Introduction
Vascular endothelial growth factor A (VEGF) is a
clinically significant angiogenic agent used for its potent
function in angiogenesis in the treatment of numerous ischemic
diseases, including coronary heart disease as well as chronic wound
and diabetic lower limb ischemias (1,2).
However, VEGF has restricted clinical application due to its short
half-life and the potential side effects of an overdose, which
include edema, inflammation, hemorrhage, hypotension and hemangioma
(3–5). Therefore, the identification or
design of a novel endothelial cell-specific proangiogenic factor
with fewer side effects is required.
A previous study generated a chimeric VEGF called
VEGF192 (according to the nomenclature of VEGF) from the fusion of
the VEGF183 (132–158, KDRARQENKSVRGK GKGQKRKRKKSR) peptide to the
COOH-terminus of the plasmin-resistant VEGF165 (6). The peptide corresponded to the amino
acid sequence 132–158 of VEGF183 following the cleavage signal
sequence and contained the plasmin and matrix metalloproteinase
(MMP) cleavage sites, as well as extracellular matrix (ECM) binding
sequences (7). The study reported
that the VEGF192 chimeric proteins demonstrated a stronger affinity
for the ECM in a hypoxic environment, enabling them to be cleaved
by MMPs and plasmin, and therefore retain their mitogenic activity
for endothelial cells. In addition, it was reported that VEGF192
reduced vascular permeability and had an increased half-life in
vivo compared to that of VEGF165 (6). Furthermore, Tammela et al
reported that modifying VEGF isoforms through the addition of novel
domains may result in ‘designer’ VEGFs with different bioactivities
from those of the parent molecules (8,9).
In the majority of cases mutant or cleaved VEGFs
demonstrate altered angiogenic activities and vascular patterning
than those of the parental molecules (9). Keskitalo et al (10)generated a chimeric VEGF/VEGF-C silk
domain fusion protein, which resulted in a chimeric VEGF-CAC
protein that significantly enhanced capillary formation compared to
that of the VEGF parent molecules (10). In addition, Tammela et al
(8) constructed a chimeric protein
composed of VEGF-C/VEGF heparin-binding domain fusion proteins,
which was reported to stimulate distinct structural changes to
lymphatic vessels. Zheng et al (11) designed a chimeric VEGF-E
(NZ7)/placental growth factor fusion protein that was shown to be
capable of promoting angiogenesis via VEGFR-2 without significant
enhancement of vascular permeability and inflammation. Lee et
al (12) suggested that
matrix-bound or non-tethered VEGF provided different signaling and
vascular patterning activities compared to those of the parent
molecules. The present study hypothesized that due to the enhanced
binding affinity of VEGF192 to ECM, it may induce differential
vascular patterning compared with that of VEGF165.
The aim of the present study was to explore the
biological effects of VEGF192 in vivo. Murine breast cancer
EMT-6 cells were manipulated to stably overexpress VEGF165 or
VEGF192. These cells were then inoculated intradermally into BALB/c
mice in order to monitor the formation of vascular patterning in
the skin proximal to tumors.
Materials and methods
Experimental procedures were approved by and all
animal experiments were conducted in accordance with the current
regulations and standards of the Animal Ethics Study Committee of
Xinxiang Medical University (Xinxiang, China).
Cell lines and animals
The EMT-6 murine breast cancer cell line was
purchased from the American Type Culture Collection (Manassas, VA,
USA) and grown in Dulbecco’s modified Eagle’s medium (DMEM;
HyClone™; Waltham, MA, USA) supplemented with 10% fetal bovine
serum (FBS; Gibco-BRL, Carlsbad, CA, USA), 100 U/ml penicillin and
100 μg/ml streptomycin (Sangon Biotech, Shanghai, China). Female
BALB/c mice (Experimental Animal Center of Henan Province,
Zhengzhou, China), aged six to eight weeks, were used for all
experiments. Female BALB/c mice were raised in a specific
pathogen-free environment throughout the experiment with a constant
room temperature of 20–25°C and constant humidity of 35–40% with a
12-h light-dark cycle and were given free access to food and
water.
Constructs
The full-length recombinant human VEGF165 was
provided by Dr Liu Jingjing (minigene lab of China Pharmaceutical
University, Nanjing, China). The VEGF165 mutant resistant to
plasmin proteolysis was generated as described by Lauer et
al (13). Construction of
VEGF192 was performed using polymerase chain reaction
(PCR)-mediated mutagenesis as previously described (7).
The novel VEGF192 and VEGF165 constructs, harboring
their native kozak sequence, were inserted into the mammalian
expression vector pcDNA3.1plus (Invitrogen Life Technologies,
Carlsbad, CA, USA) and further characterized using DNA sequencing
(GENEWIZ, Inc., Suzhou, China).
Stable gene transfection
Endotoxin-free plasmids pcDNA3.1plus-VEGF165,
pcDNA3.1plus-VEGF192 and the control vector pcDNA3.1plus were
extracted using the E.Z.N.A.™ endo-free plasmid midi kit (Omega
Bio-Tek, Norcross, GA, USA) according to the manufacturer’s
instructions. Stable transfections of EMT-6 cells were performed
using the Lipofectamine™ 2000 reagent according to manufacturer’s
instructions (Invitrogen Life Technologies). Parental cells were
also transfected using the pcDNA 3.1plus vector (control plasmid).
Transfected cells were incubated in complete growth medium
containing 600 μg/ml G418 antibiotics (Sangon Biotech) for 1–2
weeks.
Stably transfected individual clones were generated
using serial dilutions of G418. G418-resistant clones to be
screened in complete medium plus 600 μg/ml G418, and then stable
transfected individual clones were generated by limited dilutions
maintained in complete growth medium containing 300 μg/ml of
G418.
ELISA screening test for stable clones
overexpressing VEGF isoforms
Levels of human VEGF were quantified in conditioned
medium, prepared as follows: Cells were plated in triplicate at a
density of 2.5×105 cells per well and incubated
overnight in 24-well plates. Cells were then incubated for 48 h in
fresh serum-free medium containing 100 μg/ml heparin (Sangon
Biotech) in order to release cell-associated VEGF. Conditioned
medium was then collected and cellular debris removed using
centrifugation at 10,000 × g for 5 min at 40°C. A Human VEGF ELISA
kit (Wuhan Boster Biological Technology, Ltd., Wuhan, China) was
used to measure human VEGF protein levels in conditioned medium
according to the manufacturer’s instructions. Each sample was
measured in duplicate.
RNA extraction and reverse transcription
quantitative PCR (RT-semi-qPCR)
Confluent EMT-6 cells were cultured in DMEM-10% FBS.
RNA was isolated using TRIzol® reagent according to the
manufacturer’s instructions (Sangon Biotech). Reverse transcription
of 1 μg total RNA was performed using 200 U Superscript II RNase H
reverse transcriptase with oligo deoxy-thymine (dT; Takara Bio,
Inc., Dalian, China).
In order to further semi-quantify the human VEGF
gene in the screened clones, RT-semi-qPCR was performed using the
following primers: Human VEGF forward (NM_001171622.1),
5′-CTTGCTGCTCTACCTCCACC-3′ and VEGF reverse,
5′-GCCTCGGCTTGTCACATCT-3′; GAPDH forward, 5′-AGCGAGACCCCACTAACA-3′
and GAPDH reverse, 5′-ATGAGCCCTTCCACAATG-3′. GAPDH was used as the
control.
The complementary DNA (cDNA) samples were mixed with
the above-mentioned primers and amplified using the following
cycling profile: 95°C for 5 min, 26 cycles at 95°C for 15 sec, then
either 57°C for 30 sec (VEGF) or 55°C for 30 sec (GAPDH), followed
by 72°C for 30 sec. The PCR products were visualized using
electrophoresis with 1.5% agarose containing 0.5 μg/ml ethidium
bromide solution.
In vivo assay for tumor-adjacent
angiogenesis
A total of 24 eight-week-old female BALB/c mice were
divided randomly into three groups (n=8), anesthetized by
intraperitoneal injection of 3.6% chloral hydrate solution (0.2 ml)
(Sangon Biotech), then shaved on both flanks. Each group was
injected intradermally with a single cell suspension
(2.5×105 cells in 0.1 ml DMEM) of EMT-6 cell clones
overexpressing VEGF192 (V192-N3), EMT-6 cell clones overexpressing
VEGF165 (V165-N6) or the parental cells which carried the
pcDNA3.1plus vector (cV). Each mouse received two injections of
tumor cells on each flank. When the tumors reached similar volumes
[28±3 mm3, based on the formula: Tumor volume
(mm3)=length × width2 ×0.52], the mice were
sacrificed. The inner surface of their abdominal wall skins, which
covered the implant sites, was removed and spread onto filter
paper. Images of the newly formed blood vessels growing into the
injected tumor area were captured using a Canon SX50 HS (Zhuhai,
China) camera and were used for analysis.
In vivo tumorigenesis
A total of 24 eight-week-old female BALB/c mice were
divided randomly into three groups (n=8). Then 100 μl single cell
suspension (1×107 cells/ml in RPMI 1640 medium; HyClone,
Beijing, China), of either V192-N3, V192-N4 or V165-N6 EMT-6 cells
as well as cV was injected subcutaneously into the flank of mice in
the respective groups. N4 is a clone which has a capacity to
express more VEGF protein (~200 pg/ml) than that of clone N3
(~100pg/ml VEGF), which was used to determine the effect of the
VEGF192 expression level on the proliferation of EMT-6. When tumors
became palpable, tumor sizes were measured every two days using
calipers. Tumor volumes were calculated using the following
formula: Tumor volume (cm3)=length × width ×0.52.
Histological and immunohistochemical
analysis
Intradermal tumors (6–8 mm diameter; ~28
mm3) were fixed with 4% paraformaldehyde (Zhongshan
Bio-tech, Co., Ltd., Beijing, China), dehydrated and
paraffin-embedded, then cut into 5-μm sections. Sections were
stained with hematoxylin and eosin (H.E.) (Zhongshan Bio-tech, Co.,
Ltd.) according to the manufacturer’s instructions. H.E. sections
were examined by tumor histologist, Professor Su Ning from the
School of Medicine of Southeast University (Nanjing, China). For
immunohistochemical analysis, the sections were deparaffinized and
rehydrated into phosphate-buffered saline. Antigen retrieval was
performed using citrate buffer (pH 6.5) for 30 min at 92–98°C. The
sections were stained in order to identify endothelial cells using
the following antibodies: Rabbit anti-mouse CD31 monoclonal
antibody (Sino Biological, Inc., Beijing, China); followed by goat
anti-rabbit immunoglobulin G antibody (Zhongshan Bio-tech, Co.,
Ltd.). Antibody binding was visualized using 3,3′-diaminobenzidine
(Zhongshan Bio-tech, Co., Ltd.) and signal amplification was
achieved via the avidin-biotin complex (Zhongshan Bio-tech, Co.,
Ltd.). Staining was then quantified according to the Chalkley
method (14).
Measurement of microvessel density
Tumor vascularization was assessed using the
Chalkley method. Areas of highest vascular density (‘hot spots’)
were identified at low magnification in the whole sections
(magnification, ×100). Magnification was then increased (x200) and
a point-counting grid was applied to each hot spot, a minimum of
three hot spots were counted in each representative tumor zone. The
most representative tumor zones were studied in each case. Results
of vessel density were expressed as the mean ± standard deviation
(SD) of vessel number per mm2.
Wound healing
Confluent cell cultures were grown on 24-well plates
in DMEM-10% FBS. Wounds were generated with the tip of a
micropipette (~100 μm), and cells were maintained in serum-free
DMEM for 48 h. Following 24 h, cells were fixed with 4%
paraformaldehyde and stained with crystal violet (Sangon Biotech).
Images of three fields of vision were captured and analyzed for
each well at ×100 magnification. Each experiment was performed in
triplicate.
Statistical analysis
Data are presented as the mean ± SD. One-way
analysis of variance was performed to test for differences between
groups. Individual group comparisons were performed using the
unpaired Tukey’s post-hoc test. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
Overexpression of VEGF165 and VEGF192 in
stably transfected EMT-6 cells
EMT-6 cells were stably transfected with either cDNA
of VEGF165, VEGF192 or a control vector generating EMT-6 cell
clones V165, V192 and cV, respectively, in order to compare their
effects on the vascular pattern formation and angiogenic activity
in vivo. Nine clones stably overexpressing VEGF192 and
eleven clones stably overexpressing VEGF165 were obtained. ELISA
was used to screen the 100 μg/ml heparin-treated conditioned media
from the G418-resistant clones, which were further confirmed using
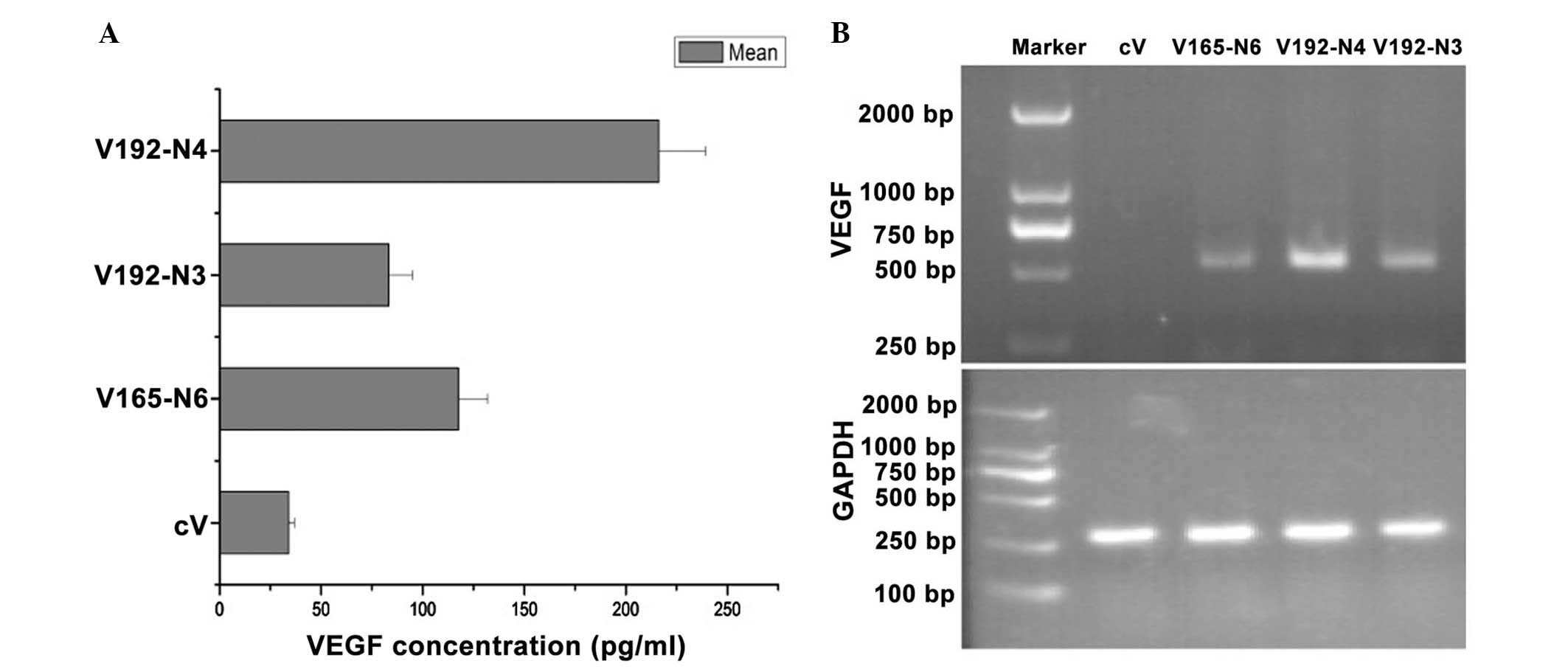
RT-semi-qPCR (Fig. 1).
Among the VEGF-expressing cells, clones V192-N3 and
V165-N6 produced comparable quantities of VEGF (Fig. 1A). These cells were then used in
the subsequent angiogenesis and tumorigenesis assays in
vivo. V192-N4, which produced a higher level of VEGF192 than
V192-N3, was also used to investigate the effects of increased
expression of VEGF192 on EMT-6 tumor growth. cV cells were used as
a control.
VEGF192 induces a distinct vascular
patterning in the skin adjacent to the tumor
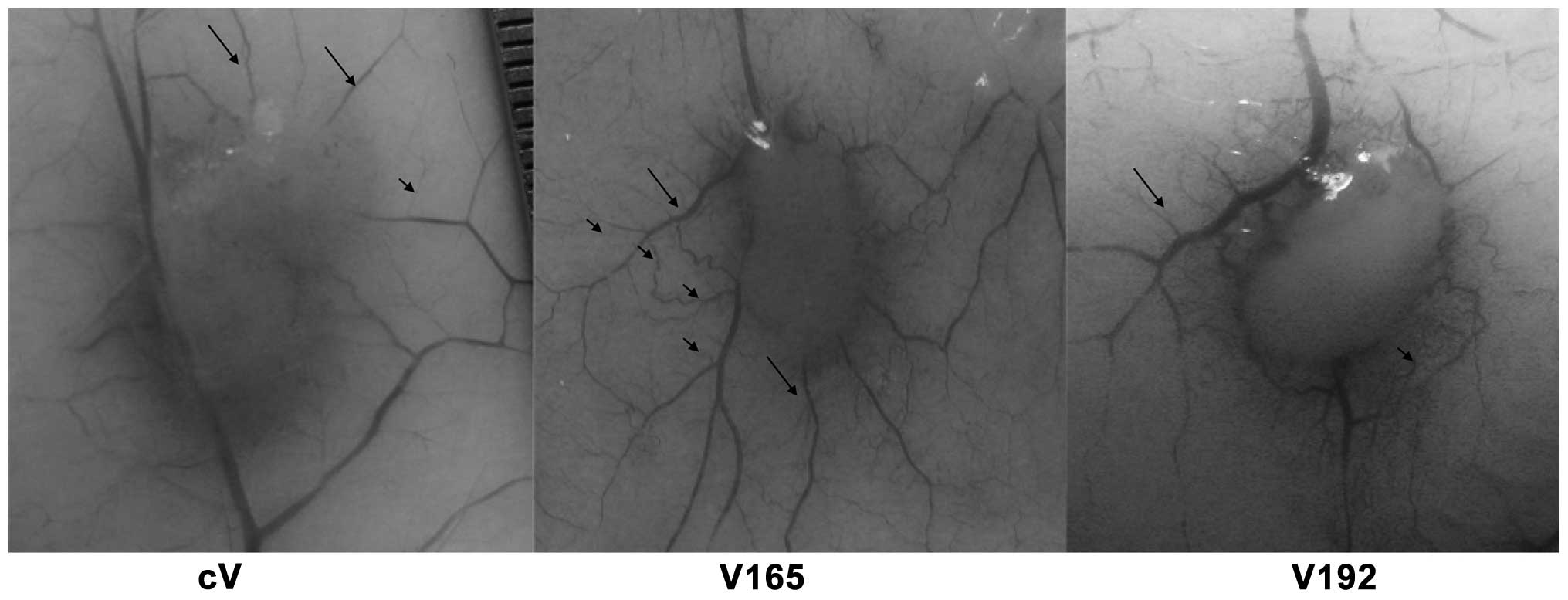
EMT-6 clone cells V192-N3, V165-N6 and cV were each
inoculated intradermally to inspect and compare the vascular
pattern characteristics and angiogenic activities of VEGF192 and
VEGF165 in vivo. When the tumors reached a certain volume
(~28±3 mm3), the mice were sacrificed and the inner
surface of their abdominal wall skins, which covered the implant
sites, was removed and spread onto filter paper. Images were then
captured for analysis. As shown in Fig. 2, the cV-forming tumors were poorly
vascularized. The average number of second and third-type vessels
surrounding cV-induced tumors were 8±2 and 16±4, respectively
(Table I). However, the tumors
formed by V165-N6 and V192-N3 induced a significantly different
vascular patterning, as shown in Fig.
2. Highly vascularized tumors were induced by V165-N6,
demonstrating a tree-like normal branching; values of second and
third-type vessels surrounding V165 were 7±2 and 45±6 mm,
respectively (Table I). Regular
vascular patterning surrounded the tumor; however, the capillaries
spread further around the tumor, with an average secondary vessel
length of 9±2 mm, compared with the corresponding cV vessels of 5±2
mm. By contrast, the vascular patterning induced by V192-N3 was
dense, irregular and disorganized; capillaries were only observed
adjacent to the tumor, with a shorter average secondary vessel
length of 3±1 mm (Table I).
 | Table IAverage number and length of the
secondary and tertiary level vessels. |
Table I
Average number and length of the
secondary and tertiary level vessels.
| Variable | cV | V165 | V192 |
|---|
| Number of secondary
vessels (n) | 8±2 | 7±2 | 16±3 |
| Number of tertiary
level vessels (n) | 16±4 | 45±6 | 37±6 |
| Length of secondary
vessels (mm) | 5±2 | 9±2 | 3±1 |
| Length of tertiary
level vessels (mm) | 3±1 | 2±0.5 | 1±0.5 |
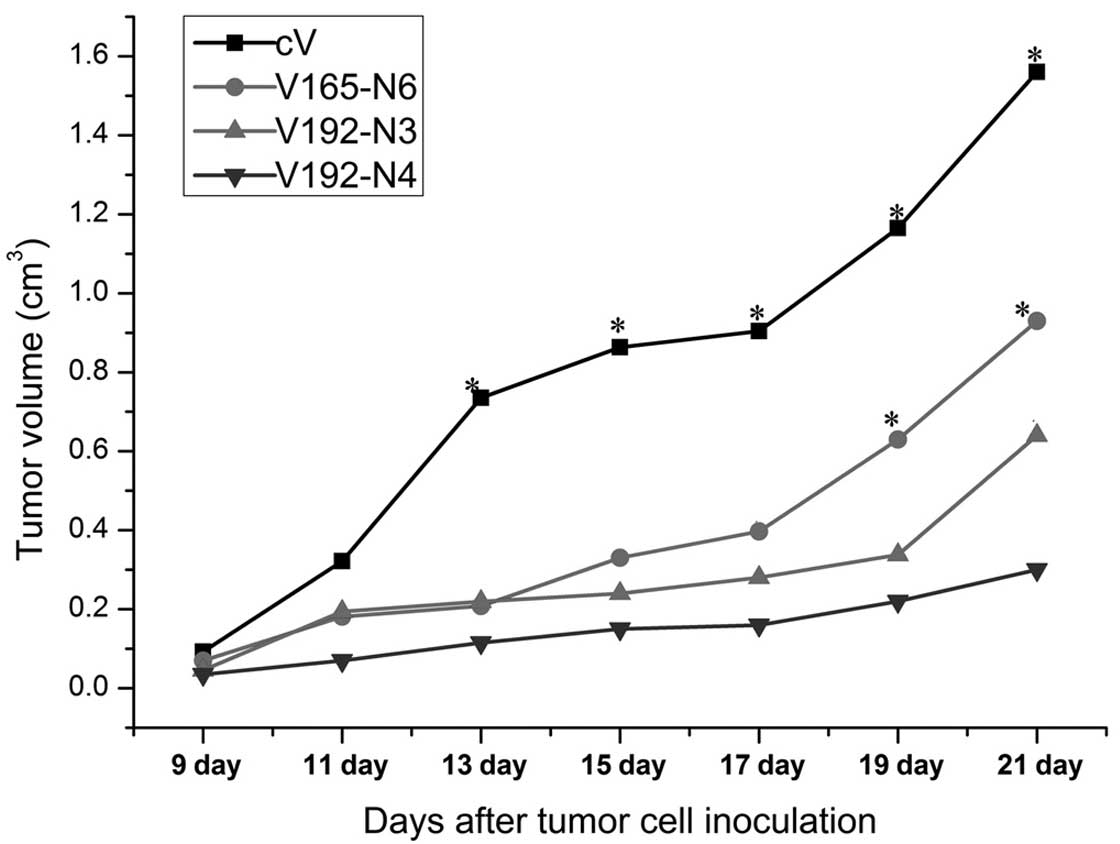
VEGF192 overexpression slows tumor growth
in vivo
The effects of VEGF192 or VEGF165 overexpression on
tumor growth were investigated using subcutaneous transplantations
of V192-N3, V192-N4, V165-N6 or cV EMT-6 clones into mice. As shown
in Fig. 3, each VEGF transfectant
demonstrated significant inhibition of tumor growth rates compared
to those of the cV control group from day 13 onwards. Furthermore,
from day 19 following inoculation the V165 tumor-forming group
demonstrated significantly higher tumor growth rates compared to
those of the two V192 groups. In addition, the V192-N4 group
appeared to have a slower growth rate compared with that of the
V192-N3 group; however, the difference was not statistically
significant. Overall the results indicated that the overexpression
of VEGF192 correlated with the strong inhibition of tumor
growth.
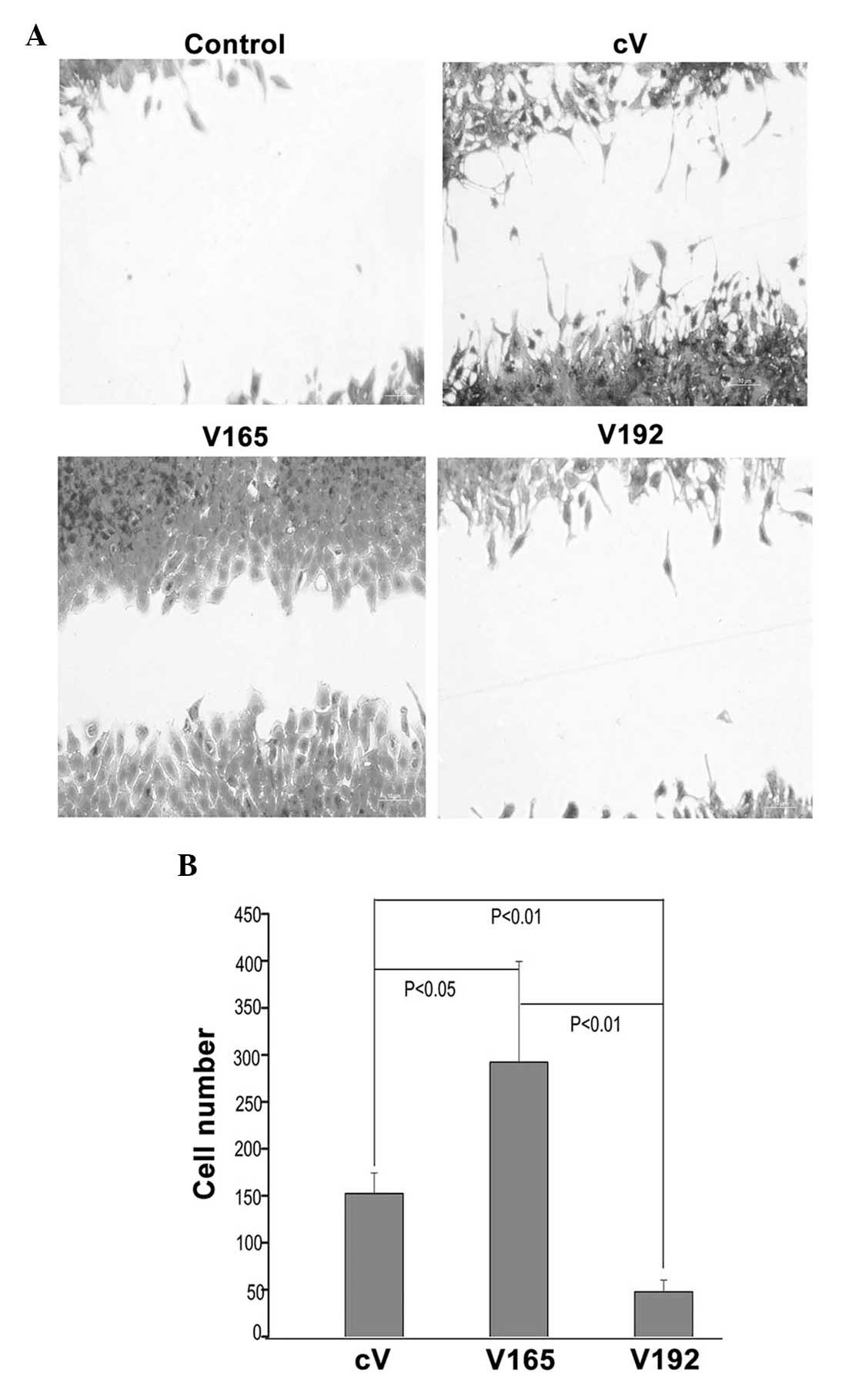
VEGF192 overexpression significantly
inhibits migration
Wound healing assays were used investigate the
effects of VEGF192 or VEGF165 overexpression on the migratory
capacity of EMT 6 cells. As shown in Fig. 4A, VEGF192-overexpressing cells
showed reduced migration compared to that of the
VEGF165-overexpressing and control cells. The number of
VEGF192-overexpressing EMT-6 cells migrating into the wound area
was significantly decreased compared to that of
VEGF165-overexpressing cells and control cells (P<0.05)
(Fig. 4B). This therefore
indicated that the overexpression of VEGF192 inhibited EMT-6 cell
migration.
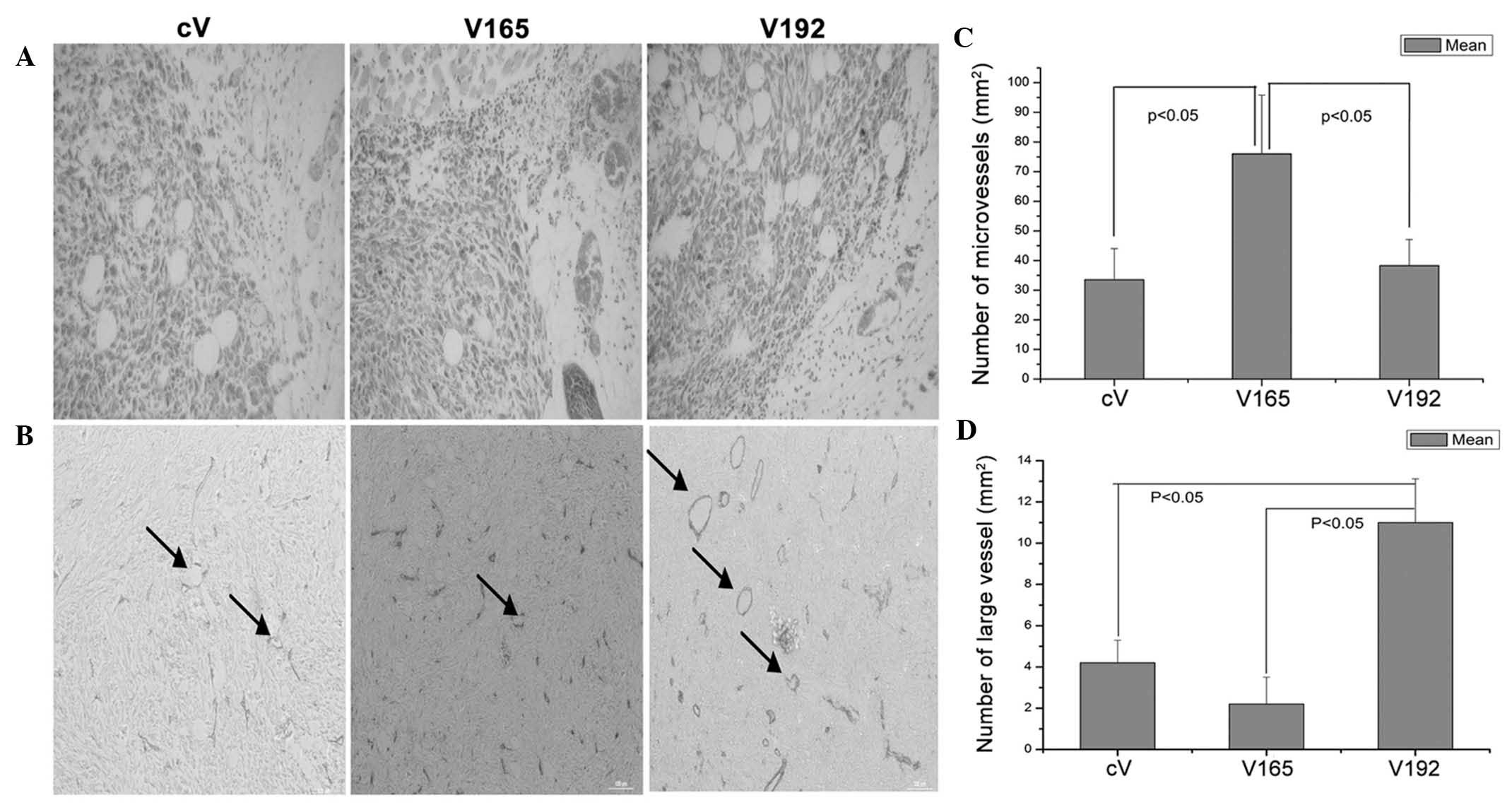
Immunohistochemical analysis of
microvessel density
As shown in Fig.
5A, there were no obvious areas of necrosis observed in the
histological sections from the cV and V165 cell tumors; however,
there was marked necrosis in sections from the V192 cell tumor. In
addition, areas of hemorrhage were observed in the V165 and V192
cell tumors.
VEGF165 and VEGF192-transfected clones producing
identical quantities of VEGF (V165-N6 and V193-N3) were selected
for immunohistochemical analysis of angiogenesis in vivo.
Mouse anti-CD31 monoclonal antibodies were used to assess the
microvessel density of allograft tumors. The results showed that
VEGF165-overexpressing tumors were significantly more vascularized
compared with the V192-overexpressing and cV tumors (Fig. 5B and C). Furthermore, no
significant difference was observed between the vascular densities
of the VEGF192-overexpressing tumors and those of the cV tumors;
however, the former exhibited a significantly enlarged vascular
caliber, with an increased total number of vessels >10 μm in
diameter, compared with that of the cV and V165-overexpressing
tumors (P<0.05) (Fig. 5B and
D).
Discussion
The aim of the present study was to investigate the
effect of VEGF192 on angiogenesis and vascular patterning through
generating EMT-6 murine breast cancer clones which stably
overexpressed VEGF192 and VEGF165. Grunstein et al (15)utilized tumor cells which
overexpressed an individual isoform of VEGF-A and expressed low
levels of endogenous VEGF in order to investigate their effects on
tumor angiogenesis. The study reported marked differences in tumor
vascularization; however, endogenous VEGF was not eliminated from
the invading stroma (15). The
results of the present study demonstrated that the overexpression
of VEGF192 in EMT-6 tumor cells resulted in increased microvessel
density, distinct vascular patterning and a significantly reduced
tumor growth rate compared with those of VEGF165-overexpressing
tumors.
Previous studies have reported that the vasculature
in the adjacent areas to VEGF-overexpressing tumors may also
undergo structural alterations (12,16).
The VEGF gene produces several isoforms (primarily VEGF121, VEGF165
and VEGF189) through the process of gene splicing. VEGF isoforms
commonly differ due to the presence or absence of exons 6a and 7;
these two exons were reported to determine the diffusible or
sequestered (ECM-bound) states of VEGF proteins (17). Numerous studies have confirmed that
VEGF isoforms have different roles in vasculogenesis; these studies
included evidence from specific VEGF isoform knock-out experiments
in mice (18–20). In addition, it was reported that
normal VEGF concentration gradients were necessary for regular
vascular pattern formation (21);
for example, VEGF120/120 mouse embryos, engineered to express a
unique isoform of VEGF-A that lacks ECM (heparin)-binding domains,
showed significantly decreased capillary branching (18). Conversely, VEGF189 with an
ECM-binding domain was reported to provide stimulatory cues that
guided endothelial cell-sprouting for the initiation of vascular
branch formation (22). The
present study hypothesized that VEGF192 may have a higher affinity
for ECM compared with that of VEGF165 and therefore may be cleaved
by plasmin to release active VEGF isoforms (7). However, it was found that the formed
VEGF concentration gradient of VEGF192 was as effective as that of
VEGF165. Conversely, VEGF165 was found to have an intermediate
affinity for the ECM, with half the VEGF molecules in soluble form,
and could therefore form a VEGF concentration gradient. This may
therefore explain the difference between the highly-branched, dense
and disorganized vascular patterning in tumors of V192-transfected
cells and the normal branched, tree-like vascular patterning due to
V165-overexpression.
The results of the present study demonstrated that
the growth rates of V192-overexpressing tumors were slower than
those of V165-overexpressing tumors. A possible explanation for
this may be that due to the strong affinity to the ECM V192 was not
able to be completely cleaved by MMP or plasmin to form an
effective VEGF concentration gradient in vivo, which
resulted in a lower vascular density (Fig. 5B). Conversely, the growth rate of
specific VEGF isoform-overexpressing tumors in vivo was
previously found to be correlated with angiogenic activity as well
as the receptor expression profile of the tumor cells (23). Three receptors, namely VEGF
receptor (VEGFR) 1, VEGFR2 and neuropilin 1 (NRP1) have been
reported to bind VEGF. The expression of these receptors was
previously thought to be limited to endothelial cells; however,
they have also been reported to be expressed in certain cancer
cells (24,25). Furthermore, VEGF has been suggested
to be involved in the initiation of cancer cell migration and
proliferation (23). Therefore, it
may be inferred that the decreased V192 growth rate in the present
study was possibly due to the ineffective binding of V192 to NRP1
on EMT-6 cells (NRP1 and VEGFR1 expression were detected in EMT-6
cells using RT-semi-qPCR, data not shown). Hervé et al
(26) demonstrated that the
interaction of VEGF189 with NRP1 stimulated the proliferation of
MDA-MB-231 cells.
The peptide VEGF183 (132–158) contains MMP and
plasmin cleavage sites, in addition to the ECM-binding sequence
(core sequence, ‘KRKRKKSR’). Previous studies have confirmed the
interference of the ‘KRKRKKSR’ sequence with interactions of other
heparin-binding growth factors and their receptors (27). In VEGF189, exon 8 is located at the
C-terminus; therefore, the ‘KRKRKKSR’ sequence is not exposed to
the C-terminus, preventing direct interference with the VEGF
receptors. It is possible that this may be the mechanism by which
VEGF189 stimulated the dose-dependent migration of breast cancer
cells and proliferation of endothelial cells (26,28).
However, VEGF192 was not protected by VEGF exon 8, which may have
resulted in the interference of the ‘KRKRKKSR’ sequence with the
binding of V192 with the corresponding VEGF receptors. This may
therefore explain how VEGF192 overexpression could decrease the
growth rate and inhibit the migration of EMT-6 cells.
The microvessel density in tumors of cells
transfected with V192 was found to be decreased compared to that of
tumors of cells transfected with V165; however, the microvessel
caliber in tumors of cells transfected with V192 was significantly
increased compared with that of tumors of cells transfected with
V165 or cV. This phenomenon could not be explained and further
research is required for this to be elucidated. Previous studies
have demonstrated that the anchorage of different VEGFs to the ECM
may convey differential signaling responses to endothelial cells
(29). This therefore indicated
that the induced dilation of intratumoral vessels may have resulted
from the higher affinity of VEGF189 for the ECM (25).
In conclusion, the results of the present study
suggested that one of the functions of the exon 8 in VEGF-A was to
prevent the ‘KRKRKKSR’ sequence from interfering with the
interactions between heparin-binding growth factors and their
respective receptors. It has therefore been suggested that the
fusion of exon 8 to the C-terminus of VEGF192 may be beneficial for
its angiogenic activities. In addition, the present study confirmed
certain suspected characteristics of VEGF192, including the
stronger affinity for the ECM, a longer half-life and release via
plasmin-cleavage. However, it is now suggested that VEGF192 may be
more beneficial for use in the field of tissue engineering with
heparinized materials. Of note, VEGF192 provided a novel insight
into VEGF design and indirect evidence for the function of exon 8
of VEGF-A.
Acknowledgements
This study was supported by grants from the Basic
Science and Frontier Technology Planning Project of Henan Province,
Educational Commission of Henan Province and Scientific Research
Fund of Xinxiang Medical University (nos. 142300410027, 12B320021
and 2013ZD110, respectively).
References
|
1
|
Hoeben A, Landuyt B, Highley MS, Wildiers
H, Van Oosterom AT and De Bruijn EA: Vascular endothelial growth
factor and angiogenesis. Pharmacol Rev. 56:549–580. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee RJ, Springer ML, Blanco-Bose WE, Shaw
R, Ursell PC and Blau HM: VEGF gene delivery to myocardium:
deleterious effects of unregulated expression. Circulation.
102:898–901. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hariawala MD, Horowitz JR, Esakof D, et
al: VEGF improves myocardial blood flow but produces EDRF-mediated
hypotension in porcine hearts. J Surg Res. 63:77–82. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Simons M: Angiogenesis: where do we stand
now? Circulation. 111:1556–1566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huiyong Z, Yong L, Yunxiao S, Wuling Z,
Jingjing L and Taiming L: Generation of a chimeric
plasmin-resistant VEGF165/VEGF183 (132–158) protein and its
comparative activity. Protein Pept Lett. 20:947–954. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Robinson CJ and Stringer SE: The splice
variants of vascular endothelial growth factor (VEGF) and their
receptors. J Cell Sci. 114(Pt 5): 853–865. 2001.PubMed/NCBI
|
|
8
|
Tammela T, He Y, Lyytikkä J, et al:
Distinct architecture of lymphatic vessels induced by chimeric
vascular endothelial growth factor-C/vascular endothelial growth
factor heparin-binding domain fusion proteins. Circ Res.
100:1468–1475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simons M: Silky, sticky chimeras-designer
VEGFs display their wares. Circ Res. 100:1402–1404. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Keskitalo S, Tammela T, Lyytikkä J, et al:
Enhanced capillary formation stimulated by a chimeric vascular
endothelial growth factor/vascular endothelial growth factor-C silk
domain fusion protein. Circ Res. 100:1460–1467. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng Y, Watanabe M, Kuraishi T, Hattori
S, Kai C and Shibuya M: Chimeric VEGF-ENZ7/PlGF specifically
binding to VEGFR-2 accelerates skin wound healing via enhancement
of neovascularization. Arterioscler Thromb Vasc Biol. 27:503–511.
2007. View Article : Google Scholar
|
|
12
|
Lee S, Jilani SM, Nikolova GV, Carpizo D
and Iruela-Arispe ML: Processing of VEGF-A by matrix
metalloproteinases regulates bioavailability and vascular
patterning in tumors. J Cell Biol. 169:681–691. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lauer G, Sollberg S, Cole M, Krieg T and
Eming SA: Generation of a novel proteolysis resistant vascular
endothelial growth factor165 variant by a site-directed mutation at
the plasmin sensitive cleavage site. FEBS Lett. 531:309–313. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suhonen KA, Anttila MA, Sillanpää SM,
Hämäläinen KM, Saarikoski SV, Juhola M and Kosma VM: Quantification
of angiogenesis by the Chalkley method and its prognostic
significance in epithelial ovarian cancer. Eur J Cancer.
43:1300–1307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grunstein J, Masbad JJ, Hickey R, Giordano
F and Johnson RS: Isoforms of vascular endothelial growth factor
act in a coordinate fashion to recruit and expand tumor
vasculature. Mol Cell Biol. 20:7282–7291. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mineur P, Colige AC, Deroanne CF, et al:
Newly identified biologically active and proteolysis-resistant
VEGF-A isoform VEGF111 is induced by genotoxic agents. J Cell Biol.
179:1261–1273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ferrara N: Vascular endothelial growth
factor. Arterioscler Thromb Vasc Biol. 29:789–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carmeliet P, Ng YS, Nuyens D, et al:
Impaired myocardial angiogenesis and ischemic cardiomyopathy in
mice lacking the vascular endothelial growth factor isoforms
VEGF164 and VEGF188. Nat Med. 5:495–502. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ruhrberg C, Gerhardt H, Golding M, et al:
Spatially restricted patterning cues provided by heparin-binding
VEGF-A control blood vessel branching morphogenesis. Genes Dev.
16:2684–2698. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maes C, Stockmans I, Moermans K, et al:
Soluble VEGF isoforms are essential for establishing epiphyseal
vascularization and regulating chondrocyte development and
survival. J Clin Invest. 113:188–199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferrara N: Binding to the extracellular
matrix and proteolytic processing: two key mechanisms regulating
vascular endothelial growth factor action. Mol Biol Cell.
21:687–690. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stalmans I, Ng YS, Rohan R, et al:
Arteriolar and venular patterning in retinas of mice selectively
expressing VEGF isoforms. J Clin Invest. 109:327–336. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schoeffner DJ, Matheny SL, Akahane T, et
al: VEGF contributes to mammary tumor growth in transgenic mice
through paracrine and autocrine mechanisms. Lab Invest. 85:608–623.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soker S, Takashima S, Miao HQ, Neufeld G
and Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor
cells as an isoform-specific receptor for vascular endothelial
growth factor. Cell. 92:735–745. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mercurio AM, Lipscomb EA and Bachelder RE:
Non-angiogenic functions of VEGF in breast cancer. J Mammary Gland
Biol Neoplasia. 10:283–290. 2005. View Article : Google Scholar
|
|
26
|
Hervé MA, Buteau-Lozano H, Vassy R, et al:
Overexpression of vascular endothelial growth factor 189 in breast
cancer cells leads to delayed tumor uptake with dilated
intratumoral vessels. Am J Pathol. 172:167–178. 2008. View Article : Google Scholar :
|
|
27
|
Jia H, Jezequel S, Löhr M, et al: Peptides
encoded by exon 6 of VEGF inhibit endothelial cell biological
responses and angiogenesis induced by VEGF. Biochem Biophys Res
Commun. 283:164–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hervé MA, Buteau-Lozano H, Mourah S, Calvo
F and Perrot-Applanat M: VEGF189 stimulates endothelial cells
proliferation and migration in vitro and up-regulates the
expression of Flk-1/KDR mRNA. Exp Cell Res. 309:24–31. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen TT, Luque A, Lee S, Anderson SM,
Segura T and Iruela-Arispe ML: Anchorage of VEGF to the
extracellular matrix conveys differential signaling responses to
endothelial cells. J Cell Biol. 188:595–609. 2010. View Article : Google Scholar : PubMed/NCBI
|