Introduction
Microglial cells, resident brain macrophages of the
central nervous system (CNS), are pivotal in the pathogenesis of
several neurodegenerative diseases (1). Microglia are activated in response to
stress, and are involved in innate and adaptive immune responses by
inducing the production of various pro-inflammatory mediators,
including nitric oxide (NO), inducible NO synthase (iNOS), tumor
necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, nuclear
factor-κB (NFκB), caspase-3 and heat shock protein (HSP) 60
(2–6), all of which contribute to
neurodegeneration (7,8). A number of microglia-targeted
pharmacotherapies, such as protein kinase C inhibitors and
microglia-inhibiting factors, have been proposed to suppress
microglial activation and promote neuronal survival in vivo
(9–11). However, the inability of these
drugs to penetrate the blood-brain barrier and the complexity of
current pharmacological agents, as well as the possible side
effects, has halted long-term clinical use in the treatment and
prevention of diseases of the CNS (12).
Dextromethorphan (DM), a derivative of morphinan, is
one of the most widely used non-opioid cough suppressants, acting
as the active ingredient in numerous antitussive formulations
(13). As an antitussive, DM is
superior to opioids used at antitussive doses in that DM lacks any
gastrointestinal side effects, such as constipation, and produces a
lower degree of depression of the CNS. DM is rapidly absorbed from
the gastrointestinal tract, where it enters the bloodstream and
crosses the blood-brain barrier (14). The anticonvulsant and
neuroprotective properties of DM have been demonstrated, and
treatment with DM has been shown to improve the cerebrovascular and
functional consequences of global cerebral ischemia (14). However, the mechanisms underlying
the neuroprotective effects of DM remain poorly understood.
Previous studies have demonstrated that naloxone,
another analogue of morphinan, protects against lipopolysaccharide
(LPS)-induced neurotoxicity in vitro and in vivo
through the inhibition of the release of proinflammatory factors
and free radicals (15–18). DM is structurally similar to
naloxone and has been shown to protect against LPS-induced dopamine
neurodegeneration in mixed neuron-glia coculture through the
inhibition of microglial overactivation, and the subsequent
reduction in the levels of proinflammatory cytokines, free radicals
and reactive oxygen species (19).
In the present study, the DeltaVision Elite microscopy imaging
system was used to analyze the effects of DM on the production of
proinflammatory mediators.
Materials and methods
Materials
The following reagents were used in this study: DM
and LPS (Sigma-Aldrich, St. Louis, MO, USA); rabbit monoclonal
antibodies against β-actin and NF-κB p65 (Abcam, Cambridge, MA,
USA); mouse monoclonal anti-HSP60 and anti-heat shock factor 1
(HSF1) antibodies (Stressgen®, San Diego, CA, USA; Enzo
Life Sciences, Inc., Farmingdale, NY, USA); rabbit polyclonal
anti-caspase-3 antibody (Cell Signaling Technology, Inc., Beverly,
MA, USA); proteinase inhibitor cocktails (Merck Chemicals,
Whitehouse Station, NJ, USA); IL-6, IL-1β and TNF-α ELISA kits
(eBioscience, San Diego, CA, USA); bicinchoninic acid (BCA) and
enhanced chemiluminescence (ECL) kits (Pierce Biotechnology, Inc.,
Rockford, IL, USA); Dulbecco’s modified Eagle’s medium (DMEM) and
fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY, USA); Griess
reagent and iNOS kits (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China); Cell Counting kit-8 (CCK-8; Beyotime Biotech,
Jiangsu, China); and fluorescein isothiocyanate (FITC)-conjugated
Affinipure goat anti-rabbit and tetramethylrhodamine
(TRITC)-conjugated Affinipure goat anti-mouse antibodies (Abcam).
All additional materials were purchased from ZSGB-BIO (Beijing,
China), unless otherwise stated.
Microglial cell culture
BV2 mouse microglial cells (Shanghai Cell Bank,
Shanghai, China) were cultured in DMEM supplemented with 10% FBS,
penicillin (100 units/ml) and streptomycin (100 g/ml). The cultures
were maintained at 37°C in a humidified incubator with 95%
O2 and 5% CO2. The cells were treated with 1
μg/ml LPS for 30 min, followed by administration of 10, 25, 50, 80
or 100 μM DM (dissolved in PBS) for 24 h.
Cell viability assay
Cell viability was measured using a CCK-8 assay. BV2
cells (5×104 cells in 100 μl/well) were seeded in
96-well plates. CCK-8 solution (10 μl) was added to each well and
the cultures were incubated at 37°C for 90 min. The absorbance at
450 nm was measured using a SmartSpec Plus Spectrophotometer
(Bio-Rad, Beijing, China). The results are plotted as the means ±
standard deviation (SD) of three separate experiments with four
determinations per experiment for each experimental condition. The
cell survival ratio was calculated, normalizing the results to the
control group (without LPS+DM).
ELISA
The levels of IL-6, IL-1β, HSP60 and TNF-α present
in the culture medium, were quantified according to the
manufacturer’s instructions for the respective ELISA kits. The
absorbance was determined at 450 nm using a Model 680 microplate
reader (Bio-Rad, Beijing, China).
Western blot analysis
Western blotting was performed according to a
standard method. Briefly, BV2 cells were washed with PBS three
times and lyzed in radioimmunoprecipitation assay buffer. The
protein concentration was determined using the BCA kit according to
the manufacturer’s instructions. Equal quantities of each protein
sample were separated by SDS-PAGE and then transferred to a
polyvinylidene difluoride membrane. The membranes were blocked with
5% milk powder and incubated with the respective primary antibodies
(anti-P65 1;1,000; anti-caspase, 1:1,000; anti-HSP60, 1:1,000;
anti-HSF-1, 1:200; and anti-β-actin, 1:5,000) in Tris-buffered
saline with Tween-20 (TBST) overnight at 4°C. Subsequent to rinsing
in milk-TBST, the blots were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:5,000). The target
proteins were detected by using the ECL detection system and X-ray
films.
Immunofluorescence
BV2 cells (5×104 cells in 100 μl/well)
were seeded on coverslips in 24-well plates. Following 24 h
incubation, the cells were treated with LPS (1 μg/ml) for 1 h and
then incubated with the indicated concentrations of DM for 24 h.
Subsequently, the cells were washed twice with PBS, fixed with 4%
paraformaldehyde in PBS for 15 min, washed three times with PBS and
then permeabilized with 0.5% Triton X-100 in PBS for 20 min. The
cells were again washed three times with PBS and blocked with 5%
bovine serum albumin for 1 h at 37°C. Following blocking, the cells
were incubated with primary antibodies (anti-P65 1;100;
anti-caspase, 1:100; anti-HSP60, 1:200; anti-HSF-1, 1:50; and
anti-iNOS, 1:100) in PBS overnight at 4°C, then rinsed with PBS,
and incubated with the FITC- and TRITC-conjugated secondary
antibodies (1:1,000) for 1 h at 37°C. The cells were then washed
with PBS, stained with DAPI and then mounted. Images were captured
at magnification, ×400 using a DeltaVision Elite microscopy imaging
system (Applied Precision; GE Healthcare, Issaquah, WA, USA).
Statistics
Statistical differences were determined using a
one-way analysis of variance test. P<0.05 was considered to
indicate a statistically significance difference. The data
represent the means ± standard error of the mean.
Results
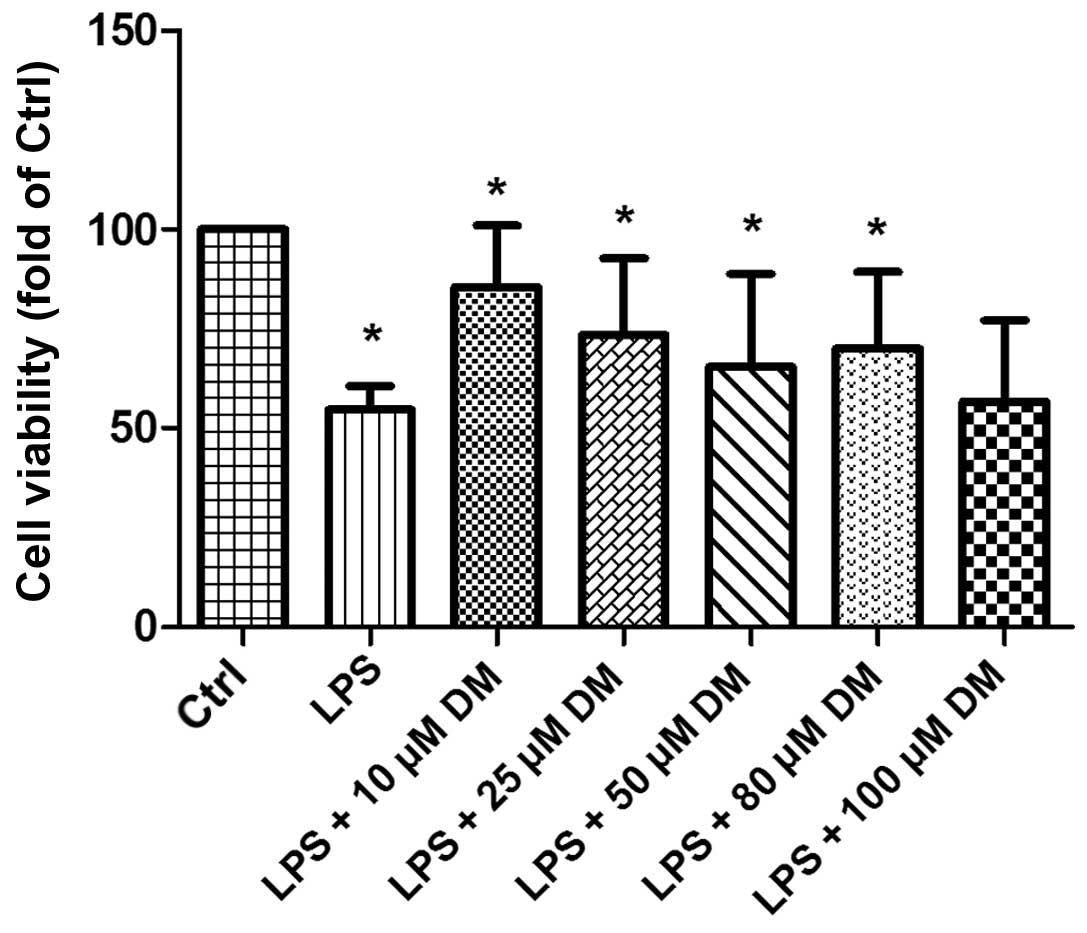
DM promotes the viability of BV2
microglia
To determine whether DM exerts an effect on the
viability of LPS-stimulated BV2 cells, a CCK-8 assay was performed.
The results demonstrated that the microglia group treated with
10–100 μM DM for 24 h exhibited significantly increased cell
viability following LPS stimulation, as compared with the group
treated with LPS alone (Fig. 1).
The cells treated with 10 μM DM exhibited the maximal viability, as
compared with the other concentrations, thus 10 μM was selected for
subsequent experiments. The results indicated that DM may exert a
positive effect on the viability of LPS-stimulated BV2
microglia.
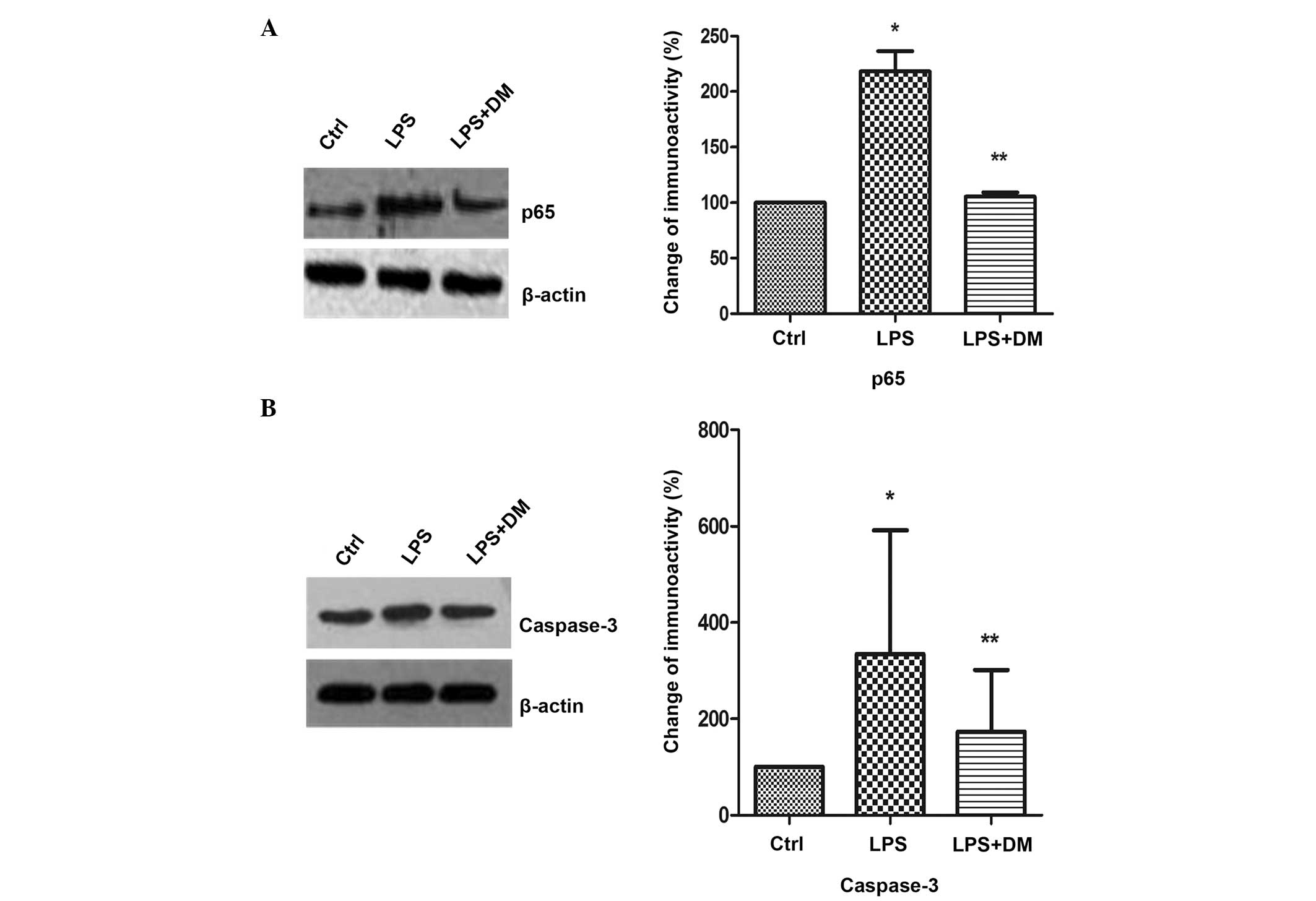
DM inhibits NFκB and caspase-3
expression
NFκB regulates the expression of numerous
proinflammatory factors and caspase-3 is an important protein in
the NFκB signaling pathway. Inhibition of caspase-3 prevents the
neuronal loss induced by activated microglia in brain diseases.
Therefore, the effects of DM on NFκB and caspase-3 expression
levels in LPS-stimulated BV2 microglia were investigated using
western blotting. The levels of the p65 subunit of NFκB were
significantly increased following LPS treatment, as compared with
the control (P<0.05), but were markedly inhibited by DM, as
compared with LPS treatment alone (P<0.05; Fig. 2A). Subsequent to LPS stimulation,
caspase-3 expression was found to be significantly suppressed
following DM treatment, as compared with LPS treatment only
(P<0.05; Fig. 2B).
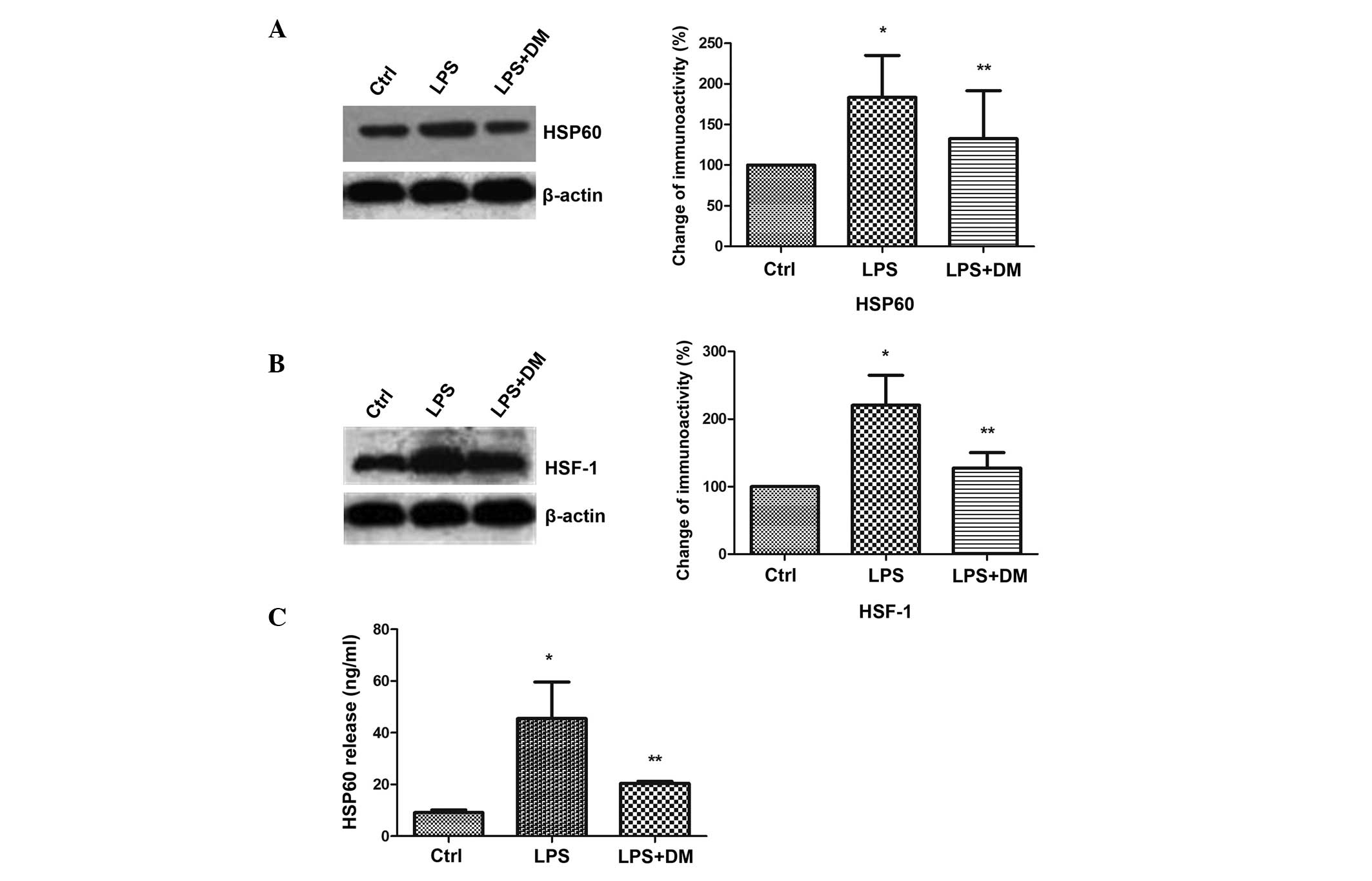
DM inhibits HSP60 protein expression and
release in LPS-stimulated BV2 microglia
The levels of HSP60 expression and release were
detected in activated BV2 cells. The western blotting results
demonstrated that LPS significantly enhanced HSP60 expression
levels, as compared with those of the control group (P<0.05),
but DM significantly inhibited this increase, as compared with LPS
treatment alone (P<0.05; Fig.
3A). HSF-1 has been shown to bind with the heat shock element
on the HSP60 promoter to regulate HSP60 gene expression (20). Therefore, the HSF-1 expression
levels were analyzed and were found to be significantly upregulated
by LPS (P<0.05), but significantly downregulated by additional
DM treatment (P<0.05). This indicated that HSP60 expression was
induced by HSF-1 (Fig. 3B). HSP60
has been reported to translocate extracellularly upon stress to
exert injury effects (21). The
ELISA results demonstrated that HSP60 was released into the culture
medium upon LPS-mediated activation of BV2, but the increased
expression levels of extracellular HSP60 was significantly
suppressed by the addition of DM (P<0.05; Fig. 3C).
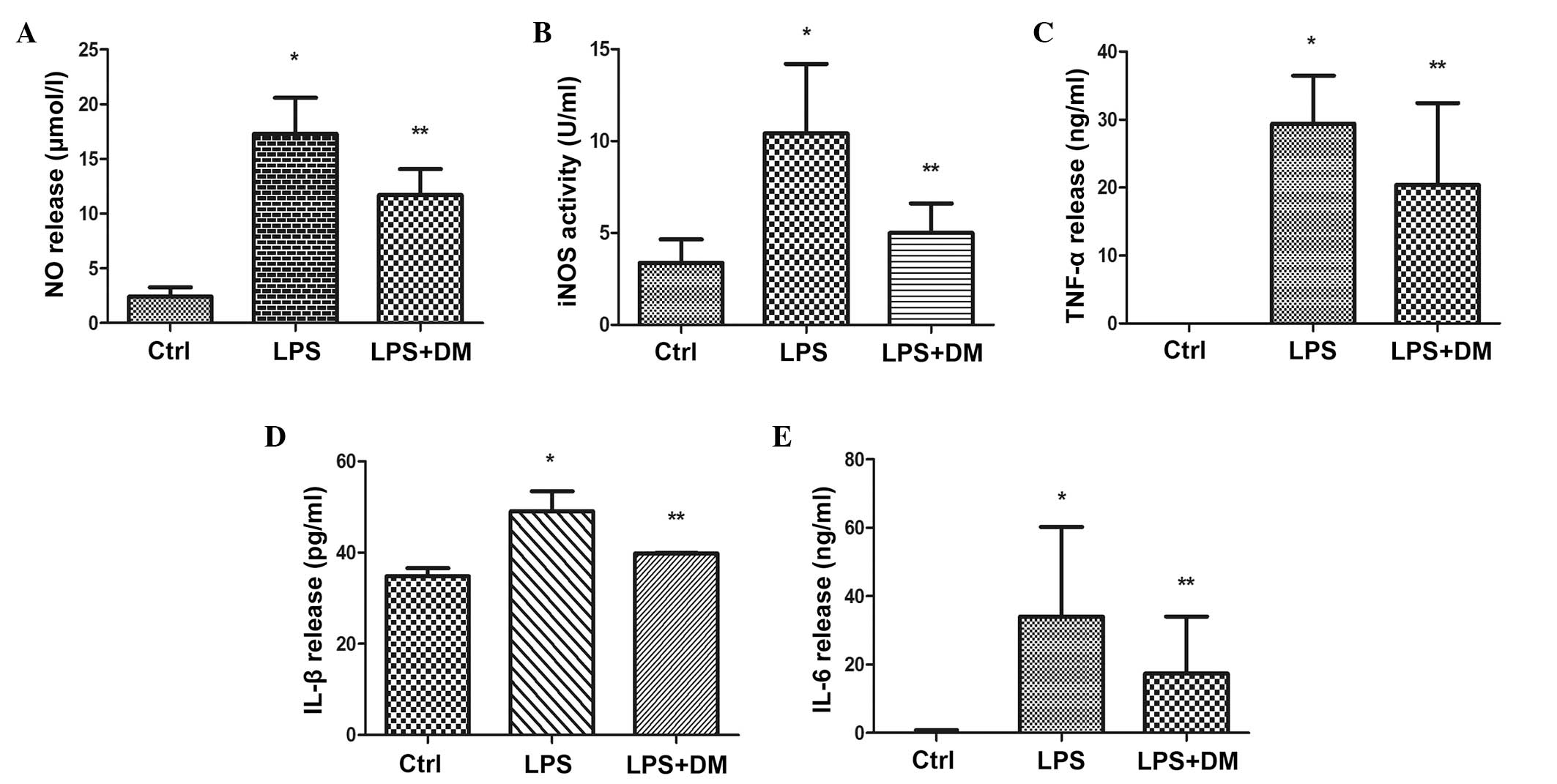
DM inhibits proinflammatory factor
production
ELISA assay was used to determine whether DM
suppresses the release of proinflammatory factors, including NO,
iNOS, TNF-α, IL-1β and IL-6 in LPS-stimulated BV2 cells. As shown
in Fig. 4, 24 h LPS treatment of
BV2 cells resulted in significant increases in the levels of the
investigated proinflammatory factors in the culture media, as
compared with the control (all P<0.05). However, additional DM
treatment significantly reduced the release of all proinflammatory
factors, as compared with LPS treatment alone (P<0.05). These
results indicated that DM effectively suppressed the production of
neurotoxic factors in overactivated microglia.
Immunofluorescence
To confirm the results from traditional molecular
techniques (western blotting and ELISA), immunofluorescence
experiments were conducted. The staining patterns of protein
localization using anti-HSP60, HSF-1, NFκB, caspase-3 and iNOS
antibodies in LPS- and LPS+DM-treated cells were markedly
different. The fluorescence intensities with those antibodies were
markedly higher in the LPS group as compared with those in the DM
group (Fig. 5A). The statistical
analysis is shown in Fig. 5B and
is consistent with the findings from the western blotting and
ELISA. From these data, DM may be considered to effectively inhibit
HSP60, HSF-1, NF-κB, caspase-3 and iNOS expression in
LPS-stimulated microglia.
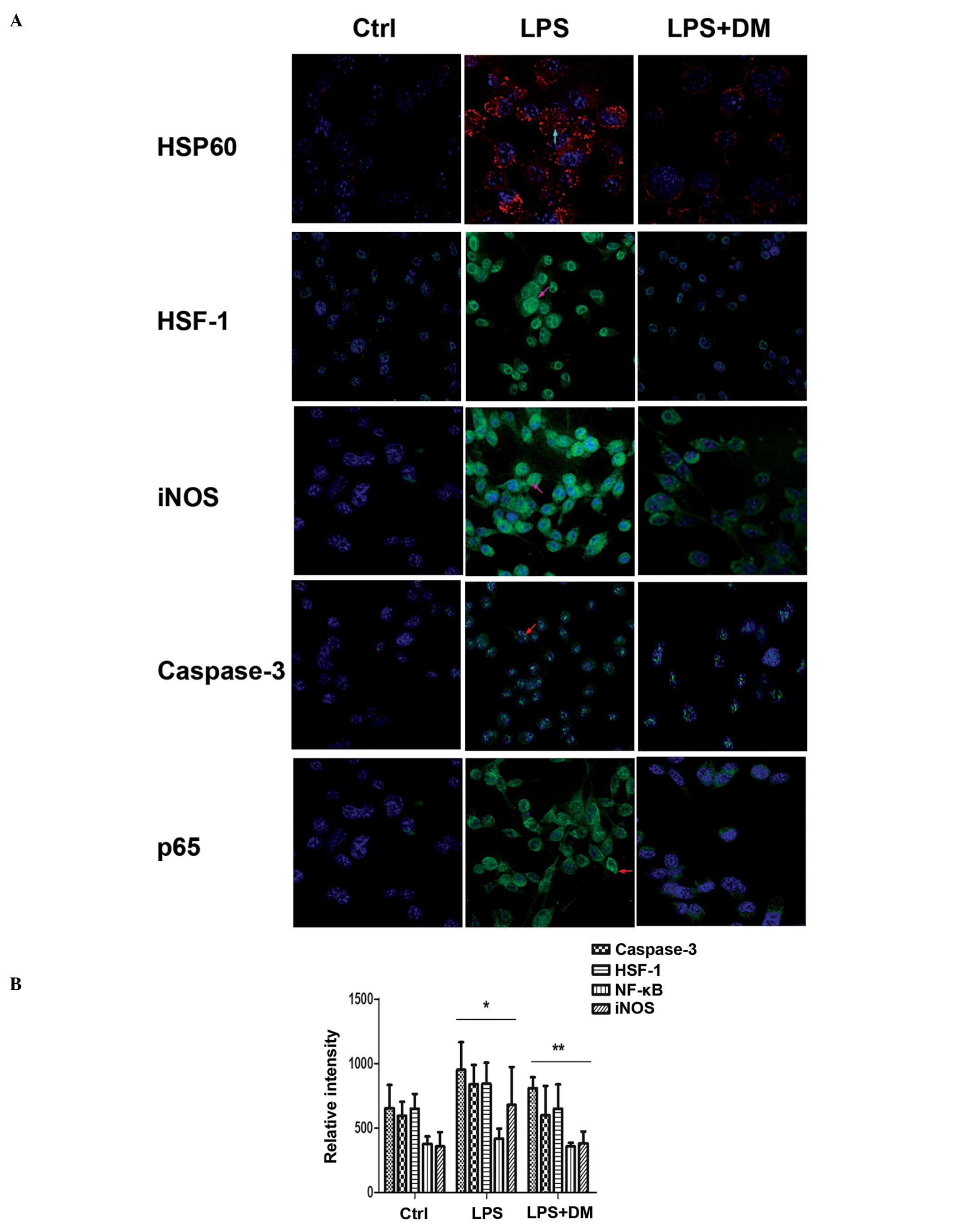 | Figure 5Dextromethorphan (DM) inhibits heat
shock protein 60 (HSP60), heat shock factor 1 (HSF-1), nuclear
factor-κB (NFκB), caspase-3 and inducible nitric oxide synthase
(iNOS) expression in LPS-stimulated BV2 microglial cells, as
detected by immunofluorescence. The cells were pretreated with
lipopolysaccharide (LPS) for 0.5 h, followed by incubation with 10
μM DM for 24 h. (A) The expression fluorescence intensity of HSP60,
HSF-1, iNOS, NFκB and caspase-3 varied between the control, LPS and
LPS + DM groups. The arrows indicate the various subcellular
localization of the respective proteins in the cells. All the
images were produced by merging the cytoplasmic and nuclear images
(digital image capture, magnification, ×400). (B) Statistical
analysis of the signal intensities of HSP60, HSF-1, NFκB, caspase-3
and iNOS. The data represent the means ± standard error of the mean
of each separate experiment performed. *P<0.05, as
compared with the Ctrl group. **P<0.05, as compared
with the LPS group. Ctrl, control. |
Discussion
In the present study, treatment with 10 μM DM was
demonstrated to effectively inhibit LPS-induced activation of
microglia. DM reduced the expression levels of NFκB, caspase-3,
HSP60, HSF-1 and iNOS in microglia, and effectively suppressed the
release into the culture medium of HSP60, NO and several
proinflammatory cytokines, including TNF-α, IL-6 and IL-1β, in
microglia stimulated by LPS. Therefore, the data suggest that DM
may be a useful therapeutic agent in the treatment of inflammatory
diseases.
The activation of microglia is important in neural
parenchymal defence against infectious diseases, as well as
inflammation, trauma, ischaemia, brain tumors and neurodegeneration
(22). The activated microglia
secrete various proinflammatory and neurotoxic factors that are
hypothesized to induce neurodegeneration (23). Therefore, the identification of
novel strategies to reduce microglial overactivation is of
therapeutic importance, and the inhibition of proinflammatory
enzymes and cytokines may be an effective therapeutic approach
against these neurodegenerative disorders. Previous studies have
investigated the neuroprotective effects of several substances,
including polysaccharides from Lycium barbarum (11), curcumin (24), fucoidan (25) and naloxone (unpublished data). The
present study suggests that DM is also a potential therapeutic
tool.
Numerous studies have reported that DM exerts
neuroprotective effects (26–28).
DM is considered to be an antagonist of the N-methyl-D-aspartate
(NMDA) receptor complex. Therefore the mechanism responsible for
the neuroprotective activity of DM has been hypothesized to occur
through antagonistic effects on the NMDA receptor (29). However, explaining the numerous
observed beneficial effects becomes difficult due to the
identification of high (nanomolar) and low (micromolar) affinity
binding sites for DM in the CNS (30,31).
In the present study, the neuroprotective effects of DM in the
inflammation-mediated neurodegenerative disorders were found to
have been most likely mediated by the inhibition of LPS-induced
microglial activation and the suppression of neurotoxic factor
production, but not by glutamate-mediated excitatory
neurotoxicity.
The activation of NFκB is considered to be pivotal
in the inflammatory response resulting from microglial activation.
NFκB is a multifunctional transcription factor and is an important
target in controlling inflammation, as the transcription of
numerous proinflammatory molecules depends on the activation of
NFκB (32,33). Therefore, several anti-inflammatory
therapies have aimed to inhibit NFκB activity in LPS models or
inflammatory diseases (34).
Caspase-3 is crucial in cell death and CNS inflammation (35). LPS-stimulated microglia have been
demonstrated to be non-toxic to neighboring neurons when
caspase-3/7 is inhibited (36).
The activation of NFκB by caspase-3 is also critical in
inflammation. Thus, in the present study, the effects of DM on NFκB
and caspase-3 expression levels were detected. The results
demonstrated that DM treatment following LPS stimulation markedly
inhibited caspase-3 and the NFκB downstream mediator p65,
suggesting that the anti-inflammatory effects of DM may be a result
of inhibition of the NFκB signaling pathway.
HSP60 is primarily considered to be a mitochondrial
protein, but may translocate to the plasma membrane, even being
released extracellularly upon stress, which is a process that has
been demonstrated to be induced by the NFκB-p65 cascade (21). Extracellular HSP60 may become toxic
by targeting self-reactive T cells in inflammatory diseases
(37). HSP60 gene expression is
regulated by the corresponding transcription factor, HSF-1, which
binds to the HSP60 gene promoter. NFκB has also been shown to
initiate transcription of the HSP60 stress gene, which elicits a
potent proinflammatory response in innate immune cells (38). TNF-α is a mediator of NFκB
signaling and induces an increase in the expression levels of
HSP60, which has been shown to be reversed by p65 inhibition
(38). Therefore, in the present
study, HSP60 expression in BV2 cells and extracellular release, as
well as TNF-α levels in the culture medium were measured, following
NFκB activation by LPS. The results reveal that DM suppressed HSP60
expression and release, and also reduced extracellular TNF-α
levels.
Microglial activation is widely known to produce
proinflammatory cytokines, NO and iNOS. This was confirmed in the
present study by measuring the levels of IL-1β, IL-6, NO and iNOS
in the culture medium of BV2 cells stimulated by LPS. The iNOS gene
is under the transcriptional control of various inflammatory
mediators, including cytokines and LPS. NO, a product of iNOS, has
been found to be important as a signaling molecule in a number of
areas of the body, as well as acting as a cytotoxic or regulatory
effector molecule of the innate immune response (39). However, in the present study,
following DM treatment, IL-1β, IL-6, NO and iNOS expression levels
were markedly inhibited.
Combined with results from previous studies, the
findings from the present study indicate that DM may exert
neuroprotective action through the inhibition of the NFκB signaling
pathway to prevent the overactivation of microglia.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 31060140 and 31260243), the
Project-sponsored by SRF for ROCS and State Education Ministry.
Additional funding was provided to Dr Yin Wang by the Program for
New Century Excellent Talents in University.
References
|
1
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: uncovering the molecular
mechanisms. Nature Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar
|
|
2
|
Hanisch UK and Kettenmann H: Microglia:
active sensor and versatile effector cells in the normal and
pathologic brain. Nat Neurosci. 10:1387–1394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gehrmann J, Matsumoto Y and Kreutzberg GW:
Microglia: intrinsic immuneffector cell of the brain. Brain Res
Rev. 20:269–287. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Innamorato NG, Lastres-Becker I and
Cuadrado A: Role of microglial redox balance in modulation of
neuroinflammation. Curr Opin Neurol. 22:308–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lynch MA: The multifaceted profile of
activated microglia. Mol Neurobiol. 40:139–156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li YH, Teng P, Wang Y, et al: Expression
and regulation of HSP60 in activated microglia cells. J Ningxia Med
Coll. 8:712–714. 2011.
|
|
7
|
Zhang D, Sun L, Zhu H, et al: Microglial
LOX-1 reacts with extracellular HSP60 to bridge neuroinflammation
and neurotoxicity. Neurochem Int. 61:1021–1035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lehnardt S, Schott E, Trimbuch T, et al: A
vicious cycle involving release of heat shock protein 60 from
injured cells and activation of toll-like receptor 4 mediates
neurodegeneration in the CNS. J Neurosci. 28:2320–2331. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thanos S, Mey J and Wild M: Treatment of
the adult retina with microglia-suppressing factors retards
axotomy-induced neuronal degradation and enhances axonal
regeneration in vivo and in vitro. J Neurosci. 13:455–466.
1993.PubMed/NCBI
|
|
10
|
Thanos S: The relationship of microglial
cells to dying neurons during natural neuronal cell death and
axotomy-induced degeneration of the rat retina. Eur J Neurosci.
3:1189–1207. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Teng P, Li YH, Cheng WJ, et al:
Neuroprotective effects of Lycium barbarum polysaccharides in
lipopolysaccharide-induced BV2 microglia cells. Mol Med Rep.
7:1977–1981. 2013.PubMed/NCBI
|
|
12
|
Rangarajan P, Eng-Ang L and Dheen ST:
Potential drugs targeting microglia: current knowledge and future
prospects. CNS Neurol Disord Drug Targets. 12:799–806. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shin EJ, Bach JH, Lee SY, Kim JM, Lee JJ,
Hong JS, et al: Neuropsychotoxic and neuroprotective potentials of
dextromethorphan and its analogs. J Pharmacol Sci. 116:137–148.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mousavi SA, Saadatnia M, Khorvash F,
Hoseini T and Sariaslani P: Evaluation of the neuroprotective
effect of dextromethorphan in the acute phase of ischaemic stroke.
Arch Med Sci. 7:465–469. 2011. View Article : Google Scholar
|
|
15
|
Liu B, Du L and Hong JS: Naloxone protects
rat dopaminergic neurons against inflammatory damage through
inhibition of microglia activation and superoxide generation. J
Pharmacol Exp Ther. 293:607–617. 2000.PubMed/NCBI
|
|
16
|
Liu Y, Qin L, Wilson BC, et al: Inhibition
by naloxone stereoisomers of beta-amyloid peptide (1–42)-induced
superoxide production in microglia and degeneration of cortical and
mesencephalic neurons. J Pharmacol Exp Ther. 302:1212–1219. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang RC, Rota C, Glover RE, Mason RP and
Hong JS: A novel effect of an opioid receptor antagonist, naloxone,
on the production of reactive oxygen species by microglia: a study
by electron paramagnetic resonance spectroscopy. Brain Res.
854:224–229. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu B, Du L, Kong LY, et al: Reduction by
naloxone of lipopolysaccharide-induced neurotoxicity in mouse
cortical neuron-glia co-cultures. Neuroscience. 97:749–756. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Y, Qin L, Li G, et al:
Dextromethorphan protects dopaminergic neurons against
inflammation-mediated degeneration through inhibition of microglial
activation. J Pharmacol Exp Ther. 305:212–218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li G, Liu Y, Tzeng NS, et al: Protective
effect of dextromethorphan against endotoxic shock in mice. Biochem
Pharmacol. 69:233–240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hansen JJ, Bross P, Westergaard M, et al:
Genomic structure of the human mitochondrial chaperonin genes:
HSP60 and HSP10 are localised head to head on chromosome 2
separated by a bidirectional promoter. Hum Genet. 112:71–77. 2003.
View Article : Google Scholar
|
|
22
|
Lin L, Kim SC, Wang Y, Gupta S, et al:
HSP60 in heart failure: abnormal distribution and role in cardiac
myocyte apoptosis. Am J Physiol Heart Circ Physiol.
293:H2238–H2247. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kreutzberg GW: Microglia: a sensor for
pathological events in the CNS. Trends Neurosci. 19:312–318. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park E, Kim DY and Chun HS: Resveratrol
inhibits lipopolysaccharide-induced phagocytotic activity in BV2
cells. J Korean Soc Appl Biol Chem. 55:803–807. 2012. View Article : Google Scholar
|
|
25
|
Karlstetter M, Lippe E, Walczak Y, et al:
Curcumin is a potent modulator of microglial gene expression and
migration. J Neuroinflammation. 8:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park HY, Han MH, Park C, et al:
Anti-inflammatory effects of fucoidan through inhibition of NF-κB,
MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia
cells. Food Chem Toxicol. 49:1745–1752. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Britton P, Lu XC, Laskosky MS and Tortella
FC: Dextromethorphan protects against cerebral injury following
transient, but not permanent, focal ischemia in rats. Life Sci.
60:1729–1740. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lesage AS, De Loore KL, Peeters L and
Leysen JE: Neuroprotective sigma ligands interfere with the
glutamate-activated NOS pathway in hippocampal cell culture.
Synapse. 20:156–164. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prince DA and Feeser HR: Dextromethorphan
protects against cerebral infarction in a rat model of
hypoxia-ischemia. Neurosci Lett. 85:291–296. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Berman FW and Murray TF: Characterization
of [3H]MK-801 binding to N-methyl-D-aspartate receptors
in cultured rat cerebellar granule neurons and involvement in
glutamate-mediated toxicity. J Biochem Toxicol. 11:217–226. 1996.
View Article : Google Scholar
|
|
31
|
Craviso GL and Musacchio JM: High-affinity
dextromethorphan binding sites in guinea pig brain. I Initial
characterization. Mol Pharmacol. 23:619–628. 1983a.
|
|
32
|
Craviso GL and Musacchio JM: High-affinity
dextromethorphan binding sites in guinea pig brain. II Competition
experiments. Mol Pharmacol. 23:629–640. 1983b.
|
|
33
|
Khasnavis S, Jana A, Roy A, et al:
Suppression of nuclear factor-κB activation and inflammation in
microglia by physically modified saline. J Biol Chem.
287:29529–29542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gu JH, Ge JB, Li M, et al: Inhibition of
NF-κB activation is associated with anti-inflammatory and
anti-apoptotic effects of Ginkgolide B in a mouse model of cerebral
ischemia/reperfusion injury. Eur J Pharm Sci. 47:652–660. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Woods DC, White YA, Dau C and Johnson AL:
TLR4 activates NF-κB in human ovarian granulosa tumor cells.
Biochem Biophys Res Commun. 409:675–680. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Soria JA, Arroyo DS, Gaviglio EA, et al:
Interleukin 4 induces the apoptosis of mouse microglial cells by a
caspase-dependent mechanism. Neurobiol Dis. 43:616–624. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Burguillos MA, Deierborg T, Kavanagh E, et
al: Caspase signalling controls microglia activation and
neurotoxicity. Nature. 472:319–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim SC, Stice JP, Chen L, et al:
Extracellular heat shock protein 60, cardiac myocytes and
apoptosis. Circ Res. 105:1186–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Chen L, Hagiwara N and Knowlton
AA: Regulation of heat shock protein 60 and 72 expression in the
failing heart. J Mol Cell Cardiol. 48:360–366. 2010. View Article : Google Scholar :
|
|
40
|
Kröncke KD, Fehsel K and Kolb-Bachofen V:
Inducible nitric oxide synthase and its product nitric oxide, a
small molecule with complex biological activities. Biol Chem Hoppe
Seyler. 376:327–343. 1995.PubMed/NCBI
|