Introduction
Due to a wide range of industrial applications,
cadmium (Cd) is substantially distributed in various environments
(1). Absorption of Cd can be
achieved through numerous routes, including ingestion, smoking and
inhalation of polluted air, and Cd may accumulate in the liver,
kidney, lung and heart for 20–30 years (2). Cd exposure is associated with various
diseases, such as renal failure, anemia, itai-itai disease and
cancer (3–5), and has therefore long been considered
a threat to human health (6,7). Cd
elicits toxicity through various pathways, including oxidative
stress and initiation of apoptosis (8,9).
Numerous studies have documented the cytotoxicity of Cd under
various settings. However, there has thus far been no research that
has evaluated the biological effects of Cd at non-toxic
concentrations, such as chronic low-dose environmental exposure,
which is likely coupled to cellular dysfunction and even morbidity.
As a divalent metal ion, cellular intrusion of Cd has been
suspected to result in systemic disorder of iron homeostasis;
however, the mechanisms underlying Cd-induced effects on iron
metabolism remain unclear.
Iron is a necessary metal for all cells, and a
fine-tuned regulatory system has evolved to maintain an elaborate
balance for systemic iron homeostasis (10). The hepcidin-ferroportin axis has a
central role in regulating iron flow (11). Hepcidin binds and induces
degradation of its receptor ferroportin, the iron exporter, which
leads to intracellular iron retention (12). Ferroportin is the only known iron
exporter in mammals, which is mainly expressed in enterocytes and
macrophages, and its concentration in splenic macrophages has been
shown to determine the iron levels in serum and other organs
(11,12). Ferroportin concentration is largely
regulated at the post-transcriptional level, through the iron
responsive element and iron regulatory protein (IRE-IRP) regulatory
system (13–15); however, it may also be regulated at
the transcriptional level (16,17).
Ferroportin mutations or dysfunction results in iron metabolism
disorders, known as ferroportin diseases, which are often
associated with iron overload (18,19).
Type B ferroportin disease is characterized by increased
concentrations of ferroportin, due to failure to respond to
hepcidin, which causes continuous iron export from the cells into
the plasma; this is associated with hyperferritinemia and excess
iron in hepatocytes (20). The aim
of the present study was to elucidate the regulation of ferroportin
in macrophages by Cd at non-toxic concentrations.
Materials and methods
Cell culture
The THP-1 human macrophage, J774A.1 mouse macrophage
and HEK293T human embryonic kidney cell lines were purchased from
the Shanghai Cell Bank of Type Culture Collection (Shanghai,
China). The cells were cultured in RPMI-1640 medium (Gibco Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (Hyclone Laboratories, Inc., Logan, UT, USA) and 100 U/ml
penicillin/streptomycin (Hyclone Laboratories, Inc.). Activation of
the THP-1 cells was initiated in complete medium with 1 μg/ml PMA
(Promega Corporation, Madison, WI, USA) for 18 h (21).
AlamarBlue® assay
Cell viability was measured using the alamarBlue
assay, according to the manufacturer’s instructions (Invitrogen
Life Technologies, Carlsbad, CA, USA). Briefly, the THP-1 and
J774A.1 cells were plated in 96-well plates, at a concentration of
5.0×103 cells/well. The cells were then treated with
various concentrations of CdCl2 (0–64 μM; Sigma-Aldrich,
St. Louis, MO, USA) for 24 h, followed by reading with a microplate
reader (Thermo Electron Corporation, Waltham, MA, USA) at 590 nm
with excitation at 530 nm.
Western blot analysis
The THP-1 cells were collected after washing with
cold phosphate-buffered saline (PBS; Solarbio Science &
Technology Co., Ltd., Beijing, China), and total proteins were
extracted using lysis buffer (Solarbio Science & Technology
Co., Ltd., Beijing, China) that was pre-mixed with a protease
inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Protein
lysates (30–50 μg total proteins) were then separated by 10%
SDS-PAGE and further analyzed by western blotting, as previously
described (22). Briefly, proteins
were transferred onto pure nitrocellulose membranes, followed by
primary antibody (in 1% milk) incubation overnight at 4°C. The
primary antibodies used in the present study were as follows:
Anti-GAPDH (1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) and anti-ferroportin (1:500; Sigma-Aldrich). Subsequently, the
secondary antibodies (in 1% milk) were applied for detection of the
target proteins (for 1 h at 37°C). The secondary antibodies used in
the present study were as follows: Goat anti-rabbit-HRP (for GAPDH;
1:10,000; ComWin Biotech Co., Ltd., Beijing, China) and goat
anti-rat-HRP (for FPN; 1:5,000; ComWin Biotech Co., Ltd.). Bands
were analyzed by Image J software (version 1.48; National
Institutes of Health, Bethesda, MD, USA) following coloration.
GAPDH was used as an internal control.
For the inhibition of transcription or translation,
THP-1 cells were treated with CdCl2 in the presence or
absence of 1 μg/ml actinomycin D (a transcriptional inhibitor;
ComWin Biotech Co., Ltd.) or cycloheximide (an inhibitor of protein
biosynthesis; ComWin Biotech Co., Ltd.) for 12 h. Subsequently,
cells were cultured with new medium containing CdCl2 for
another 12 h.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Following treatment with 4 and 8 μM CdCl2
for 24 h (where 100 μM ferric ammonium citrate was used as a
positive control), total RNAs were extracted from the THP-1 cells
using TRIzol®, according to the manufacturer’s
instructions (Invitrogen Life Technologies). For the reverse
transcription reaction, 2 μg total RNAs were used to synthesize
first strand cDNA, using oligo (dT) primers. qPCR was conducted to
determine gene expression levels using SYBR® Green qPCR
Master mix (Qiagen, Hilden, Germany) on a Mx3005P qPCR system
(Agilent Technologies, Inc., Santa Clara, CA, USA). The primer
sequences used in the present study were as follows: Forward:
5′-CGGTGTCTGTGTTTCTGGTAGA-3′ and reverse:
5′-CTGGGCCACTTTAAGTCTAGC-3′ for Ferroportin; and forward:
5′-GAAGGTGAAGGTCGGAGT-3′ and reverse: 5′-GAAGATGGTGATGGGATTTC-3′
for GAPDH. GAPDH was used as a housekeeping gene for normalization.
The qRT-PCR reaction was run at 95°C for 5 min (pre-denaturation)
followed by 45 cycles at 95°C for 15 sec, at 55°C for 30 sec, and
at 72°C for 30 sec. Following the reaction, a melting curve
analysis from 60–95°C was applied to all reactions to ensure
consistency and specificity of the amplified products. Bands formed
during agarose gel electrophoresis were quantified using the Image
J software.
Luciferase reporter assay
A DNA fragment of the IRE sequence encoding the
5′-UTR of human ferroportin mRNA was cloned and then subcloned into
the 5′-UTR of the luciferase gene, within the pGL3-Promoter
luciferase reporter vector (Promega Corporation). The
HindIII (Invitrogen Life Technologies) and NCOI (Invitrogen
Life Technologies) enzymes were used in the construction of the
plasmid. The constructed plasmid was validated by DNA sequencing
using RVprimer3 primer (Invitrogen Life Technologies) and sequence
alignment was performed with ClustalX software (version 2.1; Conway
Institute University College Dublin, Dublin, Ireland). In the
transfection experiments, 0.8 μg target plasmid and 80 ng
Renilla luciferase plasmid were co-transfected into HEK293T
cells using Lipofectamine® 2000 (Invitrogen Life
Technologies) in 24-well plates. Following a 24 h incubation, the
cells were washed with cold PBS, and then subjected to luciferase
activity determination, using a Dual-Luciferase Reporter Assay
system (Promega Corporation). Relative firefly luciferase
activities were calculated by normalization to those of
Renilla luciferase.
Labile iron pool (LIP) measurement
Following treatment of THP-1 and J774A.1 cells with
8 μM CdCl2 for 24 h, intracellular LIP levels were
evaluated according to the standard calcein acetoxymethyl ester
staining method (Sigma-Aldrich), as described previously (23). Briefly, cells were washed twice
with PBS and treated with 0.5 μM calcein (Sigma-Aldrich) for 15 min
at 37°C. Subsequently, cells were washed twice with PBS and divided
into two parts. An aliquot was treated with 100 μM desferoxamine
(Sigma-Aldrich) for 1 h at 37°C and the other was left untreated.
The intracellular fluorescence was then measured by FACS analysis
with excitation at 488 nm and reading at 525 nm with a flow
cytometer (FACS Calibur®; Becton Dickinson, Franklin
Lakes, NJ, USA). The LIP levels were determined from deduction of
the cellular fluorescence of deferoxamine-treated cells, compared
with the fluorescence of untreated cells.
Statistical analysis
Two-tailed Student’s t-test and one-way analysis of
variance were used to analyze the experimental data. The SPSS
Statistics 17.0 package (SPSS, Inc., Chicago, IL, USA) was utilized
to analyze the data. The data are represented as the mean ±
standard error. P<0.05 was considered to indicate a
statistically significant difference.
Results and Discussion
Ferroportin is the only known iron exporter in
mammalian cells, and it is mainly expressed on macrophages and
duodenal enterocytes (24–26). Macrophages have a key role in
maintaining iron homeostasis, through governing iron egress into
plasma (27,28). The present study focused on the
biological effects of Cd on the concentration of ferroportin in
macrophages. To accurately determine the potential effects of Cd on
ferroportin, sublethal concentrations of CdCl2 were
identified that did not induce significant toxicity in macrophages,
in order to determine the effects of relatively low non-toxic
concentrations of Cd. Cell viability was evaluated following
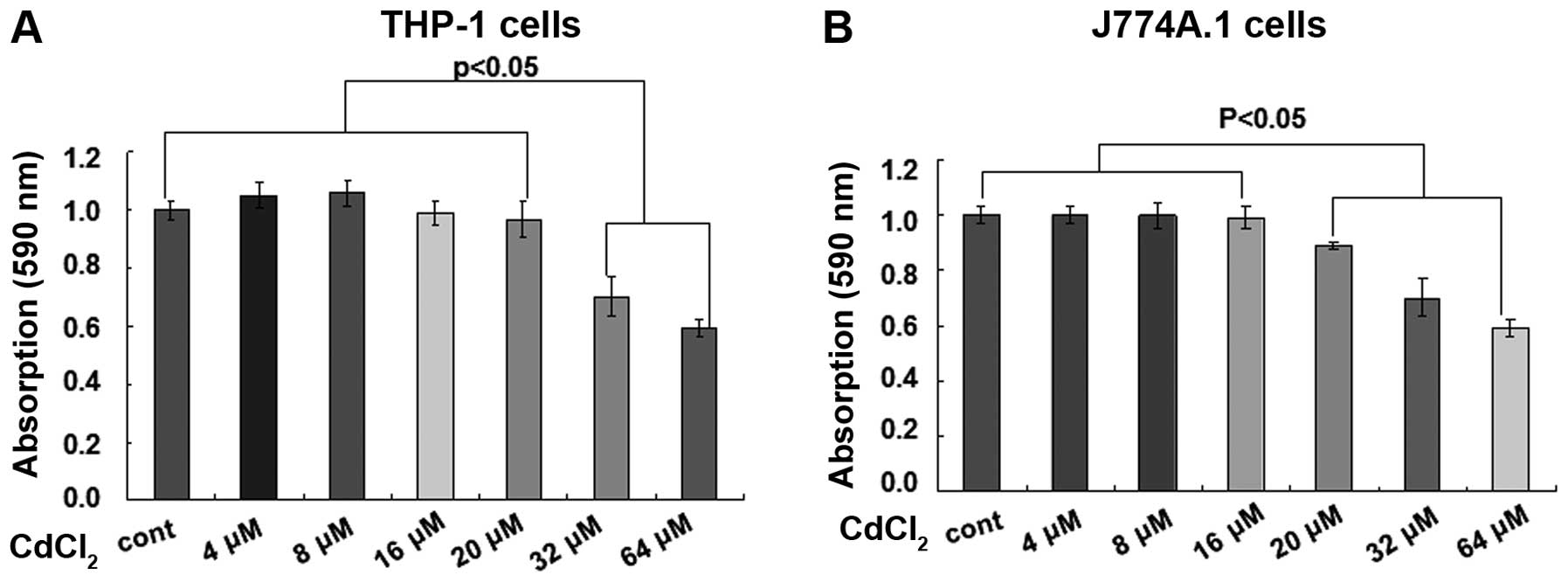
treatment with various concentrations of CdCl2 in THP-1
and J774A.1 cells. Cell viability of THP-1 cells was significantly
reduced in response to treatment with CdCl2 at
concentrations ≥32 μM for 24 h, as determined by an alamarBlue
assay (Fig. 1A; P<0.05). The
cell viability of J774A.1 cells was significantly decreased in
response to treatment with CdCl2 at concentrations ≥20
μM for 24 h (Fig. 1B; P<0.05).
Therefore, two non-toxic concentrations, 4 and 8 μM, were selected
for use in the following experiments.
The possible influence of CdCl2 treatment
was then determined on the expression of ferroportin. Since, to the
best of our knowledge, only a human ferroportin antibody has been
reported to function effectively, and no efficient mouse
ferroportin antibody is commercially available, the protein
expression of ferroportin was only examined in the THP-1 cells.
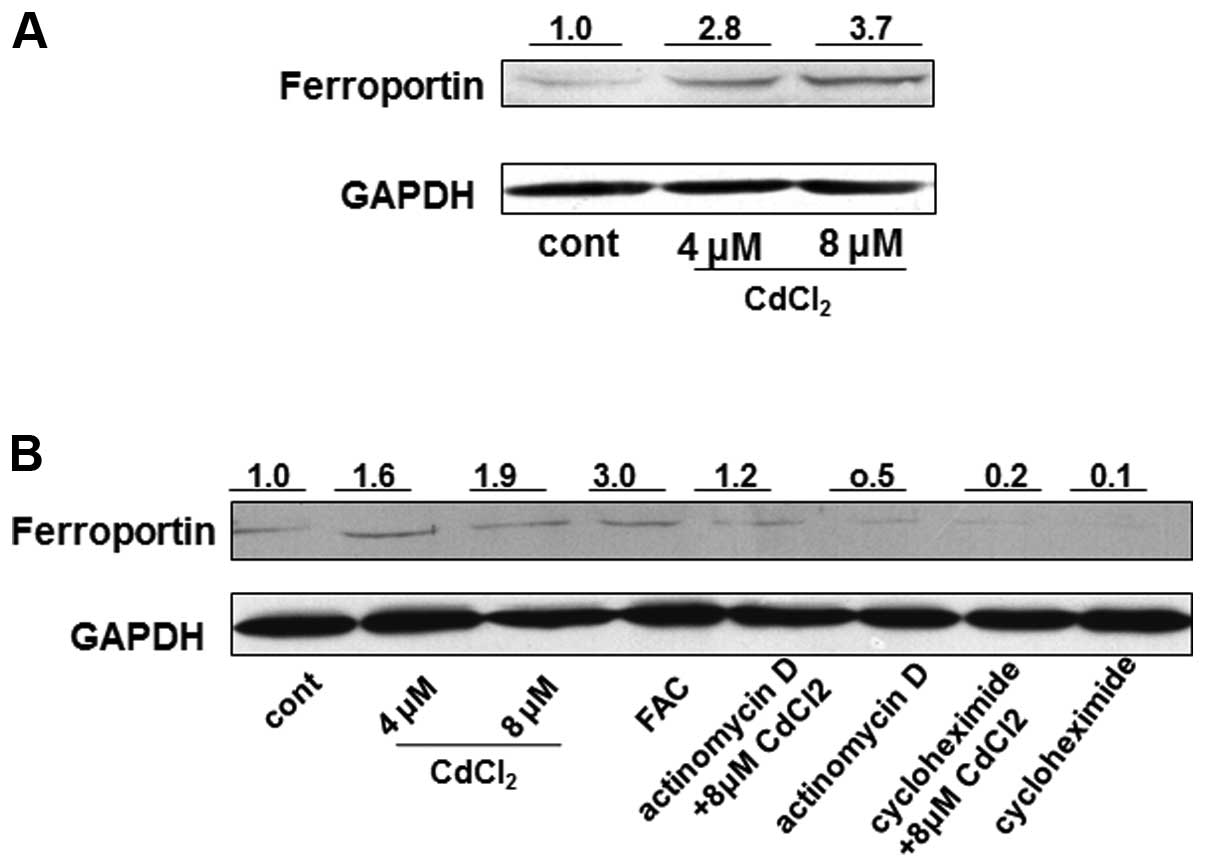
Following treatment of the cells with 4 and 8 μM CdCl2
for 24 h, the cells were collected for western blot analysis.
Ferroportin protein expression was markedly increased, by
>2-fold, in the 4 and 8 μM Cd-treated cells, as compared with
the untreated cells (Fig. 2A). To
investigate the mechanisms responsible for the Cd-induced increase
in ferroportin protein, THP-1 cells were treated with
CdCl2 in the presence or absence of 1 μg/ml actinomycin
D (a transcriptional inhibitor) and 10 μg/ml cycloheximide (an
inhibitor of protein biosynthesis) for 12 h. The protein expression
of ferroportin in the cells simultaneously treated with Cd and
actinomycin D was reduced by ~40%, as compared with the cells
treated with Cd alone; however, the protein expression was still
greater (~20%) in the Cd-treated cells, as compared with the
untreated cells (Fig. 2B).
Furthermore, the protein expression of ferroportin was markedly
reduced, by ~90%, in the cells treated with Cd and cycloheximide,
as compared with the cells treated with Cd alone (Fig. 2B). These results indicate that the
regulation of ferroportin by Cd may occur at the transcriptional
and post-translational levels, but appears more likely to occur at
the post-translational level. Ferric ammonium citrate (100 μM) was
used as a positive control to promote ferroportin concentration
(Fig. 2B). In addition, the mRNA
expression levels of ferroportin were determined in the cells
treated with 8 μM CdCl2 by RT-qPCR. There were no
significant differences in the ferroportin mRNA expression levels
between the Cd-treated and untreated cells (P>0.05, data not
shown). These results suggest that the regulation of ferroportin by
Cd primarily occurs at the post-transcriptional level.
In regards to the post-transcriptional regulation of
ferroportin, an IRE within its 5′-UTR has been recognized as being
under the control of IRPs (15,17).
As a divalent metal ion, Cd possesses similar properties to iron
(29), and it has long been
postulated that Cd may interfere with the transport and metabolism
of numerous essential metals, including iron, copper and zinc,
presumably through competition between Cd and the other metals
(30,31). Therefore, the present study
investigated the possible interruption of IRP-IRE actions on the
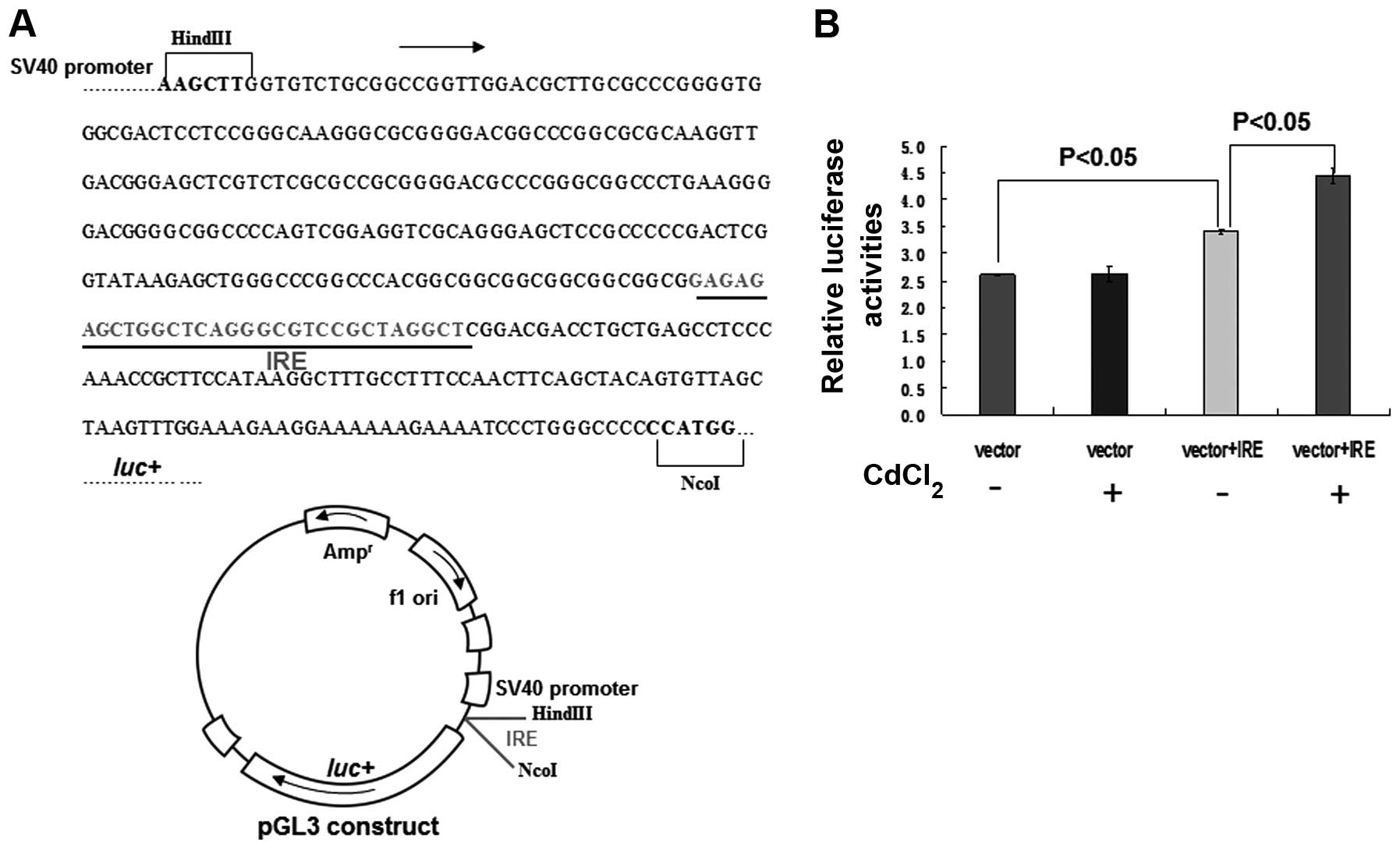
5′-UTR of ferroportin, by Cd. A construct was generated by
inserting a fragment of the IRE DNA sequence ahead of the 5′-UTR of
luciferase, within a pGL3-promoter luciferase reporter vector
(Fig. 3A). Treatment with
CdCl2 significantly enhanced luciferase activity by
>27% in the cells transfected with vector+IRE, as compared with
the untreated cells (Fig. 3B;
P<0.05). Conversely, there were no significant differences in
luciferase activity observed in the cells transfected with the
construct devoid of IRE, with or without CdCl2 treatment
(Fig. 3B). Notably, the luciferase
activity of the cells transfected with vector+IRE was increased by
33%, as compared with the cells transfected with vector only
(Fig. 3B, P<0.05), thus
implying that IRE is required for maintaining ferroportin levels.
These results collectively demonstrate that Cd regulates
ferroportin protein expression predominantly through the IRE-IRP
regulatory system, and Cd presumably replaced iron in driving IRP
removal from IRE in the 5′-UTR of ferroportin.
LIP is a sensitive marker of iron storage and
bioavailability, which dynamically binds to low-affinity ligands
depending on different physiological settings (32,33).
LIP levels are concertedly regulated and maintained within a strict
range that meets cellular demand for iron, but prevents excess
iron-triggered damage (23). To
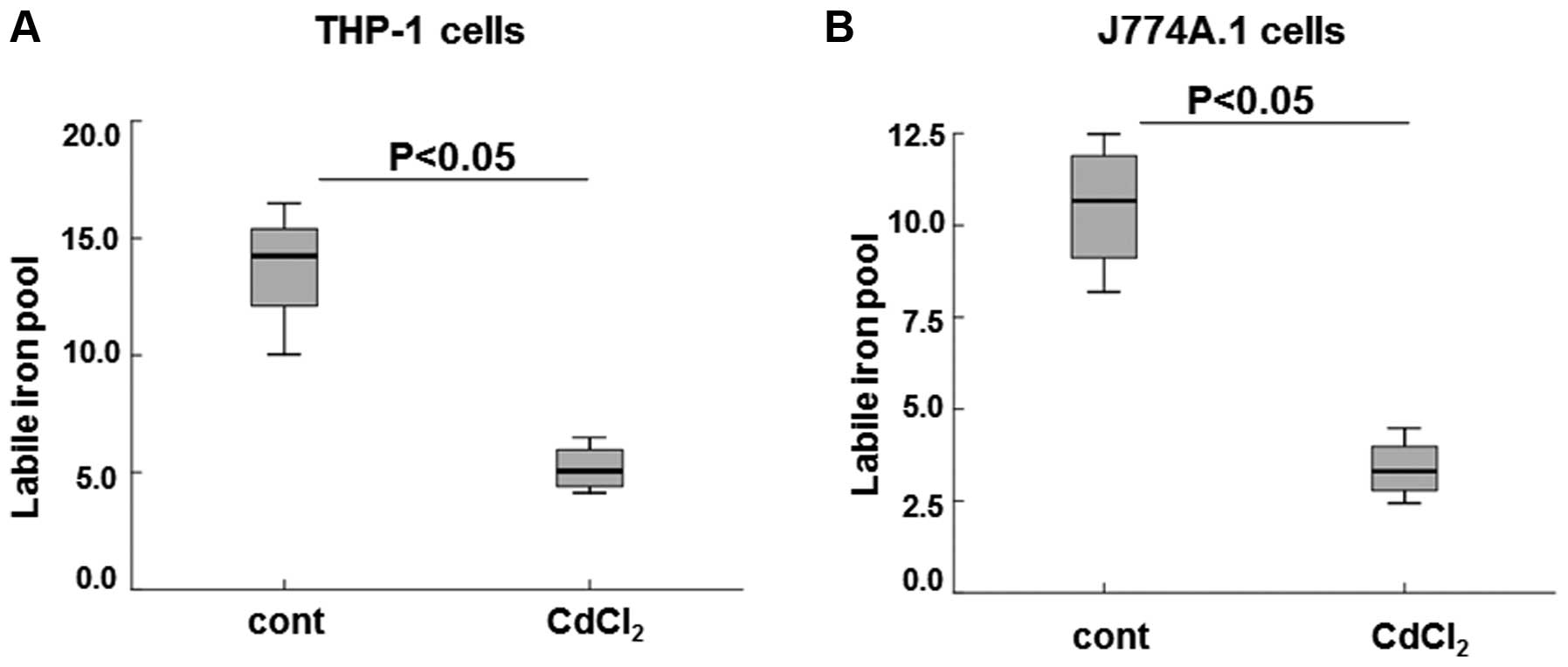
assess the effects of Cd-induced increased ferroportin on cellular
iron storage and bioavailability, LIP levels were measured in the
THP-1 and J774A.1 cells treated with 8 μM CdCl2.
Treatment with Cd significantly decreased LIP levels by >3-fold
in the THP-1 cells, as compared with the control cells (Fig. 4A; P<0.05). A similar decrease in
LIP levels was observed in the J774A.1 cells treated with Cd, as
compared with the untreated cells (Fig. 4B, P<0.05). These results suggest
that increased LIP levels were correlated with increased
ferroportin expression, in response to treatment with Cd.
Numerous previous studies have revealed deleterious
actions of Cd in diverse systems and models (2–5);
however, relatively few studies have attempted to determine the
potential biological effects of exposure to chronic sublethal Cd
levels. Increasing evidence has suggested that Cd may affect the
homeostasis of essential metals, through competition or other
unknown mechanisms (34,35). Cd burden was previously shown to
stimulate the expression of divalent metal transporter 1 in
enterocytes (36). Furthermore, a
previous study demonstrated that Cd could attenuate erythropoietin
production in the kidney, leading to an increase of hepcidin, which
is often coupled with iron disorders (37). Park and Chung (38) also reported that Cd could disturb
iron homeostasis by producing reactive oxygen species (38). However, research aiming to fully
elucidate the effects of Cd on iron-associated genes and proteins
remains limited, and further studies are required, in order to
improve knowledge.
In conclusion, the present study identified a novel
regulation of ferroportin by Cd. Ferroportin protein expression was
upregulated in response to sublethal treatment of Cd in
macrophages, resulting in attenuation of LIP. Notably, the results
suggest that Cd may regulate ferroportin translation through the
5′-UTR IRE of ferroportin. These results therefore may have
identified the molecular basis by which Cd impairs cellular iron
storage and bioavailability, under non-toxic concentrations in
macrophages.
Acknowledgements
The present study was supported by a grant from the
National ‘973’ program (grant no. 2014CB932000) and the National
Natural Science Foundation of China (grant no. 21377159). The
authors of the present study would also like to thank the
laboratory members for their assistance with the experiments and
reagents.
References
|
1
|
Hetherington L, Brown T, Benham A, Bide T,
Lusty P, Hards V, Hannis S and Idoine N: World Mineral Production
2002–06. Keyworth, Nottingham: British Geological Survey; 2008
|
|
2
|
Satarug S, Garrett SH, Sens MA and Sens
DA: Cadmium, environmental exposure, and health outcomes. Cien
Saude Colet. 16:2587–2602. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Veljkovic AR, Nikolic RS, Kocic GM, et al:
Protective effects of glutathione and lipoic acid against
cadmium-induced oxidative stress in rat’s kidney. Ren Fail.
34:1281–1287. 2012. View Article : Google Scholar
|
|
4
|
Bernhoft RA: Cadmium toxicity and
treatment. Scientific World Journal. 2013:3946522013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cen YL, Tang LY, Lin Y, Su FX, Wu BH and
Ren ZF: Association between urinary cadmium and clinicopathological
characteristics of breast cancer. Zhonghua Zhong Liu Za Zhi.
35:632–635. 2013.(In Chinese). PubMed/NCBI
|
|
6
|
Riederer AM, Belova A, George BJ and
Anastas PT: Urinary cadmium in the 1999–2008 U.S. National Health
and Nutrition Examination Survey (NHANES). Environ Sci Technol.
47:1137–1147. 2013. View Article : Google Scholar
|
|
7
|
Satarug S and Moore MR: Adverse health
effects of chronic exposure to low-level cadmium in foodstuffs and
cigarette smoke. Environ Health Perspect. 112:1099–1103. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brama M, Politi L, Santini P, Migliaccio S
and Scandurra R: Cadmium-induced apoptosis and necrosis in human
osteoblasts: role of caspases and mitogen-activated protein kinases
pathways. J Endocrinol Invest. 35:198–208. 2012.
|
|
9
|
Angenard G, Muczynski V, Coffigny H, et
al: Cadmium increases human fetal germ cell apoptosis. Environ
Health Perspect. 118:331–337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gordon WL: The absorption, transport and
storage of iron. Queens Med Mag (1972). 38:30–35. 1945.
|
|
11
|
Ganz T: Hepcidin and iron regulation, 10
years later. Blood. 117:4425–4433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nemeth E, Tuttle MS, Powelson J, et al:
Hepcidin regulates cellular iron efflux by binding to ferroportin
and inducing its internalization. Science. 306:2090–2093. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galy B, Ferring-Appel D, Becker C, et al:
Iron regulatory proteins control a mucosal block to intestinal iron
absorption. Cell Rep. 3:844–857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Casey JL, Hentze MW, Koeller DM, et al:
Iron-responsive elements: regulatory RNA sequences that control
mRNA levels and translation. Science. 240:924–928. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lymboussaki A, Pignatti E, Montosi G,
Garuti C, Haile DJ and Pietrangelo A: The role of the iron
responsive element in the control of ferroportin1/IREG1/MTP1 gene
expression. J Hepatol. 39:710–715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Drakesmith H and Prentice AM: Hepcidin and
the iron-infection axis. Science. 338:768–772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evstatiev R and Gasche C: Iron sensing and
signalling. Gut. 61:933–952. 2012. View Article : Google Scholar
|
|
18
|
Cazzola M: Genetic disorders of iron
overload and the novel “ferroportin disease”. Haematologica.
88:721–724. 2003.PubMed/NCBI
|
|
19
|
Beutler E, Barton JC, Felitti VJ, et al:
Ferroportin 1 (SCL40A1) variant associated with iron overload in
African-Americans. Blood Cells Mol Dis. 31:305–309. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Griffiths WJ, Mayr R, McFarlane I, et al:
Clinical presentation and molecular pathophysiology of autosomal
dominant hemochromatosis caused by a novel ferroportin mutation.
Hepatology. 51:788–795. 2010.
|
|
21
|
Maeß MB, Wittig B, Cignarella A and
Lorkowski S: Reduced PMA enhances the responsiveness of transfected
THP-1 macrophages to polarizing stimuli. J Immunol Method.
402:76–81. 2014. View Article : Google Scholar
|
|
22
|
Qu G, Zhang C, Yuan L, et al: Quantum dots
impair macrophagic morphology and the ability of phagocytosis by
inhibiting the Rho-associated kinase signaling. Nanoscale.
4:2239–2244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prus E and Fibach E: Flow cytometry
measurement of the labile iron pool in human hematopoietic cells.
Cytometry A. 73:22–27. 2008. View Article : Google Scholar
|
|
24
|
Abboud S and Haile DJ: A novel mammalian
iron-regulated protein involved in intracellular iron metabolism. J
Biol Chem. 275:19906–19912. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Donovan A, Brownlie A, Zhou Y, et al:
Positional cloning of zebrafish ferroportin1 identifies a conserved
vertebrate iron exporter. Nature. 403:776–781. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Donovan A, Lima CA, Pinkus JL, et al: The
iron exporter ferroportin/Slc40a1 is essential for iron
homeostasis. Cell Metab. 1:191–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weiss G: Iron and immunity: a double-edged
sword. Eur J Clin Invest. 32(Suppl 1): 70–78. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ganz T: Macrophages and systemic iron
homeostasis. J Innate Immun. 4:446–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martelli A and Moulis JM: Zinc and cadmium
specifically interfere with RNA-binding activity of human iron
regulatory protein 1. J Inorg Biochem. 98:1413–1420. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
De Smet H, Blust R and Moens L:
Cadmium-binding to transferrin in the plasma of the common carp
Cyprinus carpio. Comp Biochem Physiol C Toxicol Pharmacol.
128:45–53. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brzóska MM and Rogalska J: Protective
effect of zinc supplementation against cadmium-induced oxidative
stress and the RANK/RANKL/OPG system imbalance in the bone tissue
of rats. Toxicol Appl Pharmacol. 272:208–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Breuer W, Shvartsman M and Cabantchik ZI:
Intracellular labile iron. Int J Biochem Cell Biol. 40:350–354.
2008. View Article : Google Scholar
|
|
33
|
Pinnix ZK, Miller LD, Wang W, et al:
Ferroportin and iron regulation in breast cancer progression and
prognosis. Sci Transl Med. 2:43ra562010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen J, Shi YH and Li MY: Changes in
transferrin and hepcidin genes expression in the liver of the fish
Pseudosciaena crocea following exposure to cadmium. Arch Toxicol.
82:525–530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Buerge-Weirich D, Hari R, Xue H, Behra P
and Sigg L: Adsorption of Cu, Cd, and Ni on goethite in the
presence of natural groundwater ligands. Environ Sci Technol.
36:328–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gunshin H, Mackenzie B, Berger UV, et al:
Cloning and characterization of a mammalian proton-coupled
metal-ion transporter. Nature. 388:482–488. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Horiguchi H: Anemia induced by cadmium
intoxication. Nihon Eiseigaku Zasshi. 62:888–904. 2007.(In
Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park BY and Chung J: Cadmium increases
ferroportin-1 gene expression in J774 macrophage cells via the
production of reactive oxygen species. Nutr Res Pract. 3:192–199.
2009. View Article : Google Scholar
|