Introduction
Cisplatin (cis-diamminedichloroplatinum II; CDDP) is
one of the most effective chemotherapeutic agents and is widely
used for the treatment of solid tumors. The side effects and
acquired drug resistance that occur during cisplatin treatment
limit its clinical use (1–3). The primary cellular target of
cisplatin is considered to be nuclear DNA. Cisplatin-induced DNA
damage activates various signaling pathways to promote cell death
predominantly by inducing apoptosis (4–6). A
number of studies have identified that cisplatin can induce
endoplasmic reticulum (ER) stress and nucleus-independent apoptotic
signaling (7–10).
Various physiological and pathological conditions
may lead to ER stress, which results in an accumulation of unfolded
or misfolded proteins in the ER lumen (11,12).
This cellular stress subsequently causes an activation of the
unfolded protein response (UPR), which induces the expression of
chaperones and proteins involved in the recovery process. Severe ER
stress can lead to cell death, commonly by the activation of
intrinsic apoptosis (13,14). The accumulated unfolded or
misfolded proteins in the ER are marked for degradation by the
ubiquitin-proteasome or autophagy-lysosome pathway (15,16).
Previous studies have demonstrated that inhibitors of autophagy,
such as 3-methyladenine and chloroquine, effectively enhance the
cytotoxicity of chemotherapeutic agents such as cisplatin and S1 by
increasing ER stress (17–20). Thus, in the present study, the
effect of the proteasome inhibitor lactacystin (LAC) on cisplatin
cytotoxicity was assessed. LAC covalently binds to the N-terminal
threonine of the 20S proteasome subunit X, and irreversibly
modifies all catalytic β subunits. LAC inhibits proteases such as
cathepsin A and tripeptidyl peptidase II (21–24).
In the present study, it was hypothesized that the
use of LAC would increase ER stress-associated apoptosis induced by
cisplatin in HeLa human cervical cancer (HCC) cells. HeLa cells
were treated with cisplatin, LAC or a combinational therapy
incorporating the two drugs simultaneously, and the subsequent
effects were analyzed by MTT assay, protein and RNA expression
analyses.
Materials and methods
Cell culture
HeLa human cervical cancer cells were cultured at
37°C in an atmosphere of 5% CO2 and 95% air, in Iscove’s
modified Dulbecco’s medium (IMDM; Life Technologies, Grand Island,
NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco Life
Technologies, Carlsbad, CA, USA)and 100 U/ml penicillin and 100
μg/ml streptomycin, prior to use in all experiments. The cells were
divided into four groups as follows: Non-treated cells;
cisplatin-treated cells (5 μg/ml); LAC-treated cells (10 μM);
Cisplatin (5 μg/ml) and LAC (10 μM)-treated cells. Cisplatin and
LAC were purchased from Sigma-Aldrich (St. Louis, MO, USA)
MTT assay
Cell viability was determined by MTT assay. HeLa
cells, during the exponential growth phase, were seeded into
96-well culture plates in 100 μl IMDM at a density of
1×104 cells/well. After 24-h incubation, the indicated
dose of cisplatin (5 μg/ml) and/or LAC (10 μM) was added for 12-h
incubation in four parallel wells. MTT assays (Beyotime Institute
of Biotechnology, Haimen, China) were performed as follows: 20 μl
MTT solution (5 mg/ml in PBS) was added to the cells for 4 h, after
which, 150 μl dimethyl sulfoxide (Beijing Chemical Industry Co.,
Ltd., Beijing, China) was added to each well. The cells were
agitated for 10 min, prior to absorbance measurements at 570 nm
using a Microplate Reader (Bio-Rad Laboratories, Hercules, CA,
USA). The growth inhibition rate was calculated as % inhibition = 1
− absorbance of experimental group/absorbance of control group ×
100. The mean value of the four replica wells was calculated for
each treatment group.
Western blotting
Whole-cell protein extracts from HeLa cells were
prepared with cell lysis buffer (50 mM Tris-HCl, pH 7.5; 150 mM
NaCl; 1 mM Na2EDTA; 1 mM EDTA; 1% Triton; 2.5 mM sodium
pyrophosphate; 1 mM β-glycerophosphate; 1 mM
Na3VO4; 1 mM NaF; 1 μg/ml leupeptin; and 1 mM
PMSF) for western blotting. The protein extracts were quantified
using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories). For
Western blot analysis, protein lysates (30–50 μg) were separated by
12% SDS-PAGE and transferred onto Immobilon-P Membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
non-fat dry milk in buffer (10 mM Tris-HCl, pH 7.6; 100 mM NaCl;
and 0.1% Tween 20) for 2 h at room temperature and then incubated
with the appropriate primary antibodies, including the monoclonal
rabbit anti-human Ub and monoclonal rabbit anti-human caspase-3
(1:1,000 dilutions; Epitomics, Burlingame, CA, USA), monoclonal
mouse anti-human PDI, monoclonal mouse anti-human p62, monoclonal
mouse anti-human Grp78, monoclonal mouse anti-human CHOP,
polyclonal rabbit anti-human caspase-4, monoclonal rabbit
anti-human caspase-3 and monoclonal mouse anti-human β-actin
(1:1,000 dilutions; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA) overnight at 4°C, followed by incubation with horseradish
peroxidase-conjugated secondary antibody (Hangzhou HuaAn
Biotechnology Co.. Ltd., HangZhou, China) at 1:2,000 dilution for
1.5 h at room temperature. The immunoreactive bands were visualized
by the diaminobenzidine (Sigma-Aldrich) coloration method. The
representative bands were measured using a Tanon Gel Imaging System
(Tanon Science and Technology Co., Ltd., Shanghai, China) and
analyzed. The protein expression levels were normalized to actin
and the ratios of the normalized protein are presented as the means
± standard deviation from three independent experiments. The
protein levels were quantified by densitometry using Quantity One
1-D Analysis Software (Bio-Rad Laboratories).
Immunofluorescence staining and confocal
laser microscopy
HeLa cells were cultured on coverslips overnight,
and were then treated with cisplatin (5 μg/ml) and/or LAC (10 μM)
for 12 h. The cells were then fixed with 4% paraformaldehyde
(Beijing Chemical Industry Co., Ltd.), stained with the Hoechst
33342 nuclear stain (2 μg/ml; Sigma-Aldrich) for 30 min, washed
with phosphate-buffered saline (PBS; Beijing Zhongshan Golden
Bridge Biological Technology Co., Ltd., Beijing, China), and
examined using an Olympus FV1000 confocal laser microscope (Olympus
Corporation, Tokyo, Japan) to reveal chromatin condensation. The
expression levels of active caspase-3 and γ-H2AX were
examined by indirect immunofluorescence methods. Briefly, cells
were cultured on coverslips overnight and treated with cisplatin (5
μg/ml) and/or LAC (10 μM) for 12 h. The cells were then rinsed 3
times with PBS prior to fixation with 4% paraformaldehyde for 20
min. The cells were then permeabilized with 0.1% Triton X-100
(Beijing Dingguo Changsheng Biotechnology Co., Ltd., Beijing,
China) for 5 min and blocked with bovine serum albumen (Beijing
Dingguo Changsheng Biotechnology Co., Ltd.), prior to incubation
with primary antibodies against active caspase-3 (Epitomics) and
γ-H2AX (Cell Signaling Technology, Inc., Danvers, MA,
USA) (1:100 dilution) overnight at 4°C. The cells were then
incubated with Alexa Fluor543/488-conjugated secondary antibody
(1:400; Invitrogen Life Technologies, Carlsbad, CA, USA) for 1 h,
then stained with the Hoechst 33342 (2 μg/ml) for 2 min, and washed
with PBS 3 times. The cells were mounted and examined by confocal
laser microscopy.
Statistical analysis
Data are representative of three independent
experiments each conducted in triplicate. Statistical analysis of
the data was performed using one-way analysis of variance. Tukey’s
post-hoc test was used to determine the significance for all
pairwise comparisons of interest. P<0.05 was considered to
indicate a statistically significant difference.
Results
LAC potentiates cell growth inhibition
induced by cisplatin
Based on previous studies, HeLa cells were treated
with the indicated doses of cisplatin and/or LAC for 12 h, and then
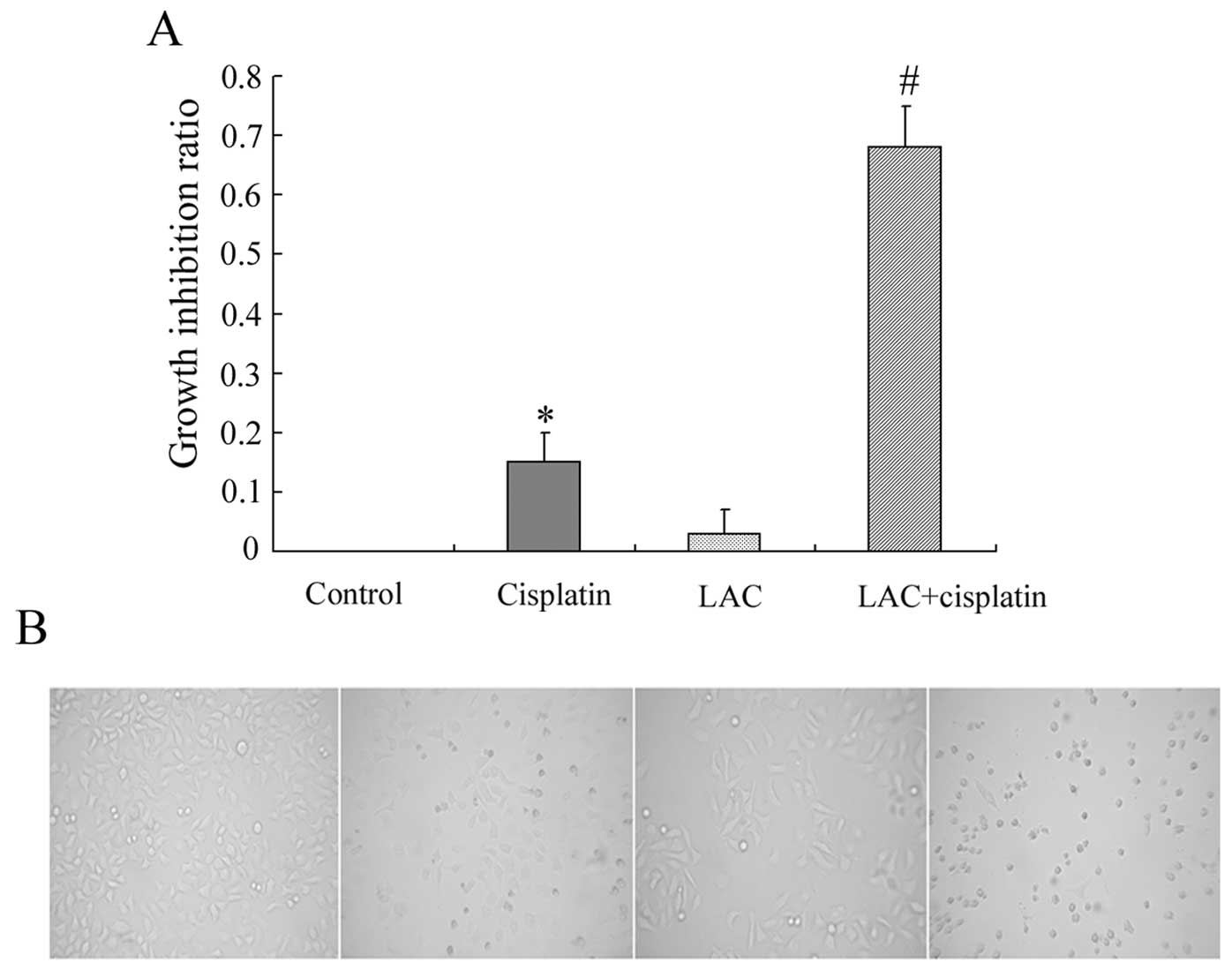
the growth inhibition was examined by MTT assay. It was observed
that cisplatin inhibited the growth of HeLa cells. MTT assay
indicated that LAC alone exerted no significant effect on cell
viability, and LAC treatment enhanced the cytotoxic effect of
cisplatin when administered in combination (Fig. 1A). Changes to cellular morphology
were observed under a inverted phase contrast microscope. Compared
with the controls, round and fragile cells were detected in the
cisplatin treatment group. The number of round and fragile cells
was increased in the group treated with cisplatin combined with LAC
(Fig. 1B).
LAC increases cisplatin-induced cell
apoptosis
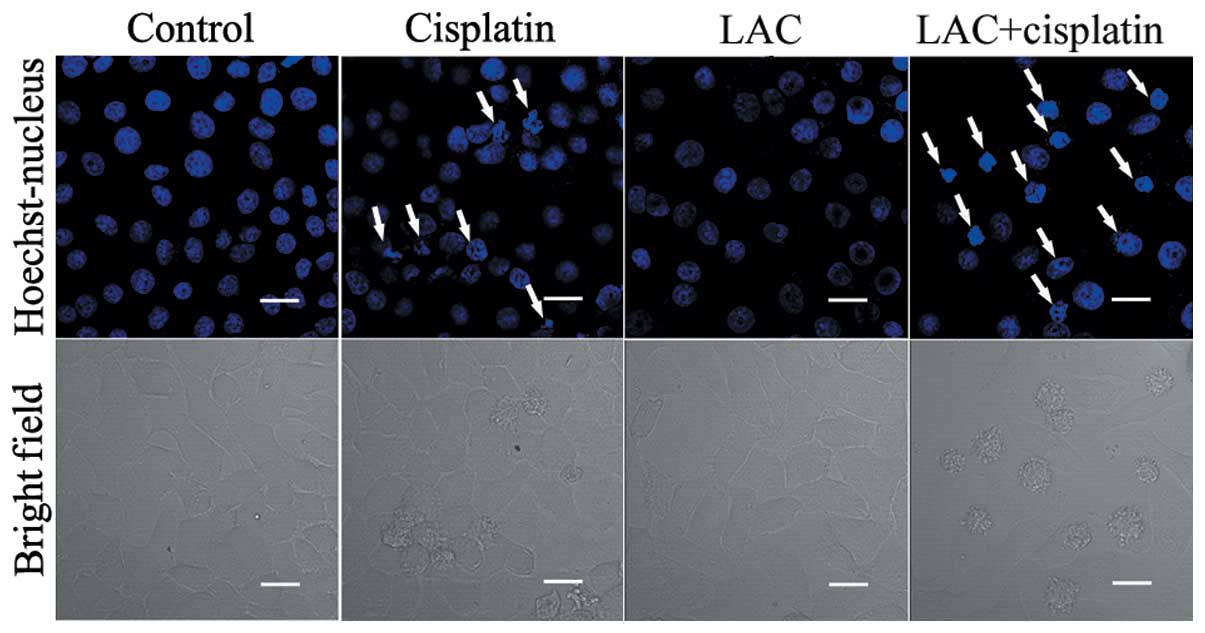
The levels of apoptosis were analyzed in order to
whether LAC may potentiate the apoptosis induced by cisplatin in
HeLa cells. Apoptotic chromatin condensation was analyzed with
Hoechst 33342 staining and confocal microscopy. As compared with
the control cells, cisplatin-induced apoptotic chromatin
condensation was clearly observed. As compared with the
cisplatin-treated group, the cells treated with both cisplatin and
LAC exhibited a marked increase in the levels of apoptotic
chromatin condensation (Fig.
2).
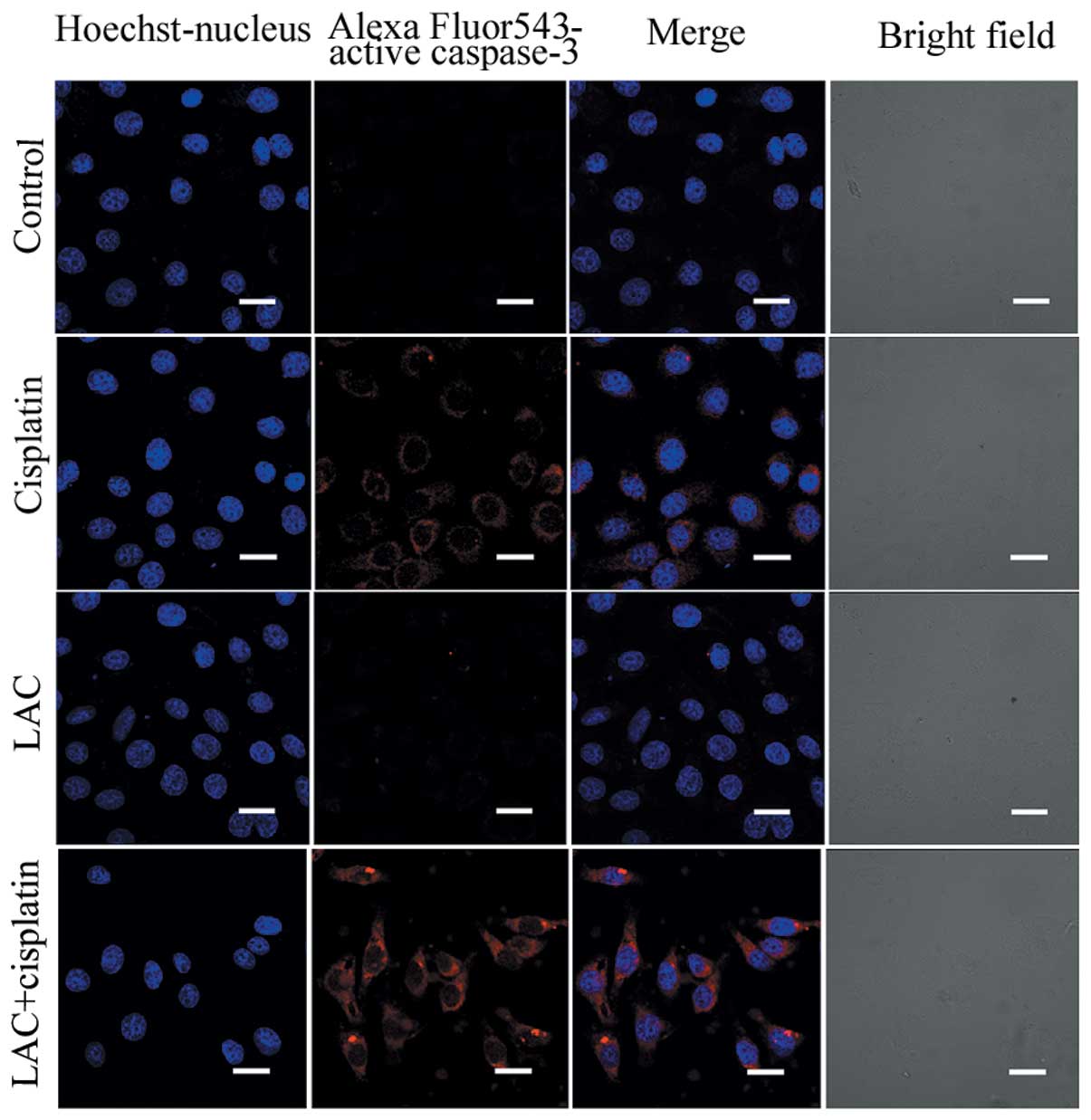
Caspase-3 functions as an executioner molecule
during apoptosis, and its activation reflects the initiation of
apoptosis. Using confocal microscopy, the activation of caspase-3
was detected in the cisplatin-treated cells and those treated with
cisplatin combined with LAC (Fig.
3). The expression of caspase-3, indicated by indirect red
fluorescence, was stronger in the cells treated with cisplatin
combined with LAC compared with cells treated with cisplatin alone,
indicating that the combined treatment increased caspase-3
activation. These results indicate that LAC can efficiently
increase the apoptosis induced by cisplatin in HeLa cells.
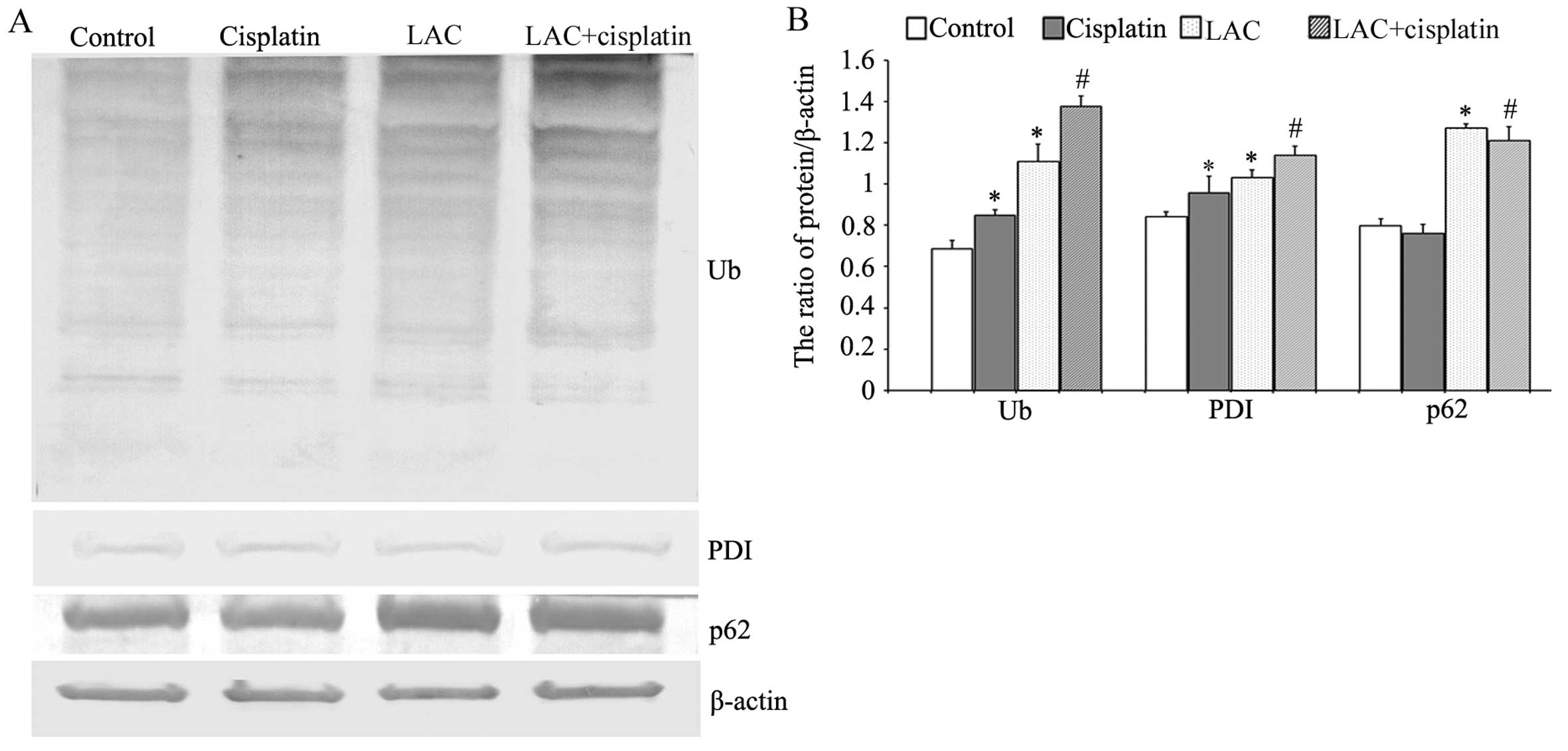
LAC increases cisplatin-induced ER
stress
Previous studies have indicated that cisplatin can
induce ER stress by misfolded protein accumulation. Misfolded
proteins may be ubiquitinated, marking them for degradation
(17). Therefore the expression
levels of ubiquitin (Ub), ER chaperone protein disulfide isomerase
(PDI; which reflects the occurrence of ER stress), and p62 (an
adaptor that transports the ubiquitinated proteins) were analyzed
by western blotting. It was observed that cisplatin and LAC
treatments induced a higher level of Ub. The combination of
cisplatin and LAC markedly increased the ubiquitination of
proteins. Cisplatin and LAC increased the expression of PDI
compared with the control cells, and combination treatment
increased the expression of PDI compared with the cisplatin group.
In addition, the protein level of p62 showed a slight reduction
following cisplatin treatment, whilst LAC treatment increased its
level in cells treated with LAC alone or when combined with
cisplatin (Fig. 4). These results
indicate that LAC can increase cisplatin-induced ER stress in HeLa
cells.
LAC increases cisplatin-induced ER
stress-associated apoptosis
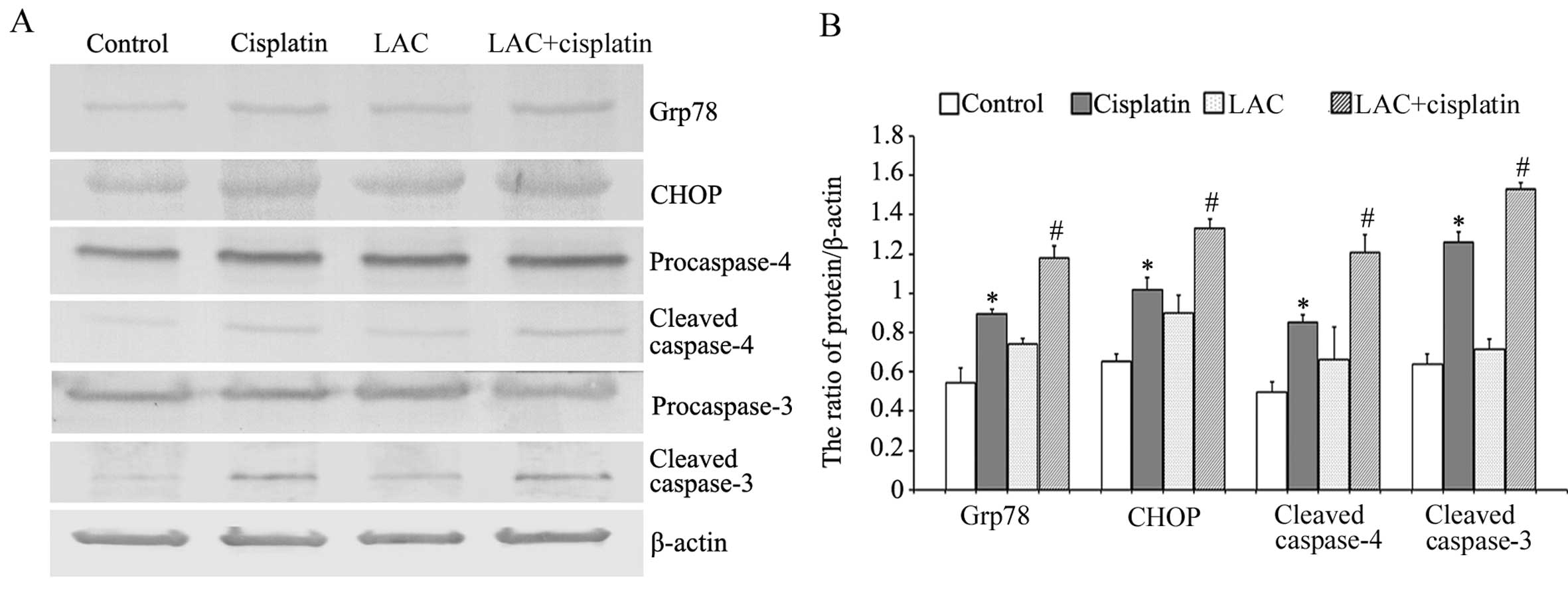
Glucose-regulated protein-78 (Grp78) is an ER
chaperone protein, which can be upregulated by ER stress (18). The growth-arrest and
DNA-damage-inducible gene, 153/C/EBP homology protein
(GADD153/CHOP), is involved in ER stress-induced cell death;
sustained ER stress induces caspase-mediated apoptosis (17,18).
Caspase-4 is an ER-resident caspase that, similar to murin
caspase-12, is processed in response to ER stress and is activated
during ER stress-induced apoptosis. Using western blot analysis, it
was observed that cisplatin induced the upregulation of Grp78,
CHOP, and cleaved caspases 3 and 4. Compared with cisplatin, LAC
increased the expression levels of all these proteins (Fig. 5). These results indicate that LAC
increased cisplatin-induced ER stress-associated apoptosis.
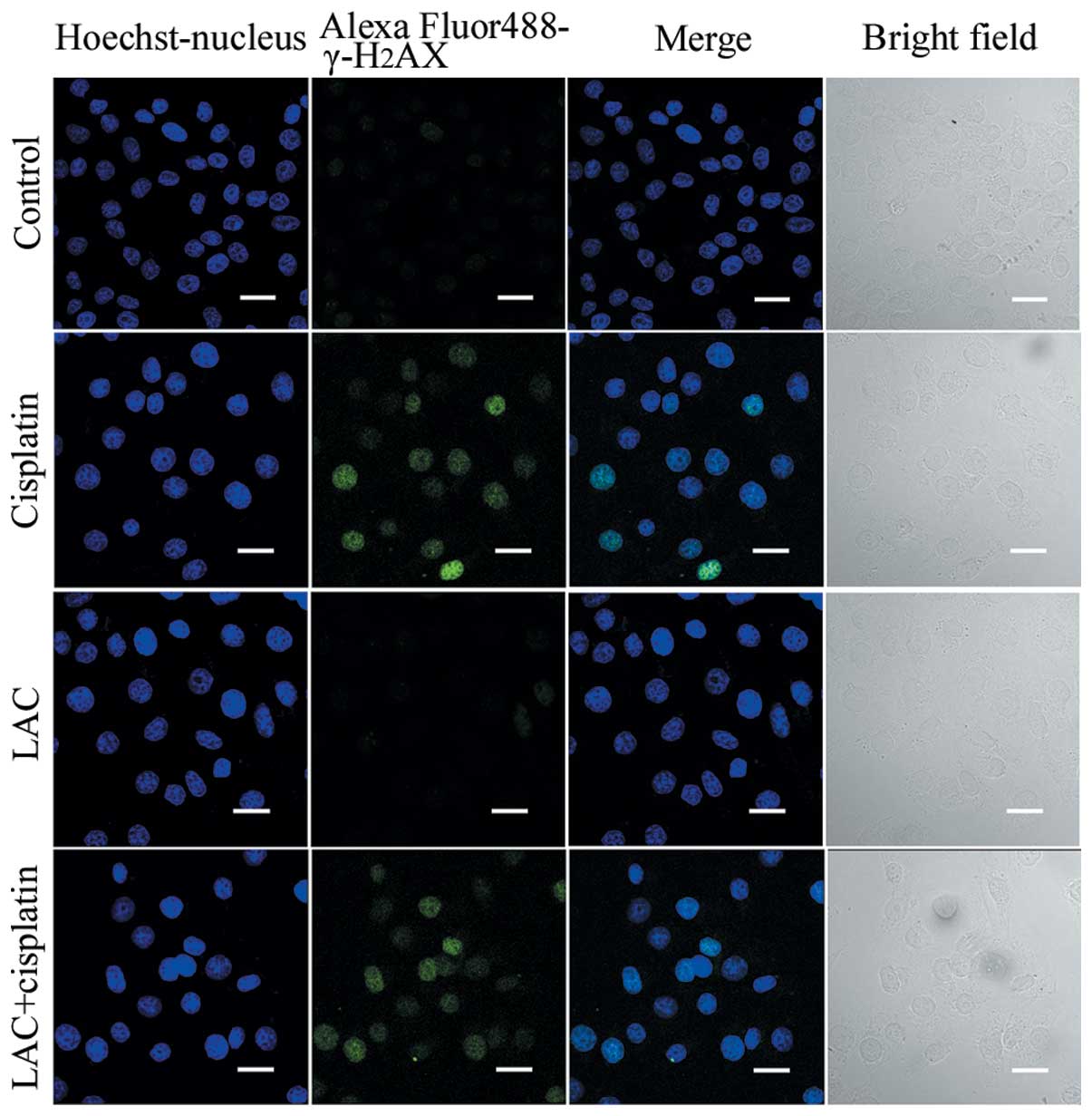
LAC does not enhance the level of
cisplatin-induced DNA double-strand breaks (DSB)
Cisplatin acts to damage DNA and inhibit DNA
synthesis, thus resulting in cancer-cell death (6). Therefore it was hypothesized in the
current study that LAC may increase DNA breakage induced by
cisplatin. DNA DSB are able to induce phosphorylation of
H2AX at a conserved serine 139 residue in the C terminal
region, leading to the formation of γ-H2AX. This
molecule is commonly used as a DNA DSB marker. Using confocal
microscopy, the expression of γ-H2AX, reported by
indirect green fluorescence, was visualized in both the cells
treated for 12 h with cisplatin and cisplatin combined with LAC
(Fig. 6). These results indicate
that LAC does not enhance the DSB induced by cisplatin in HeLa
cells.
Discussion
Cisplatin has been used as a chemotherapeutic agent
in the clinical setting for many years, with positive effects
against numerous types of solid tumors, including cervical cancer
(1). The produced side-effects and
drug resistance of cisplatin limit its use. The predominant pathway
of cell death that is induced by cisplatin is a caspase-dependent
intrinsic apoptotic pathway involving the mitochondria and ER
(6,10,25).
The ER has been previously reported to be a target
of cisplatin-induced apoptosis (17,18).
It was demonstrated that cisplatin was able to induce ubiquitinated
protein accumulation and lead to ER stress in HeLa and SKOV3 cells
(17,18). The following three ER sensors have
been identified: PKR-like ER kinase, inositol requiring enzyme 1
and activating transcription factor 6 in UPR induction (26). Upon induction of ER stress is
induced, the cell upregulates several ER resident chaperones, such
as GRP78 and PDI, to re-establish ER homeostasis. Simultaneously,
the misfolded or unfolded proteins are transported to be degraded
by the ubiquitin-proteasome or autophagy-lysosome pathways
(11,12,26).
P62 is a multifunctional molecule, functioning as an adaptor to
bind ubiquitinated proteins and transport them for degradation.
Once the proteins are degraded, bound p62 molecules are also
degraded (27,28). Sustained and unabated ER stress
induces the activation of apoptosis. CHOP (GADD153) is a member of
the C/EBP family of bZIP transcription factors, and its expression
is upregulated by ER stress. CHOP participates in
ER-stress-corrective actions through induction or suppression of a
number of genes. Prolonged activation of CHOP can trigger apoptosis
in cells under certain conditions (29,30).
In the present study, it was demonstrated that
cisplatin treatment inhibited cell growth and induced cell
apoptosis in HeLa cells. In addition, exposure to cisplatin
increased the expression of Ub, PDI and GRP78 and upregulated the
level of CHOP and cisplatin treatment induced the activation of
caspase-4 and caspase-3. Together, these findings indicate that
cisplatin can induce ER stress-associated apoptosis in human
cervical cancer HeLa cells. LAC treatment combined with cisplatin
potentiated the effects of cisplatin alone. DNA damage is
considered an indicator of apoptosis in cisplatin cytotoxicity.
H2AX phosphorylation occurs in response to
cisplatin-induced DNA lesions. The formation of γ-H2AX
is a useful indicator of cisplatin-induced DNA damage (31). Thus, the changes to
γ-H2AX formation in cells treated with cisplatin
combined with LAC was investigated in the present study. However,
there was no difference between cells treated with cisplatin alone
and those treated with cisplatin combined with LAC. These results
indicate that LAC enhanced cisplatin cytotoxicity by increasing ER
stress-associated apoptosis, rather than by increasing DNA
damage.
In conclusion, it was demonstrated that cisplatin
treatment induced ER stress-associated apoptosis in human cervical
cancer HeLa cells. LAC treatment combined with cisplatin increased
the cell growth inhibition rate, cell apoptosis and activation of
caspase-3. Additionally, LAC treatment increased the
cisplatin-induced expression levels of PDI, GRP78, CHOP, cleaved
caspase-4 and cleaved caspase-3. These data indicate that LAC is
able to enhance cisplatin cytotoxicity by increasing ER
stress-associated apoptosis, and it has the potential to improve
the results of cisplatin chemotherapy.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81372793 and
81100808), the Natural Science Foundation of Jilin Province (no.
201015240) and the Department of Education of Jilin Province
Project (no. 2013361).
References
|
1
|
Muggia F: Platinum compounds 30 years
after the introduction of cisplatin: implications for the treatment
of ovarian cancer. Gynecol Oncol. 112:275–281. 2009.PubMed/NCBI
|
|
2
|
Florea AM and Büsselberg D: Cisplatin as
an anti-tumor drug: cellular mechanisms of activity, drug
resistance and induced side effects. Cancers (Basel). 3:1351–1371.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
4
|
Eastman A: The formation, isolation and
characterization of DNA adducts produced by anticancer platinum
complexes. Pharmacol Ther. 34:155–166. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Woźniak K and Błasiak J: Recognition and
repair of DNA-cisplatin adducts. Acta Biochim Pol. 49:583–596.
2002.
|
|
6
|
Basu A and Krishnamurthy S: Cellular
responses to Cisplatin-induced DNA damage. J Nucleic Acids.
2010:pii: 201367. 2010. View Article : Google Scholar
|
|
7
|
Mandic A, Hansson J, Linder S and Shoshan
MC: Cisplatin induces endoplasmic reticulum stress and
nucleus-independent apoptotic signaling. J Biol Chem.
278:9100–9106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu H and Baliga R: Endoplasmic reticulum
stress-associated caspase 12 mediates cisplatin-induced LLC-PK1
cell apoptosis. J Am Soc Nephrol. 16:1985–1992. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peyrou M, Hanna PE and Cribb AE:
Cisplatin, gentamicin, and p-aminophenol induce markers of
endoplasmic reticulum stress in the rat kidneys. Toxicol Sci.
99:346–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu F, Megyesi J and Price PM: Cytoplasmic
initiation of cisplatin cytotoxicity. Am J Physiol Renal Physiol.
295:F44–F52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berridge MJ: The endoplasmic reticulum: a
multifunctional signaling organelle. Cell Calcium. 32:235–249.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jørgensen MM, Bross P and Gregersen N:
Protein quality control in the endoplasmic reticulum. APMIS Suppl.
109:86–91. 2003.
|
|
13
|
Rao RV, Ellerby HM and Bredesen DE:
Coupling endoplasmic reticulum stress to the cell death program.
Cell Death Differ. 11:372–380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim R, Emi M, Tanabe K and Murakami S:
Role of the unfolded protein response in cell death. Apoptosis.
11:5–13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benbrook DM and Long A: Integration of
autophagy, proteasomal degradation, unfolded protein response and
apoptosis. Exp Oncol. 34:286–297. 2012.PubMed/NCBI
|
|
16
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582.
2007.PubMed/NCBI
|
|
17
|
Yu H, Su J, Xu Y, Kang J, Li H, Zhang L,
Yi H, Xiang X, Liu F and Sun L: p62/SQSTM1 involved in cisplatin
resistance in human ovarian cancer cells by clearing ubiquitinated
proteins. Eur J Cancer. 47:1585–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu Y, Yu H, Qin H, Kang J, et al:
Inhibition of autophagy enhances cisplatin cytotoxicity through
endoplasmic reticulum stress in human cervical cancer cells. Cancer
Lett. 314:232–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhong JT, Xu Y, Yi HW, et al: The BH3
mimetic S1 induces autophagy through ER stress and disruption of
Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett.
323:180–187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu N, Xu Y, Sun JT, et al: The BH3
mimetic S1 induces endoplasmic reticulum stress-associated
apoptosis in cisplatin-resistant human ovarian cancer cells
although it activates autophagy. Oncol Rep. 30:2677–2684. 2013.
|
|
21
|
Fenteany G, Standaert RF, Lane WS, Choi S,
Corey EJ and Schreiber SL: Inhibition of proteasome activities and
subunit-specific amino-terminal threonine modification by
lactacystin. Science. 268:726–731. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Craiu A, Gaczynska M, Akopian T, et al:
Lactacystin and clasto-lactacystin beta-lactone modify multiple
proteasome beta-subunits and inhibit intracellular protein
degradation and major histocompatibility complex class I antigen
presentation. J Biol Chem. 272:13437–13445. 1997. View Article : Google Scholar
|
|
23
|
Ostrowska H, Wojcik C, Omura S and
Worowski K: Lactacystin, a specific inhibitor of the proteasome,
inhibits human platelet lysosomal cathepsin A-like enzyme. Biochem
Biophys Res Commun. 234:729–732. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Geier E, Pfeifer G, Wilm M, et al: A giant
protease with potential to substitute for some functions of the
proteasome. Science. 283:978–981. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rebillard A, Lagadic-Gossmann D and
Dimanche-Boitrel MT: Cisplatin cytotoxicity: DNA and plasma
membrane targets. Curr Med Chem. 15:2656–2663. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ogata M, Hino S, Saito A, Morikawa K, et
al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin X, Li S, Zhao Y, Ma X, Zhang K, He X
and Wang Z: Interaction domains of p62: a bridge between p62 and
selective autophagy. DNA Cell Biol. 32:220–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanida I: Autophagosome formation and
molecular mechanism of autophagy. Antioxid Redox Signal.
14:2201–2214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olive PL and Banáth JP: Kinetics of
H2AX phosphorylation after exposure to cisplatin.
Cytometry B Clin Cytom. 76:79–90. 2009.
|