further confirmed using oil red O staining (Fig. 3A). As shown in Fig. 3B, 10 and 100 nmol/l LG both reduced
lipid accumulation in steatotic L-02 cells, and the difference
between the model group and the LG100 group was significant
(P=1.67×10−4). Compared with the use of 100 nmol/l LG
alone, use of both LG (100 nmol/l) and 3-MA (autophagic inhibitor)
increased the intracellular lipid accumulation (Fig. 3B; P=9.83×10−4). Using
RAPA (autophagic enhancer) alone also reduced the lipid
accumulation in the L-02 cells (P=0.013).
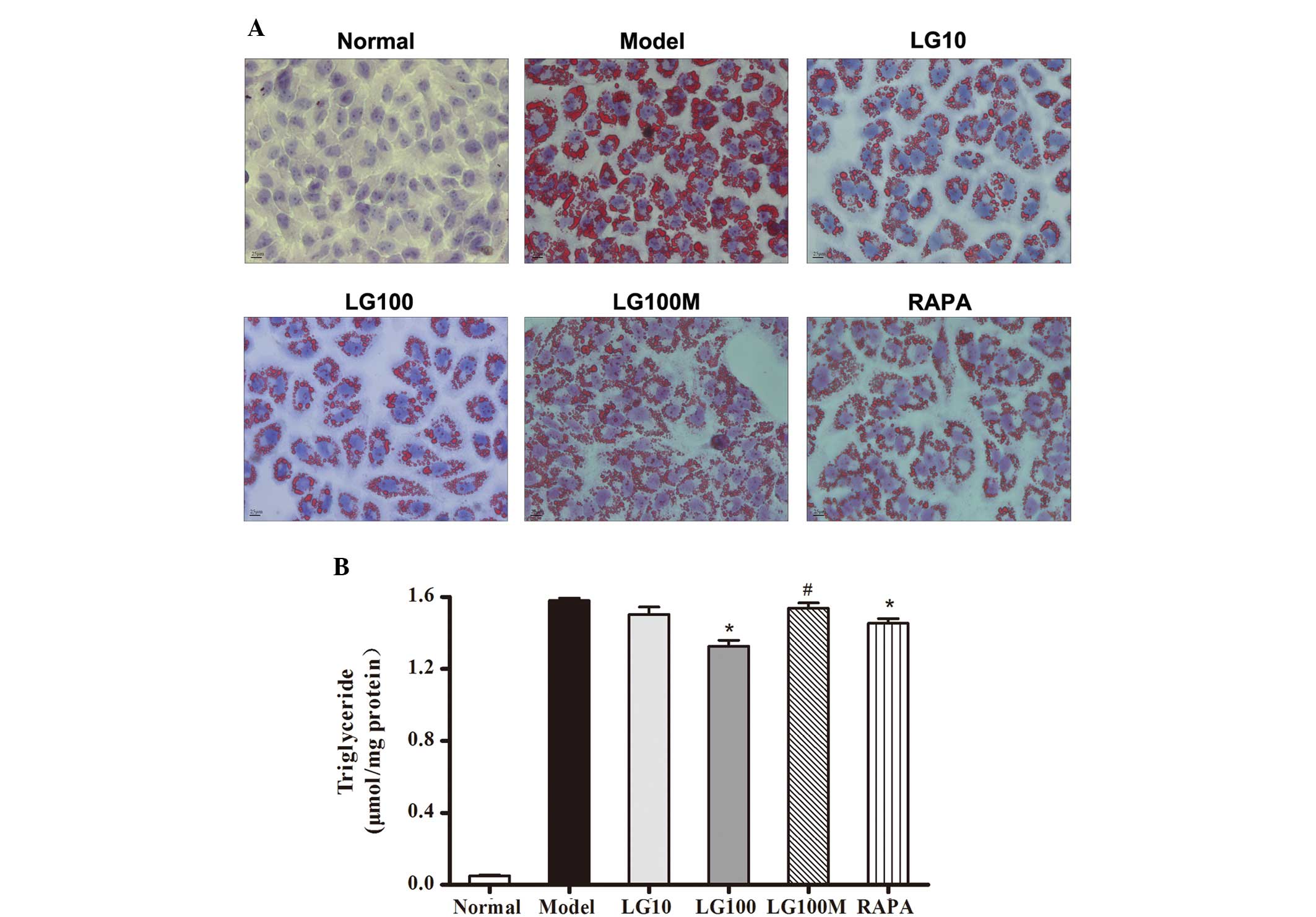 | Figure 3Liraglutide reduced lipid accumulation
in free fatty acid (FFA)-loaded L-02 cells. (A) L-02 cells were
incubated with 1 mmol/l FFA mixture for 24 h and then given various
treatments. After another 24 h, L-02 cells were stained with oil
red O. Original magnification, ×40. (B) Intracellular triglyceride
was quantified. Normal, normal group; model, model group; LG10, 10
nmol/l liraglutide group; LG100, 100 nmol/l liraglutide group;
LG100M, 100 nmol/l liraglutide + 5 mmol/l 3-methyladenine
(autophagic inhibitor) group; RAPA, 1 μmol/l rapamycin (autophagic
enhancer) group. Results are expressed as the mean ± SE from three
independent experiments. *P<0.05, versus the model
group; and #P<0.05, versus the LG100 group. |
LG enhanced autophagy in steatotic L-02
cells
LC3B-II is an important protein marker that is
reliably associated with phagophores, sealed autophagosomes and
mature autophagosomes/autolysosomes (18). To evaluate the intensity of
autophagy in the respective groups, L-02 cells were collected
following treatment, and then qPCR, western blot analysis and
electron microscopy were conducted. As shown in Fig. 4A, the LC3B mRNA expression levels
in the LG100 group were significantly higher than those in the
model group (P=1.18×10−5). When 100 nmol/l LG and 3-MA
were both used, the LC3B mRNA expression levels were notably
decreased compared with those in the LG100 group
(P=6.73×10−4). The LC3B mRNA expression levels in the
RAPA group were significantly higher than those in the model group
(P=0.0044).
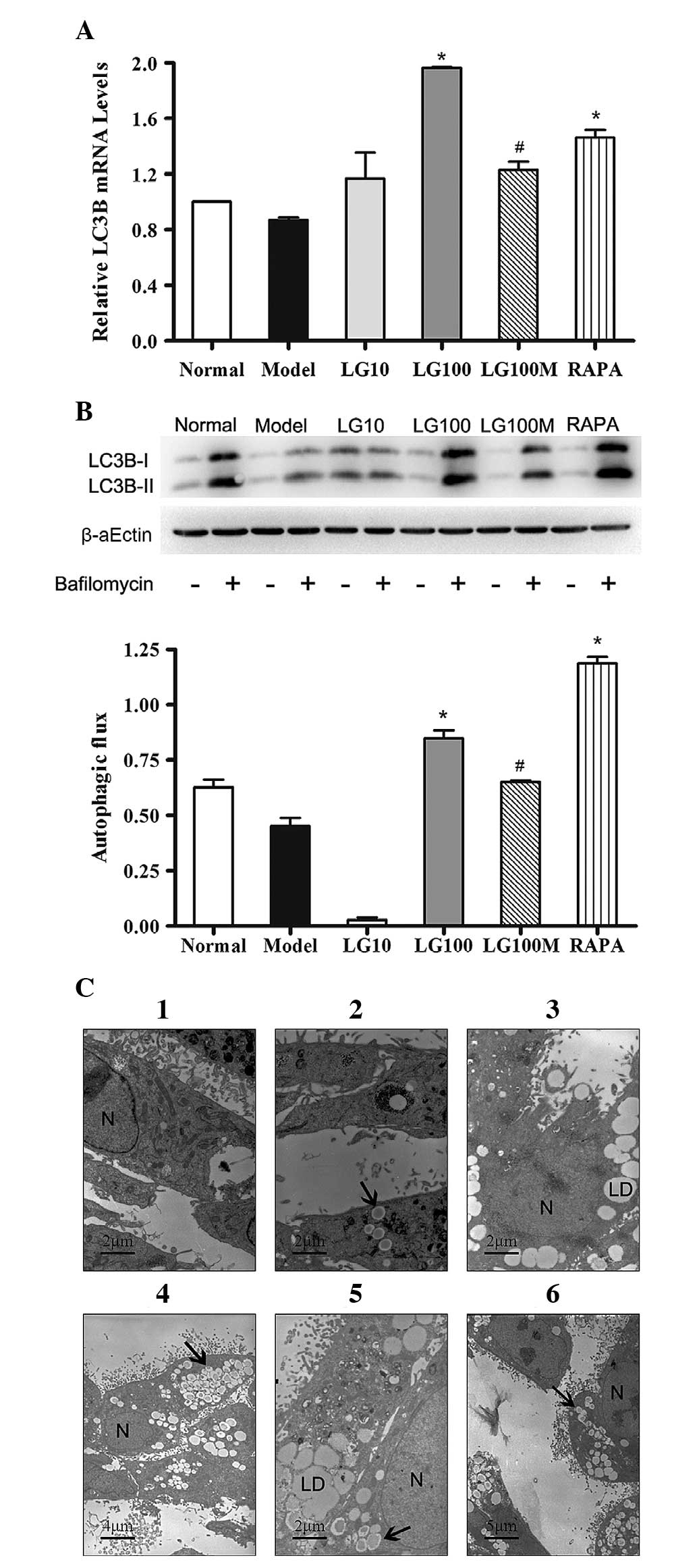 | Figure 4Liraglutide enhanced autophagy in L-02
cells treated with FFA mixture for 24 h. (A) LC3B mRNA expression
level in the groups given various treatments. β-actin was used as a
loading control. (B) Immunoblot for LC3B in the respective groups
in the presence or absence of bafilomycin (100 nmol/l). Autophagic
flux is depicted as the values obtained by subtracting the
densitometry values of LC3B-II in bafilomycin-free samples from
their respective bafilomycin-treated counterparts. Normal, normal
group; model, model group; LG10, 10 nmol/l liraglutide group;
LG100, 100 nmol/l liraglutide group; LG100M, 100 nmol/l liraglutide
+ 5 mmol/l 3-methyladenine (autophagic inhibitor) group; RAPA, 1
μmol/l rapamycin (autophagic enhancer) group. Results are expressed
as the mean ± SE from three independent experiments.
*P<0.05, versus the model control; and
#P<0.05, versus the 100 nmol/l liraglutide group. (C)
Electron microscopy of L-02 cells in the groups given various
treatments. (1) and (2) normal group; (3) model group; (4) 100
nmol/l liraglutide group; (5) 100 nmol/l liraglutide+5 mmol/l 3-MA
group; and (6) RAPA, 1 μmol/l rapamycin group. N, nucleus; LD,
lipid droplets; black arrows, autophagosomes or
autophagolysosomes. |
Since LC3B-II is localized both in the lumenal and
the cytosolic site of the autophagosome and undergoes rapid
degradation within the lysosomes, the increase in LC3B-II may be
the consequence of its increased formation and/or its attenuated
degradation (18). In the present
study, we estimated the autophagic flux by inferring LC3B-II
turnover in the presence and absence of bafilomycin (a
vacuolar-type H+-ATPase inhibitor). The L-02 cells in
the respective groups were incubated either alone or with 100
nmol/l bafilomycin. Since bafilomycin inhibits the fusion of
autophagosomes to lysosomes, the values obtained by subtracting the
densitometry values of LC3B-II in bafilomycin-free samples from
their respective bafilomycin-treated counterparts were taken as the
autophagic flux (18). Fig. 4B reveals that the autophagic flux
in the LG100 group was significantly higher than that in the model
group (P=8.11×10−6). The same trend was detected in the
RAPA group (P=7.87×10−9). Compared with the LG100 group,
the LG100M group demonstrated a significant decrease in autophagic
flux (P=0.0064). These results were in accordance with the change
in the LC3B mRNA expression levels.
Electron microscopy is well recognized as the golden
standard to observe the autophagosome. Autophagosomes and
autolysosomes were difficult to discern as they both present as
autophagic vacuoles (AVs). As shown in Fig. 4C (1–2),
numerous mitochondria and few AVs were present in normal L-02
cells. Following treatment with 1 mmol/l FFA mixture, many lipid
droplets were observed in the cytoplasm around the nucleus, as
shown in Fig. 4C (3). A large amount of AVs were observed in
steatotic L-02 cells treated with 100 nmol/l LG for 24 h (Fig. 4C-4). In steatotic L-02 cells
treated with 100 nmol/l LG and 3-MA, there were numerous lipid
droplets and few AVs (Fig. 4C-5).
A few AVs and lipid droplets coexisted in cells treated with RAPA
(Fig. 4C-6). In summary, treatment
with 100 nmol/l LG enhanced the intensity of autophagy in steatotic
L-02 cells.
Discussion
Excess lipid accumulation in hepatocytes is usually
recognized as the first step in the progression of NAFLD (17). GLP-1 and related peptides have the
potential to reduce lipid content in hepatocytes (11). Autophagy is involved in hepatic
lipid metabolism, and intracellular lipid accumulation
significantly increases when autophagy is inhibited (13). Our present study demonstrated that
LG, a long-acting GLP-1 analog, reduced lipid accumulation in the
steatotic L-02 cell model, and could mimic the pathogenic features
of NAFLD (known as simple hepatic steatosis) in humans. In
addition, our study provides potential mechanistic insights that LG
improves hepatic steatosis by enhancing autophagy.
The pathogenesis of NAFLD has not been clarified
until now and the ‘two hits theory’ is widely accepted. Excess
lipid accumulation in hepatocytes is usually recognized as the
first hit (19). PA and OA are the
most abundant FFAs in liver TGs in both normal subjects and
patients with NAFLD. A previous study showed that human hepatocytes
and HepG2 cells can be induced into steatosis by incubating with
FFA mixture for 24 h. FFA mixture (1 mmol/l; OA:PA ratio=2:1) is
associated with minor toxic and apoptotic effects, thus
representing a cellular model of steatosis that mimics benign
chronic steatosis (20). However,
due to the scarcity of human hepatocytes and the uncertainty in
using liver cancer cell lines, such as HepG2 or Huh-7, we chose the
normal human hepatocyte-derived cell line L-02 in our study.
Steatotic L-02 cells, obtained by incubating L-02 cells with 1 mM
FFA for 24 h, had apparent steatosis with minor cytotoxic,
oxidative and apoptotic effects. Hence, the steatotic L-02 cell
model reproduces the key features of simple hepatic steatosis in
human and is suitable for studies on the pathogenesis and treatment
of NAFLD.
GLP-1-related drugs were previously used for the
treatment of diabetes (21);
however, recent studies have revealed the potential function of
GLP-1-related drugs in improving NAFLD. Ding et al (8) demonstrated that exentin-4, a peptide
agonist of GLP-1R, effectively reversed hepatic steatosis in ob/ob
mice by improving insulin sensitivity. Gupta et al (7) induced steatosis by incubating Huh7
cells with 0.4 mmol/l PA and 0.4 mmol/l OA for 12 h, and then
treated the cells with 20 nmol/l exentin-4 for 6 h. A significant
reduction in lipid content was observed, and the same tendency in
HepG2 cells cultured in methionine-choline-deficient media was
revealed. In the present study, we treated steatotic L-02 cells
with LG and conducted oil red O staining and intracellular TG
quantification. The results revealed a significant reduction in
lipid accumulation in steatotic L-02 cells (Fig. 3). Since the steatotic L-02 cell
model was able to mimic the features of human simple hepatic
steatosis, our results suggest a new role for GLP-1-related drugs
in simple hepatic steatosis.
The hallmark of NAFLD is hepatic lipid (mainly TG)
accumulation in the absence of significant ethanol consumption,
viral infection or other specific etiologies. Hepatic lipid
accumulation results from an imbalance between lipid availability
(from circulating lipid uptake or de novo lipogenesis) and lipid
disposal (via FFA oxidation or very low density lipoprotein
secretion) (22). Previous studies
have made great efforts to elucidate the mechanism underlying the
anti-steatosis effects of GLP-1-related drugs. These studies
suggested that GLP-1-related drugs such as exendin-4 were able to
reduce lipid accumulation through reducing weight and improving
insulin sensitivity (6–8). In addition, there is much evidence to
show that GLP-1-related drugs attenuate hepatic steatosis through
targeting the hepatic lipid metabolism, including inhibiting
lipogenesis (10,23), promoting fatty acid β-oxidation
(10,24), increasing fatty acid transport
(24) and enhancing lipid droplet
fission (25). The phosphorylation
of cAMP activated kinase (AMPK) was recognized as being crucial in
the above pathways (6,10,23).
Previous studies have implicated a role of autophagy
in hepatic lipid metabolism. Škop et al (26) suggested that the inhibition of the
autophagic flux or lysosomal activity decreased the secretion of
very low density lipoprotein and formation of FFA oxidative
products, while the stimulation of autophagy by RAPA increased some
of these indicators. The study by Singh et al (13) demonstrated that the inhibition of
autophagy (by pharmacological or genetic approaches) in cultured
hepatocytes and mouse liver increased TG storage in lipid droplets.
The ablation of liver-specific autophagy led to excessive hepatic
lipid accumulation and the development of fatty liver. These
findings suggested that the upregulation of autophagy in
hepatocytes could increase the breakdown of lipid stores and may
serve as a novel approach in treating NAFLD.
The present study demonstrated that LG reduces lipid
accumulation in steatotic L-02 cells. The role of autophagy in the
anti-steatosis effect of LG was investigated, and the results
revealed that LG increased the mRNA and protein expression level of
LC3B-II, which serves as a widely used marker for autophagosomes
(18). Since autophagy is a
dynamic process and LC3B-II is degraded in this process, we further
analyzed the autophagic flux. Compared with that in the model
group, the intensity of the autophagic flux in the LG (100 nmol/l)
group was significantly enhanced. To evaluate autophagy more
precisely, electron microscopy was employed to observe
autophagosomes and autolysosomes (both taken as AVs). This revealed
that the number of AVs was notably increased following treatment
with 100 nmol/l LG. 3-MA inhibits autophagosome formation in
vitro by inhibiting class III PI3-kinase (18). In the current study, treatment with
3-MA weakened the effect of LG in enhancing autophagy and reducing
lipid accumulation. RAPA, as an inhibitor of mTOR, activates
autophagy (18). Our study showed
that treatment with LG enhanced autophagy and reduced lipid
accumulation in steatotic L-02 cells, which was similar to RAPA.
Therefore, the activation of autophagy plays a significant role in
the anti-steatosis effect of LG in vitro.
Two limitations of the present study need to be
addressed. Firstly, as the nature of NAFLD has been unclear until
now, it is difficult to exactly differentiate simple hepatic
steatosis from NASH. L-02 cell steatosis induced by exposure to FFA
mixture may not always mimic simple hepatic steatosis. Secondly, as
a result of the complexity in measuring autophagy, certain
experiments are warranted, such as modulating the intensity of
autophagy by knockdown or knockout of ATG genes.
In conclusion, the cell model, built by incubating
L-02 cells with an FFA mixture (OA:PA ratio, 2:1) for 24 h,
replicates the pathological features of human simple hepatic
steatosis. Treatment with LG reduces lipid accumulation in
steatotic L-02 cells and the activation of autophagy plays a
significant role in the process. Thus, our data indicate that GLP-1
analogs are a promising treatment approach and the activation of
autophagy may be a potential mechanism to improve hepatic
steatosis.
Acknowledgements
The authors thank Dr Ying-Hong Shi, Dr Zhi Dai and
Dr Wei-Ren Liu, from the Institute of Liver cancer, Zhongshan
Hospital, Fudan University, Shanghai, China, for their help in the
process of performing this study. The kind donation of LG from Novo
Nordisk was greatly appreciated. The authors also thank Dr Bing Li
and Dr Yuan Yuan for their careful modification of this paper.
References
|
1
|
Cohen JC, Horton JD and Hobbs HH: Human
fatty liver disease: old questions and new insights. Science.
332:1519–1523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferre P and Foufelle F: Hepatic steatosis:
a role for de novo lipogenesis and the transcription factor
SREBP-1c. Diabetes Obes Metab. 12 Suppl 2:83–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gentile CL, Frye MA and Pagliassotti MJ:
Fatty acids and the endoplasmic reticulum in nonalcoholic fatty
liver disease. Biofactors. 37:8–16. 2011. View Article : Google Scholar
|
|
4
|
LL and Drucker DJ: Biology of incretins:
GLP-1 and GIP. Gastroenterology. 132:2131–2157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abu-Hamdah R, Rabiee A, Meneilly GS,
Shannon RP, Andersen DK and Elahi D: Clinical review: The
extrapancreatic effects of glucagon-like peptide-1 and related
peptides. J Clin Endocrinol Metab. 94:1843–1852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Svegliati-Baroni G, Saccomanno S,
Rychlicki C, et al: Glucagon-like peptide-1 receptor activation
stimulates hepatic lipid oxidation and restores hepatic signalling
alteration induced by a high-fat diet in nonalcoholic
steatohepatitis. Liver Int. 31:1285–1297. 2011. View Article : Google Scholar
|
|
7
|
Gupta NA, Mells J, Dunham RM, et al:
Glucagon-like peptide-1 receptor is present on human hepatocytes
and has a direct role in decreasing hepatic steatosis in vitro by
modulating elements of the insulin signaling pathway. Hepatology.
51:1584–1592. 2010. View Article : Google Scholar
|
|
8
|
Ding X, Saxena NK, Lin S, Gupta NA and
Anania FA: Exendin-4, a glucagon-like protein-1 (GLP-1) receptor
agonist, reverses hepatic steatosis in ob/ob mice. Hepatology.
43:173–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma S, Mells JE, Fu PP, Saxena NK and
Anania FA: GLP-1 analogs reduce hepatocyte steatosis and improve
survival by enhancing the unfolded protein response and promoting
macroautophagy. PLoS One. 6:e252692011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ben-Shlomo S, Zvibel I, Shnell M, et al:
Glucagon-like peptide-1 reduces hepatic lipogenesis via activation
of AMP-activated protein kinase. J Hepatol. 54:1214–1223. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Samson SL and Bajaj M: Potential of
incretin-based therapies for non-alcoholic fatty liver disease. J
Diabetes Complications. 27:401–406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh R, Kaushik S, Wang Y, et al:
Autophagy regulates lipid metabolism. Nature. 458:1131–1135. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding WX, Li M, Chen X, et al: Autophagy
reduces acute ethanol-induced hepatotoxicity and steatosis in mice.
Gastroenterology. 139:1740–1752. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi SE, Lee SM, Lee YJ, et al: Protective
Role of Autophagy in Palmitate-Induced INS-1 beta-Cell Death.
Endocrinology. 150:126–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
17
|
Farrell GC and Larter CZ: Nonalcoholic
fatty liver disease: from steatosis to cirrhosis. Hepatology.
43:S99–S112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N, Yoshimori T and Levine B:
Methods in Mammalian Autophagy Research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Day CP and James OF: Steatohepatitis: a
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998.
|
|
20
|
Gomez-Lechon MJ, Donato MT,
Martinez-Romero A, Jimenez N, Castell JV and O’Connor JE: A human
hepatocellular in vitro model to investigate steatosis. Chem Biol
Interact. 165:106–116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marathe CS, Rayner CK, Jones KL and
Horowitz M: Glucagon-like peptides 1 and 2 in health and disease: a
review. Peptides. 44:75–86. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Musso G, Gambino R and Cassader M: Recent
insights into hepatic lipid metabolism in non-alcoholic fatty liver
disease (NAFLD). Prog Lipid Res. 48:1–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang L, Yang M, Ren H, et al: GLP-1
analogue prevents NAFLD in ApoE KO mice with diet and Acrp30
knockdown by inhibiting c-JNK. Liver Int. 33:794–804. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mells JE, Fu PP, Sharma S, et al: Glp-1
analog, liraglutide, ameliorates hepatic steatosis and cardiac
hypertrophy in C57BL/6J mice fed a Western diet. Am J Physiol
Gastrointest Liver Physiol. 302:G225–G235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gupta NA, Kolachala VL, Jiang R, et al:
The glucagon-like peptide-1 receptor agonist Exendin 4 has a
protective role in ischemic injury of lean and steatotic liver by
inhibiting cell death and stimulating lipolysis. Am J Pathol.
181:1693–1701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Škop V, Cahová M, Papáčková Z, et al:
Autophagy-lysosomal pathway is involved in lipid degradation in rat
liver. Physiol Res. 61:287–297. 2012.PubMed/NCBI
|