Introduction
The inflammatory reaction induced by
ischemia/reperfusion is one of the most significant effects to
occur in myocardial ischemia-reperfusion injury (1). During the process of inflammation,
various cytokines are released, including tumor necrosis factor-α
(TNF-α), interleukin-6 (IL-6) and IL-8 (2). TNF-α can trigger the inflammatory
reaction caused by myocardial ischemia-reperfusion (MI/R). Vascular
endothelial cell injury, and inflammatory cells, such as
neutrophils activated by cytokines, and adhesion molecules are also
involved in the inflammatory response. TNF-α activity and the
amount of neutrophil infiltration can be considered as indicators
of the inflammatory reaction. It has been previously demonstrated
that Toll-like receptor 4 (TLR4)/NF-κB signaling is activated
during MI/R, which in turn results in TNF-α induction, thus
initiating an inflammatory reaction during MI/R (3,4).
The TLRs are a family of molecules that function in
the process of innate immunity (5,6).
Activation of the corresponding TLRs triggers an inflammatory
response, alerting the host to the presence of microbial invasion
and initiating an immune response. Previous studies have
demonstrated that some TLR family members can signal the presence
of tissue damage to the host due to activation by endogenous
molecules released from damaged tissues (7,8).
Heparan sulfate, hyaluronic acid and other endogenous molecules
have been shown to initiate inflammatory pathways through TLR4
(9–11). It has been suggested that
TLR4-mediated signaling induces myocardial dysfunction during MI/R
(12).
Resveratrol (Res) is a polyphenolic compound, which
is predominantly naturally occurring in red grapes and wine. In a
previous epidemiological study (13) investigating the association between
eating habits and coronary heart disease, a phenomenon was proposed
that in all developed countries, the French consume the most
quantity of wine on average, but have the lowest morbidity of
coronary heart disease. This phenomenon is termed the ‘French
paradox’, and has been attributed to the benefits acquired from Res
present in wine. Res has extensive pharmacological effects,
including anticancer properties (14–17),
improving ifosfamide-induced Fanconi syndrome in rats (18), treating diabetic nephropathy
(19) and neuronal protection
(20,21). Though previous studies have
suggested that Res has an anti-inflammatory effect, the role of Res
in mediating inflammation remains to be understood (22). The present study aims to
investigate the role of the TLR4/NF-κB signaling pathway in the
anti-inflammatory effect of Res in a rat model of MI/R.
Materials and methods
Reagents
Res and L-NAME were purchased from Sigma-Aldrich
(St. Louis, MO, USA). The myeloperoxidase (MPO) assay kit was
purchased from Jiancheng Bioengineering Institute (Nanjing, China).
The TNF-α ELISA kit was purchased from R&D Systems
(Minneapolis, MN, USA). The bicinchoninic acid (BCA) protein
quantification kit was purchased from Bio-Rad (Hercules, CA, USA).
NF-κB p65 mouse monoclonal antibody (L8F6) was purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The anti-TLR4
antibody (ab13556) and goat anti-rabbit immunoglobulin (IgG) were
purchased from Abcam (Cambridge, MA, USA). EnVison™ was purchased
from Gene Tech (Shanghai, China) and the cell death detection kit
was purchased from Roche Diagnostics (Mannheim, Germany).
Animals
Forty adult male Sprague Dawley rats (250–300 g)
were purchased from the Center of Experimental Animals in the
Harbin Medical University (Harbin, Heilongjiang, China). All
animals used in this study were cared for in accordance with the
Guide for the Care and Use of Laboratory Animals published by the
United States National Institute of Health (NIH publication no.
85-23, revised 1996). All procedures were approved by the Committee
of Experimental Animals of Harbin Medical University.
MI/R model and experimental protocol
Male Sprague Dawley rats (250–300 g) were
anesthetized intraperitoneally with sodium pentobarbital
(Sigma-Aldrich; 40 mg/kg). Myocardial ischemia was produced by
exteriorizing the heart with a left thoracic incision followed by a
slipknot (5-0 silk) around the left anterior descending coronary
artery (LAD). After 30 min of ischemia, the slipknot was released
and the animal received 120 min of reperfusion.
Rats were randomly assigned to four experimental
groups, with 10 rats in each group: i) sham group: silk was drilled
underneath the LAD but the LAD was not ligated; ii) MI/R group: LAD
was ligated for 30 min followed by 120 min reperfusion together
with receiving vehicle (0.9% NaCl i.v.); iii) MI/R + Res group: Res
[100 μmol/l, intravenous (i.v)] was administered 5 min prior to
reperfusion; iv) MI/R + Res + L-NAME group: L-NAME (1 mmol/l,
i.v.), a nitric oxide (NO) synthase inhibitor, was administered 20
min prior to reperfusion. Res (100 μmol/l, i.v) was administered
fifteen minutes following L-NAME treatment.
Assay of myocardial infarct area
Following reperfusion, the myocardial infarct size
was determined by a double-staining technique and a digital imaging
system (infarct area/area at risk ×100%) (23). Following reperfusion, the coronary
blood flow was again blocked and Evans blue (2%, 4 ml) was injected
and rapidly distributed by the right ventricle into the body. The
heart was then quickly removed and cryopreserved at −20°C. The
heart was cut into 1 mm slices, placed in 1%
2,3,5-triphenyltetrazolium chloride (TTC) solution, incubated for
15 min, and then placed in 4% formaldehyde solution overnight. The
Evans blue (blue staining, non-ischemic area), TTC (red staining,
ischemic area) and non-TTC-stained areas (white, infarct area) were
analyzed using a computerized digital imaging system. The
myocardial infarct area (infarct area/area at risk%, INF/AAR%) was
subsequently calculated.
Determination of myocardial
apoptosis
Following reperfusion, myocardial apoptosis was
analyzed by terminal deoxynucleotidyl transferase dUTP nick end
labeling assay (TUNEL) using an in situ cell death detection
kit. A double-staining technique was used. TUNEL staining for
apoptotic cell nuclei and DAPI staining for all myocardial cell
nuclei were performed as described previously (24). The index of apoptosis was expressed
as the number of positively stained apoptotic myocytes/the total
number of myocytes counted ×100%.
Immunohistochemical analysis of
myocardial TLR4 expression
Prior to immunohistochemical examination, 3 μm
slices from pretreated myocardium tissue were placed in a bathing
solution of 3% H2O2 and 60% methanol in
phosphate-buffered saline (PBS) for 30 min and then treated with
0.01 mol/l sodium citrate buffer at 95°C in a microwave oven for 13
min, for antigen retrieval. The specimens were then treated with 5%
normal goat serum and 5% bovine serum albumin (BSA) in PBS. Prior
to each step, the sections were rinsed three times in PBS.
Incubation with primary TLR4 antibody was performed in a PBS-based
solution of 1% BSA for 12 h at 4°C at the recommended dilutions.
After rinsing with PBS, sections were incubated with the
corresponding secondary biotinylated goat anti-rabbit EnVison™
antibodies for 1 h at room temperature. A streptavidin/horseradish
peroxidase complex was then applied as a detection system (1:100
dilution) for 1 h. Finally, staining was developed with
3,3-diaminobenzidine tetra-hydrochloride in 0.05 mol/l Tris-HCl
buffer and 0.1% H2O2. Negative control
sections were incubated without the primary antibody. All the
samples in this study were analyzed by Image Pro Plus software
(Media Cybernetics, Inc., Rockville, MD, USA).
Western blotting analysis of myocardial
NF-κB expression
Proteins were extracted from heart tissue and the
immunoblots were probed with anti-NF-κB antibodies overnight at 4°C
followed by incubation with the corresponding secondary antibodies
at room temperature for 1 h. The blots were visualized with
ECL-Plus reagent.
Determination of MPO level
Following reperfusion, the myocardial tissue was
placed at −70°C for preservation. The MPO test kit was used to
detect the level of MPO in the myocardial tissue according to the
manufacturer’s instructions.
Detection of TNF-α level
Following reperfusion, the levels of TNF-α in the
myocardial tissue homogenate and serum were detected according to
the manufacturer’s instructions. A BCA kit was used for
quantification of the protein samples.
Statistical analysis
The data are presented as the mean ± SD. The
significance of differences among groups was evaluated by a
Student’s t-test for unpaired data or Dunnett’s t-test for multiple
comparisons preceded by one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
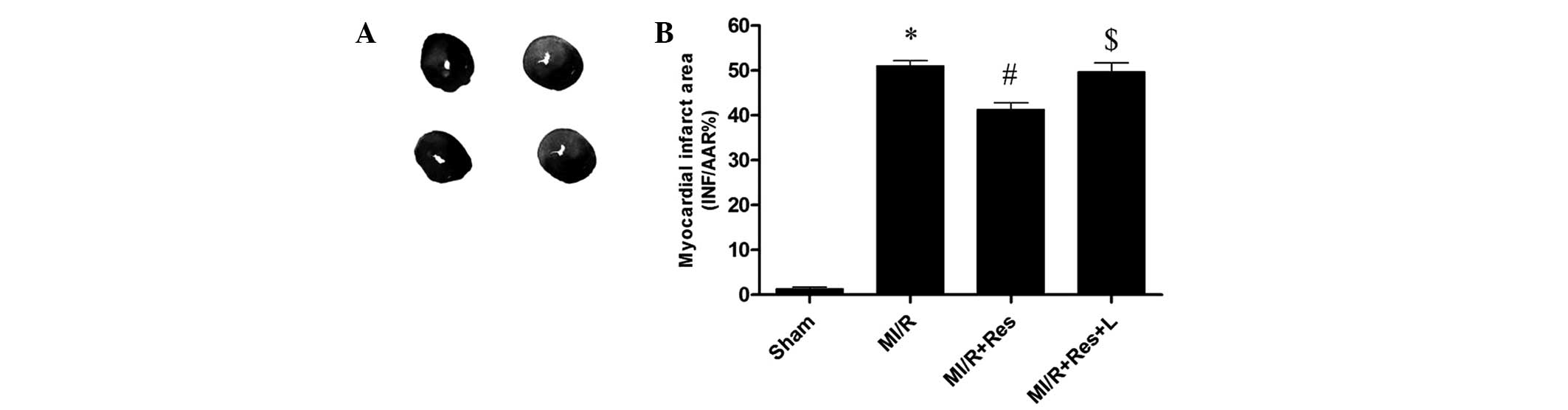
Res reduces the area of myocardial
infarction induced by MI/R
MI/R induced a significant area of infarction. As
compared with the MI/R group, Res significantly reduced the area of
myocardial infarction. The effect of Res was blocked by L-NAME, a
nitric oxide synthase (NOS) inhibitor (Fig. 1).
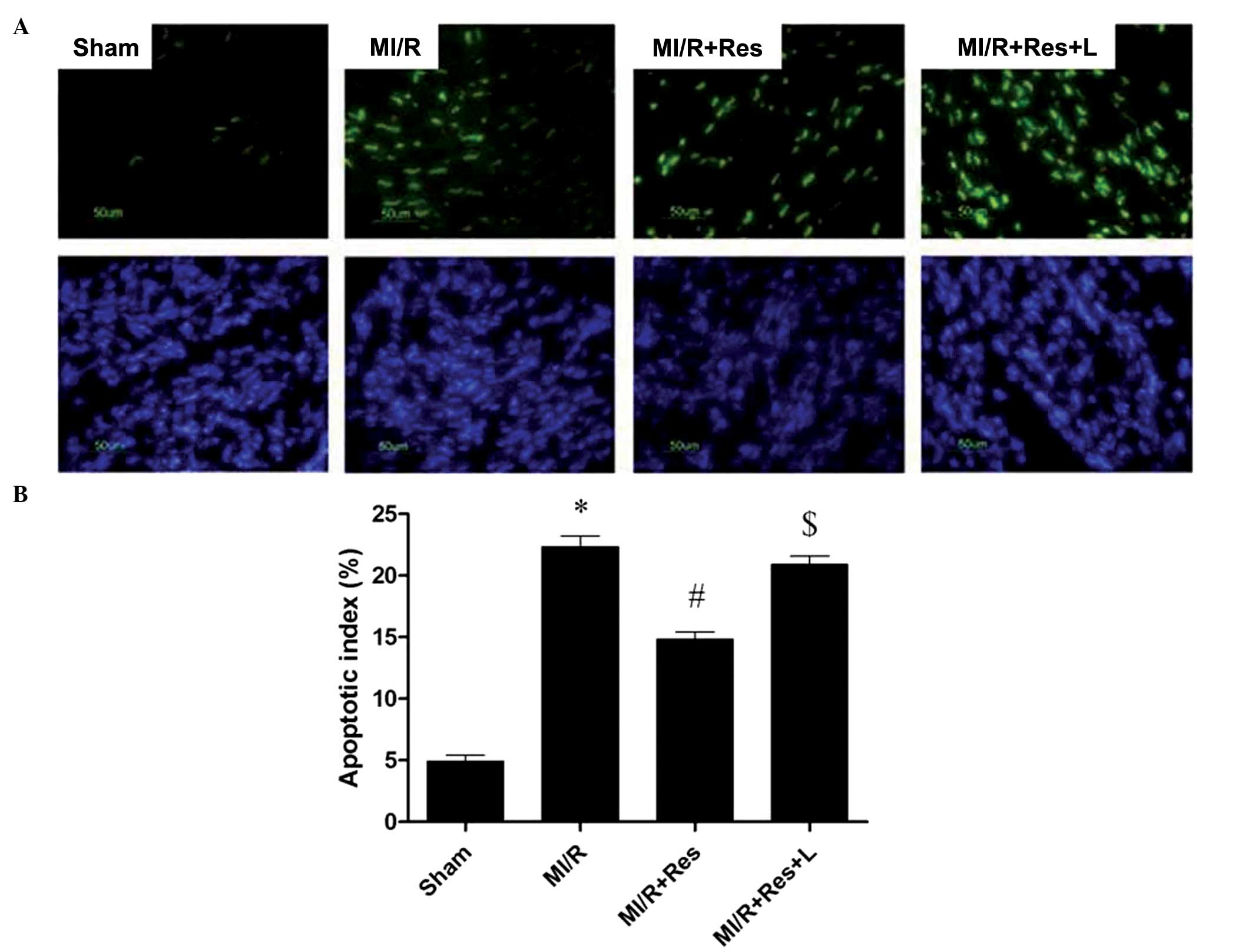
Res reduces myocardial apoptosis in rat
hearts subjected to MI/R
As shown in Fig. 2,
a low level of TUNEL-positive staining was detected in the sham
group whereas a significant number of TUNEL-positive cells were
observed in the MI/R group (*P<0.0001). Administration of Res
exerted a significant anti-apoptotic effect (#P=0.0022),
which was abolished by L-NAME ($P=0.0025).
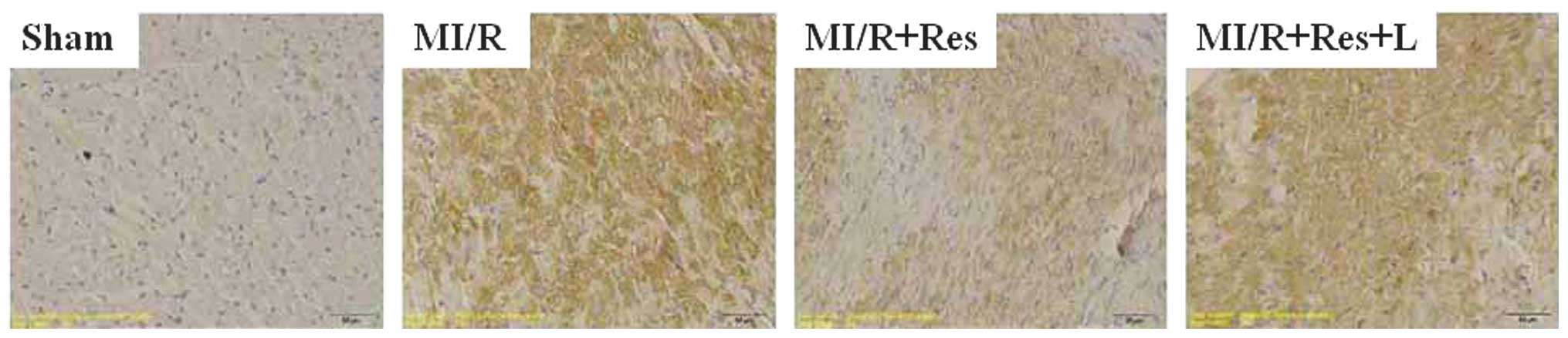
Res reduces TLR4 protein expression in
rat hearts subjected to MI/R
Immunohistochemical analysis (Fig. 3) showed that the expression of TLR4
was markedly increased in the the MI/R group (P<0.00015)
compared with the sham group. Upon administration of Res, the
expression of TLR4 was significantly reduced (P=0.0029) and this
effect was abolished by L-NAME (P=0.0035).
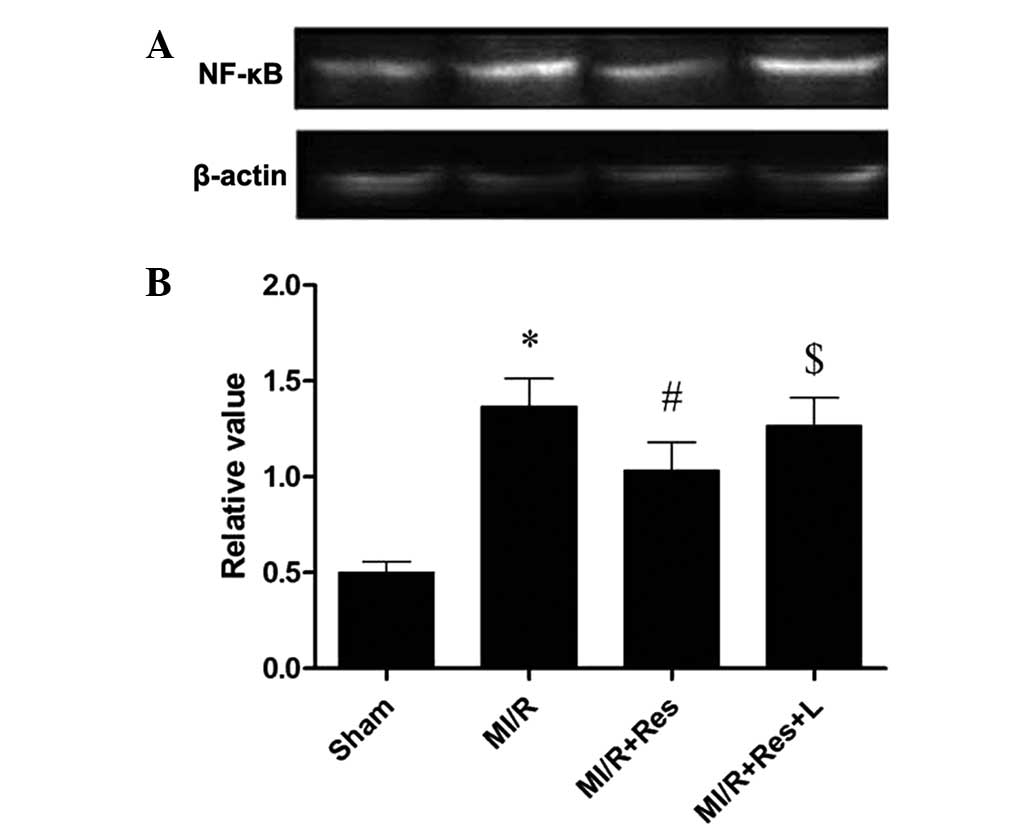
Res decreases NF-κB expression in rat
hearts subjected to MI/R
The expression of NF-κB, a downstream molecule of
TLR4, was determined in order to further study the effects of Res
on TLR4 mediated signaling. Western blot analysis (Fig. 4) indicated that the expression of
NF-κB was upregulated in MI/R group (0.0052). Administration of Res
significantly decreased NF-κB expression (#P=0.0082) and
the effect of Res was abolished by L-NAME
($P=0.0065).
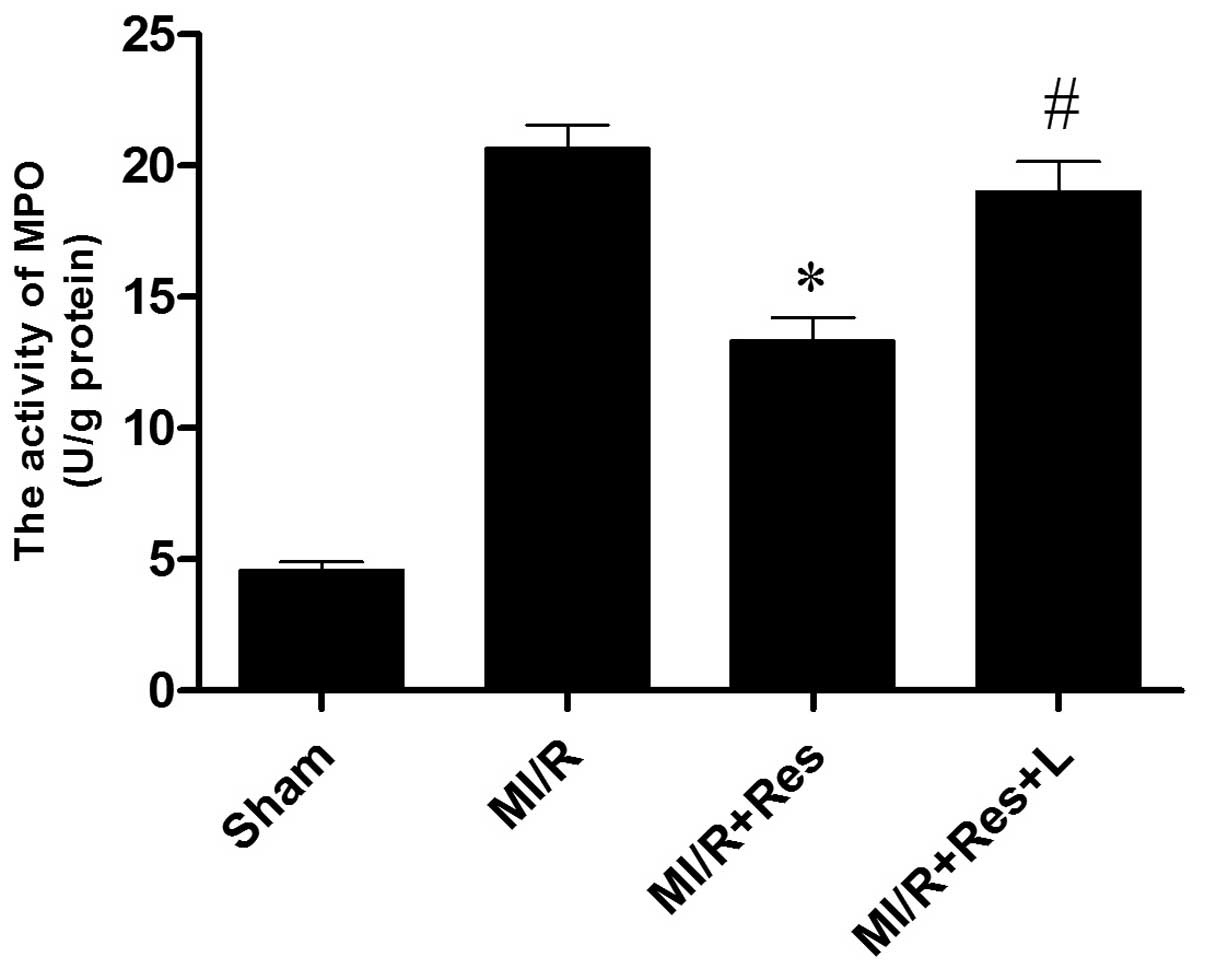
Res inhibits neutrophil infiltration in
MI/R tissue
Neutrophils contain a certain amount of MPO,
accounting for 5% of the dry cell weight. The activity of MPO in
the myocardium may therefore be considered as an indication of
neutrophil infiltration. As shown in Fig. 5, the MPO activity in the sham group
was relatively lower, whereas the MPO activity in the MI/R group
was significantly increased (P<0.0001). Res significantly
decreased myocardial MPO activity (P=0.0042), whereas L-NAME
attenuated the effects of Res (P=0.0175).
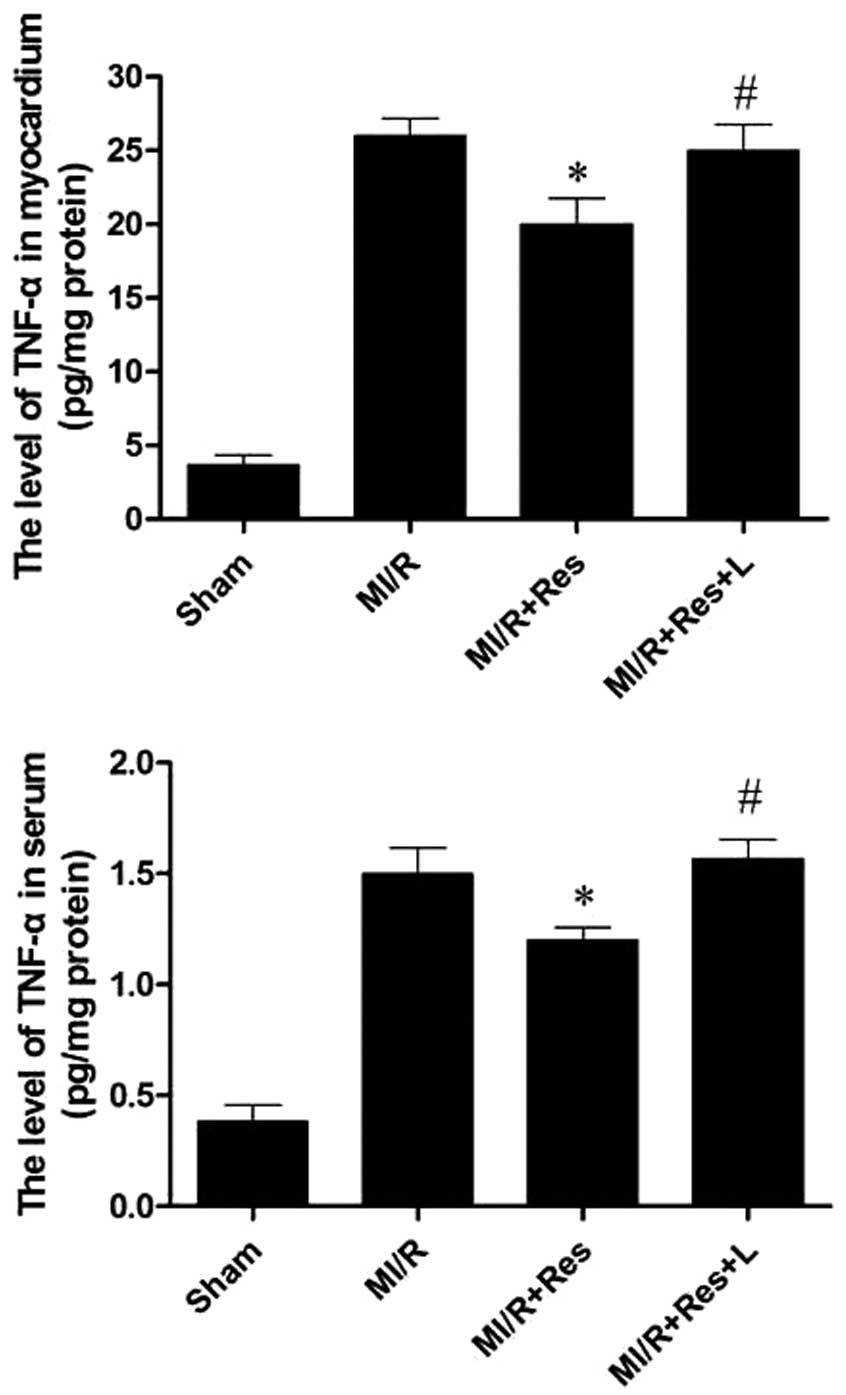
Res reduces TNF-α levels in the serum and
MI/R tissue
MI/R injury results in the production of a large
amount of TNF-α. Myocardial and serum TNF-α levels were therefore
examined. As shown in Fig. 6, as
compared with the MI/R group, Res significantly decreased the
levels of TNF-α in both the myocardium and serum, and these effects
were eliminated by L-NAME.
Discussion
The present study identified the following: i) Res
attenuates the inflammatory reaction induced by I/R injury by
inhibiting TLR4/NF-κB signaling; ii) Res attenuates the
inflammatory reaction induced by I/R injury through inhibiting
neutrophil infiltration and TNF-α production; iii) NO may function
in the protective mechanism of Res.
The inflammatory reaction has an important function
in MI/R injury (1). The release of
inflammatory cytokines and the aggregation and infiltration of
inflammatory cells are considered as the key steps in inflammation
(25).
TNF-α is predominantly secreted by macrophages, and
is likely to promote an inflammatory cascade by increasing the
release of other proinflammatory cytokines and influencing
neutrophil recruitment (26).
TNF-α is an important cytokine in inflammation and functions in the
initiation of the inflammatory response induced by MI/R (27). TNF-α can induce the release of
other inflammatory mediators, increase the expression of cell
adhesion factors, and promote neutrophil adhesion to endothelial
cells. In addition, TNF-α has a negative inotropic effect, which
can inhibit myocardial contractility, and lower blood pressure.
TNF-α can also induce cardiomyocyte apoptosis and participate in
ventricular remodeling (28).
Previous studies have suggested that the level of TNF-α
significantly increases following MI/R (29), whereas the administration of TNF-α
monoclonal antibody was shown to attenuate edema, and is conducive
to cardiac function recovery (30).
MI/R injury is induced in part by neutrophil
activation. The underlying mechanisms include: i) Cell damage
caused by the release of oxygen free radicals, proteolytic enzymes,
and cytotoxic substances; ii) the released inflammatory mediators
cause vascular endothelial cell damage, increased vascular
permeability, and edema; iii) additional activation of inflammatory
cells increase further the inflammatory response (31); iv) neutrophil adhesion to the
vascular endothelium and small blood vessel occlusion results in a
no-reflow phenomenon. Previous studies have identified the
mechanistic link between neutrophil activation and I/R injury.
Removal of neutrophils or drug inhibition of neutrophil activity
has been shown to reduce ischemia/reperfusion injury (32,33).
In the present study, it was identified that neutrophil
accumulation and TNF-α production in the MI/R group was
significantly increased. It was observed that Res reduced
neutrophil accumulation and TNF-α production, therefore indicating
that Res inhibited neutrophil accumulation and TNF-α production and
attenuated neutrophil-mediated I/R injury.
TLRs are present in both immune and non-immune
cells, and their expression is rapidly altered in response to
pathogens, cytokines, and environmental stressors (34). The functions of TLRs, including
TLR4, are responsible for the genesis of various cardiovascular
disorders as well as the activation of NF-κB and inflammatory
cytokines, including TNF-α, which results in cell death (35,36).
TLR4 is expressed in numerous tissues including the heart, where
the receptor becomes upregulated in response to ischemia. TLR4
expression levels in cardiac myocytes are enhanced by either
lipopolysaccharide or IL-1 (37).
The mechanism and function of TLR4-mediated signaling has been well
established, and it is considered that the upregulation of TLR4
results in activation of NF-κB in cardiomyocytes. This can augment
the release of cytokines, including TNF-α, IL-1 and MCF, as well as
the infiltration of leukocytes (38).
It is considered that NO is closely associated to
MI/R induced inflammation (39).
Endothelial-derived NO can inhibit cell adhesion factors, such as
P-selectin and ICAM-1 levels, thereby inhibiting leukocyte adhesion
and inward membrane migration (40). Endothelial-derived NO can inhibit
the expression of TNF-α and other pro-inflammatory factors. In
addition, endothelial-derived NO can increase the expression levels
of IL-10 and other anti-inflammatory factors, and indirectly
inhibit aggregation of inflammatory cells during local
inflammation, thereby reducing the inflammatory response (41). In the present study, administration
of L-NAME, a nitric oxide synthase inhibitor, abolished the
protective effect of Res. This suggested that NO functions in the
protective role of Res. Similarly, when methylene blue, a cGMP
inhibitor, was added, the protective action of Res was blocked,
indicating that cGMP pathway also functions in the protective role
of Res.
Previous research has shown that SIRT1 confers
protection in various models of cardiovascular oxidative stress
(42–44). SIRT1 plays a critical role in
endothelial homeostasis through regulation of the endothelial NOS
(eNOS). Endothelial-derived NO regulates blood vessel relaxation
and provides atheroprotective effects. Res, a polyphenolic
activator of SIRT1, has been shown to increase the expression of
eNOS (45) and the combination of
Res with the HMG-CoA reductase inhibitors (statins) increased the
activation of eNOS resulting in an increased functional recovery in
a model of acute myocardial infarction (46). Additionally, chronic Res treatment
improved endothelium-dependent relaxation in spontaneous
hypertensive rats; however, it did not increase eNOS expression
(47). The role of SIRT1 in the
cardioprotective process of Res remains controversial following
recent reports that have suggested that Res is not a direct
activator of SIRT1 (48). The
underlying mechanisms by which Res enhances SIRT1 activity remains
poorly defined and requires further study (49).
In conclusion, the present study demonstrated that
Res can attenuate inflammation induced by MI/R injury through the
TLR4/NF-κB signaling pathway. The protective effects of Res are
closely associated with the increase of NO production, the
inhibition of neutrophil infiltration and TNF-α production.
Acknowledgements
This work was supported by funding from the National
Natural Science Foundation of China (no. 81350026); the Scientific
Research Fund of the Heilongjiang Provincial Education Department
(no. 11551204); and the Natural Science Foundation of Heilongjiang
Province, China (no. H201344).
References
|
1
|
Xiong J, Xue FS, Yuan YJ, et al:
Cholinergic anti-inflammatory pathway: a possible approach to
protect against myocardial ischemia reperfusion injury. Chin Med J
(Engl). 123:2720–2726. 2010.
|
|
2
|
Naidu BV, Farivar AS, Woolley SM, et al:
Novel broad-spectrum chemokine inhibitor protects against lung
ischemia-reperfusion injury. J Heart Lung Transplant. 23:128–134.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Batista ML Jr, Rosa JC, Lopes RD, et al:
Exercise training changes IL-10/TNF-alpha ratio in the skeletal
muscle of post-MI rats. Cytokine. 49:102–108. 2010. View Article : Google Scholar
|
|
4
|
Shimamoto A, Chong AJ, Yada M, et al:
Inhibition of Toll-like receptor 4 with eritoran attenuates
myocardial ischemia-reperfusion injury. Circulation. 114:I270–I274.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kawai T and Akira S: Signaling to
NF-kappaB by Toll-like receptors. Trends Mol Med. 13:460–469. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaisho T and Akira S: Toll-like receptor
function and signaling. J Allergy Clin Immunol. 117:979–987. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arumugam TV, Okun E, Tang SC, et al:
Toll-like receptors in ischemia-reperfusion injury. Shock. 32:4–16.
2009. View Article : Google Scholar
|
|
8
|
Kaczorowski DJ, Nakao A, McCurry KR and
Billiar TR: Toll-like receptors and myocardial
ischemia/reperfusion, inflammation, and injury. Curr Cardiol Rev.
5:196–202. 2009. View Article : Google Scholar
|
|
9
|
Smiley ST, King JA and Hancock WW:
Fibrinogen stimulates macrophage chemokine secretion through
Toll-like receptor 4. J Immunol. 167:2887–2894. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu M, Wang H, Ding A, et al: HMGB1 signals
through Toll-like receptor (TLR) 4 and TLR2. Shock. 26:174–179.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Imai Y, Kuba K, Neely GG, et al:
Identification of oxidative stress and Toll-like receptor 4
signaling as a key pathway of acute lung injury. Cell. 133:235–249.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frantz S, Kobzik L, Kim YD, et al: Toll4
(TLR4) expression in cardiac myocytes in normal and failing
myocardium. J Clin Invest. 104:271–280. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Renaud S and de Lorgeril M: Wine, alcohol,
platelets, and the French paradox for coronary heart disease.
Lancet. 339:1523–1526. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Athar M, Back JH, Tang X, et al:
Resveratrol: A review of preclinical studies for human cancer
prevention. Toxicol Appl Pharmacol. 224:274–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aluyen JK, Ton QN, Tran T, et al:
Resveratrol: potential as anticancer agent. J Diet Suppl. 9:45–56.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Piotrowska H, Myszkowski K, Ziółkowska A,
et al: Resveratrol analogue 3, 4, 4′, 5-tetramethoxystilbene
inhibits growth, arrests cell cycle and induces apoptosis in
ovarian SKOV-3 and A-2780 cancer cells. Toxicol Appl Pharmacol.
263:53–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Afaq F, Adhami VM and Ahmad N: Prevention
of short-term ultraviolet B radiation-mediated damages by
resveratrol in SKH-1 hairless mice. Toxicol Appl Pharmacol.
186:28–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sehirli O, Sakarcan A, Velioğlu-Oğünç A,
et al: Resveratrol improves ifosfamide-induced Fanconi syndrome in
rats. Toxicol Appl Pharmacol. 222:33–41. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu Y, Nie L, Yin YG, et al: Resveratrol
protects against hyperglycemia-induced oxidative damage to
mitochondria by activating SIRT1 in rat mesangial cells. Toxicol
Appl Pharmacol. 259:395–401. 2012. View Article : Google Scholar
|
|
20
|
Li F, Gong Q, Dong H and Shi J:
Resveratrol, a neuroprotective supplement for Alzheimer’s disease.
Curr Pharm Des. 18:27–33. 2012. View Article : Google Scholar
|
|
21
|
López-Miranda V, Soto-Montenegro ML, Vera
G, et al: Resveratrol: a neuroprotective polyphenol in the
Mediterranean diet. Rev Neurol. 54:349–356. 2012.(In Spanish).
|
|
22
|
Borriello A1, Bencivenga D, Caldarelli I,
et al: Resveratrol: from basic studies to bedside. Borriello A,
Bencivenga D, Caldarelli I, Tramontano A, Borgia A, Zappia V, Della
Ragione F. Cancer Treat Res. 159:167–184. 2014. View Article : Google Scholar
|
|
23
|
Black SC and Rodger IW: Methods for
studying experimental myocardial ischemic and reperfusion injury. J
Pharmacol Toxicol Methods. 35:179–190. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Su H, Sun X, Ma H, et al: Acute
hyperglycemia exacerbates myocardial ischemia/reperfusion injury
and blunts cardioprotective effect of GIK. Am J Physiol-Endoc
Metab. 293:E629–E635. 2007. View Article : Google Scholar
|
|
25
|
Speyer CL and Ward PA: Role of endothelial
chemokines and their receptors during inflammation. J Invest Surg.
24:18–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Khimenko PL, Bagby GJ, Fuseler J and
Taylor AE: Tumor necrosis factor-alpha in ischemia and reperfusion
injury in rat lungs. J Appl Physiol (1985). 85:2005–2011. 1998.
|
|
27
|
Batista ML, Rosa JC, Lopes RD, et al:
Exercise training changes IL-10/TNF-alpha ratio in the skeletal
muscle of post-MI rats. Cytokine. 49:102–108. 2010. View Article : Google Scholar
|
|
28
|
Zhu J, Liu M, Kennedy RH and Liu SJ:
TNF-alpha-induced impairment of mitochondrial integrity and
apoptosis mediated by caspase-8 in adult ventricular myocytes.
Cytokine. 34:96–105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meldrum DR, Cleveland JC Jr, Cain BS, Meng
X and Harken AH: Increased myocardial tumor necrosis factor-alpha
in a crystalloid-perfused model of cardiac ischemia-reperfusion
injury. Ann Thorac Surg. 65:439–443. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gurevitch J, Frolkis I, Yuhas Y, et al:
Antitumor necrosis factor-alpha improves myocardial recovery after
ischemia and reperfusion. J Am Coll Cardiol. 30:1554–1561. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lefer AM, Ma XL, Weyrich A and Lefer DJ:
Endothelial dysfunction and neutrophil adherence as critical events
in the development of reperfusion injury. Agents Actions Suppl.
41:127–135. 1993.PubMed/NCBI
|
|
32
|
Ma XL, Lefer DJ, Lefer AM and Rothlein R:
Coronary endothelial and cardiac protective effects of a monoclonal
antibody to intercellular adhesion molecule-1 in myocardial
ischemia and reperfusion. Circulation. 86:937–946. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chandrasekar B, Smith JB and Freeman GL:
Ischemia-reperfusion of rat myocardium activates nuclear
factor-KappaB and induces neutrophil infiltration via
lipopolysaccharide-induced CXC chemokine. Circulation.
103:2296–2302. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Otsui K, Inoue N, Kobayashi S, et al:
Enhanced expression of TLR4 in smooth muscle cells in human
atherosclerotic coronary arteries. Heart Vessels. 22:416–422. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ha T, Li Y, Hua F, et al: Reduced cardiac
hypertrophy in Toll-like receptor 4-deficient mice following
pressure overload. Cardiovasc Res. 68:224–234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takahashi T: Toll-like receptors and
myocardial contractile dysfunction. Cardiovasc Res. 78:3–4. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hoshino K, Takeuchi O, Kawai T, et al:
Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are
hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps
gene product. J Immunol. 162:3749–3752. 1999.PubMed/NCBI
|
|
38
|
Liu YY, Cai WF, Yang HZ, et al: Bacillus
Calmette-Guérin and TLR4 agonist prevent cardiovascular hypertrophy
and fibrosis by regulating immune microenvironment. J Immunol.
180:7349–7357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu P, Hock CE, Nagele R and Wong PY:
Formation of nitric oxide, superoxide, and peroxynitrite in
myocardial ischemia-reperfusion injury in rats. Am J Physiol.
272:H2327–H2336. 1997.PubMed/NCBI
|
|
40
|
Li J, Wu F, Zhang H, et al: Insulin
inhibits leukocyte-endothelium adherence via an Akt-NO-dependent
mechanism in myocardial ischemia/reperfusion. J Mol Cell Cardiol.
47:512–519. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Zhang H, Wu F, et al: Insulin
inhibits tumor necrosis factor-alpha induction in myocardial
ischemia/reperfusion: role of Akt and endothelial nitric oxide
synthase phosphorylation. Crit Care Med. 36:1551–1558. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alcendor RR, Gao S, Zhai P, et al: Sirt1
regulates aging and resistance to oxidative stress in the heart.
Circ Res. 100:1512–1521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Danz ED, Skramsted J, Henry N, Bennett JA
and Keller RS: Resveratrol prevents doxorubicin cardiotoxicity
through mitochondrial stabilization and the Sirt1 pathway. Free
Radic Biol Med. 46:1589–1597. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pillai JB, Isbatan A, Imai S and Gupta MP:
Poly (ADP-ribose) polymerase-1-dependent cardiac myocyte cell death
during heart failure is mediated by NAD+ depletion and reduced
Sir2alpha deacetylase activity. J Biol Chem. 280:43121–43130. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wallerath T, Li H, Gödtel-Ambrust U,
Schwarz PM and Forstermann UA: Blend of polyphenolic compounds
explains the stimulatory effect of red wine on human endothelial NO
synthase. Nitric Oxide. 12:97–104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Penumathsa SV, Thirunavukkarasu M, Koneru
S, et al: Statin and resveratrol in combination induces
cardioprotection against myocardial infarction in
hypercholesterolemic rat. J Mol Cell Cardiol. 42:508–516. 2007.
View Article : Google Scholar :
|
|
47
|
Rush JW, Quadrilatero J, Levy AS and Ford
RJ: Chronic resveratrol enhances endothelium-dependent relaxation
but does not alter eNOS levels in aorta of spontaneously
hypertensive rats. Exp Biol Med (Maywood). 232:814–822. 2007.
|
|
48
|
Beher D, Wu J, Cumine S, et al:
Resveratrol is not a direct activator of SIRT1 enzyme activity.
Chem Biol Drug Des. 74:619–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chakrabarty SP, Balaram H and
Chandrasekaran S: Sirtuins: multifaceted drug targets. Curr Mol
Med. 11:709–718. 2011. View Article : Google Scholar : PubMed/NCBI
Danz ED, Skramsted J, Henry N, Bennett JA
and Keller RS: Resveratrol prevents doxorubicin cardiotoxicity
through mitochondrial stabilization and the Sirt1 pathway. Free
Radic Biol Med. 46:1589–1597. 2009. View Article : Google Scholar : PubMed/NCBI
|