Introduction
The human cytochrome P450 (CYP) enzyme system is a
superfamily of paralogs that are dominantly expressed in the adult
liver and gastrointestinal tract (1). They catalyze phase-I metabolism of a
wide variety of exogenous chemicals, resulting in substrates that
are more water soluble, and thus facilitating excretion or further
transformation into nontoxic compounds (2).
CYP3A4 is the predominant P450 enzyme expressed in
human liver, and was understood to metabolize >60% of clinically
prescribed drugs (3). CYP3A4 is
not detectable prior to birth, but its expression gradually
increases thereafter (4). Despite
the heterogeneity of liver CYP3A4 expression among adult humans,
CYP3A4 expression may also be affected transiently by xenobiotics,
mostly its substrate, including CYP inducers, such as rifampin,
phenobarbital, clotrimazole and dexamethasone (5), and inhibitors, such as verapamil,
erythromycin, nifedipine, testosterone, midazolam and amiodarone
(6). Changes in intestinal and
liver CYP3A4 activity may have a significant effect on drug
metabolism and thereby affect bioavailability of these compounds
(7). Thus, increasing attention
has been focused on the involvement of CYP3A4 in drug-drug
interactions (DDI) (8,9).
The CYP3A5 gene is located on chromosome 7q22.1,
upstream of CYP3A4, and shares 84% similarity in amino acid
sequence with CYP3A4 (10). CYP3A4
and 5 catalyze similar and overlapping metabolic reactions,
including nifedipine oxidation, testosterone 6β-hydroxylation,
erythromycin N-demethylation, cyclosporine oxidation and
hydroxylation of benzodiazepine, midazolam, triazolam, alprazolam
and terfenadine (11). Although it
may differ in catalytic activity and regioselectivity, some reports
have argued that CYP3A5 is more important than CYP3A4 for overall
drug clearance (12,13) and also in the pathogenesis of
certain diseases, such as hypertension (14). It exhibited comparable or greater
metabolic activity than CYP3A4 for certain substrates, including
carbamazepine (12).
Unfortunately, the majority of reported studies have not
distinguished the activity of CYP3A4 from CYP3A5, and use CYP3A4 to
reflect the activity of either enzyme. However, there are important
differences between the enzymes. CYP3A5 is highly-expressed in the
adult kidney (15) rather than the
liver, in contrast to CYP3A4. In addition expression of CYP3A5 in
human liver varies among individuals and species from being
undetectable to comprising >50% of total CYPs present in this
organ (16). Therefore the
importance of CYP3A5 in metabolism may have been
underestimated.
A number of studies have emphasized the polymorphism
of CYP3A5 and have investigated the relevance of this to clinical
issues in treating hypertension (17), and during liver (18) and kidney (19,20)
transplantation. However, conflicting results have been obtained.
For example, a study examining the effect of CYP3A5 deactivating
mutations, reported no association between expression of this
enzyme and the response of blood pressure to amlodipine among
African-Americans (21) By
contrast, CYP3A5 expression had a significant impact on the
response of blood pressure to amlodipine in healthy Korean subjects
(22). In order to resolve this
conflicting information, the present study investigated whether
external factors are involved in modulating the expression of
CYP3A5 in response to certain drugs. The effect of overexpression
of CYP3A5 on the ability of dexamethasone (DEX) to induce CYP3A4
activity was investigated. The effect of CYP3A5 on the promoter
activity of CYP3A4 in the presence of DEX was also examined.
Finally the metabolism of a representative substrate drug,
amlodipine in response to CYP3A5 overexpression was
investigated.
Materials and methods
Chemicals and plasmid construction
Lipofectamine® 2000 (18324-012) was
obtained from Invitrogen Life Technologies, (Carlsbad, CA, USA).
The protein G-Sepharose (17-0618-01) was obtained from GE
Healthcare Biosciences (Pittsburgh, PA, USA).. Monoclonal mouse
anti-human CYP3A4 (sc-53850) and CYP3A5 antibodies (sc-53616) were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Monoclonal mouse anti-α-tubulin antibody (T5168), quinidine and
amlodipine were purchased from Sigma-Aldrich (St Louis, MO, USA).
Horseradish peroxidase-conjugated anti-mouse secondary antibody was
obtained from GE Healthcare Biosciences. Other chemicals used to
prepare buffers were obtained from Shenggong Ltd. (Shanghai,
China). Dexamethasone was reconstituted in dimethyl sulfoxide
(DMSO) at concentration of 1 mM.
The GFP-CYP3A5 plasmid was constructed by in-frame
ligation of a CYP3A5 coding sequence amplified from HepG2 cDNA with
pIRES2-eGFP (Clonetech Laboratories, Mountain View, CA, USA).
pGL3-CYP3A4-promoter consisted of a pGL3-basic vector (Promega
Corporation, Madison, WI, USA) inserted with the 5′-flanking region
of CYP3A4 (from −1.6 kb to +100 bp to the start codon).
Cell culture and stable transfection of
CYP3A5
Human HepG2 cells (Institute of Biochemistry and
Cell Biology, Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences, Shanghai, China) were cultured from a nitrogen
preserved batch and maintained with Dulbecco’s modified Eagle’s
medium containing 10% fetal bovine serum, 100 U/ml penicillin, and
0.1 mg/ml streptomycin in a humidified incubator at 37°C with 5%
CO2. Cells (5×105) were seeded onto a 6-cm
dish and transfected with 10 μg purified GFP-CYP3A5 plasmid at 70%
confluence using Lipofectamine 2000 according to the manufacturer’s
instructions. Cells were then reseeded onto three 10-cm dishes with
complete medium supplemented with 500 μg/ml of G418 (Cellgro,
Manassas, VA, USA) for colony selection. Clones were selected 2
weeks post-transfection and expanded for RNA and protein
verification. Positive CYP3A5+ HepG2 clones were frozen
for subsequent experiments.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
HepG2 cells were seeded in 6-well plates at a
density of 5×105 cells per well Following indicated
treatments, samples were washed with phosphate-buffered saline
(PBS) and lysed with 1 ml TRIzol® (Invitrogen Life
Technologies). RNA extractions were performed according to the
manufacturer’s instructions. Total RNA (1 μg) from each sample was
used for cDNA synthesize using a Revert Aid First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA). qPCR
was then performed using All-in-One SYBR® Green qPCR mix
(GeneCopoeia, Rockville, MD, USA) on an Applied Biosystem 7300 qPCR
module. The sequences of CYP3A4/5 detecting primers are provided in
Table I, and human GAPDH (23) was used as internal control. The
qPCR conditions were set as follows: Pre-heating at 95°C for 10
min, followed by 40 cycles of 95°C for 15 sec and 60°C for 15 sec,
and a final melting curve stage. Fold changes in mRNA levels were
calculated using the 2−ΔΔCt method.
 | Table ISequences of primers and siRNAs. |
Table I
Sequences of primers and siRNAs.
| Primer and
siRNA | Primer/siRNA
sequences |
|---|
| CYP3A4
promoter | F:
gttcacaggaagcagcacaaa
R: gagagccatcactactttccttact |
| CYP3A5
overexpression | F:
attcagcaagaagaacaaggaca
R: tggtgttctcaggcacagat |
| CYP3A4 RT | F:
ccttacatatacacaccctttggaag
R: ggttgaagaagtcctcctaagct |
| CYP3A5 RT | F:
aggcgggaagcagagaaag
R: ggggtcttgtggattgttgag |
| CYP3A5 siRNA | Si-A:
tgcctttgttgggaaatgttttg
Si-B: tccattatttctctcaataatac
Si-C: gagttattctaaggatttctact |
Protein and western blotting
Treated HepG2 cells were washed twice with PBS and
lysed in radioimmunoprecipitation assay buffer (50 mm Tris, pH 7.5;
150 mm NaCl; 0.1% SDS, 0.5% sodium deoxycholate; 1% Triton X-100;
and 1 mm phenylmethanesulfonyl fluoride). Cell lysates were then
sonicated on ice for 30 sec and cell debris was removed by
centrifugation at 10,000 × g for 15 min. The protein concentrations
of each sample were determined by a bicinchoninic acid assay
(Pierce Biotechnology, Inc., Rockford, IL, USA) according to the
manufacturer’s instructions. Total protein (30 μg) was loaded onto
10% SDS-PAGE gels, and transferred to polyvinylidene fluoride
membranes (GE Healthcare Biosciences). Each membrane was blocked
with 5% non-fat milk and incubated with the indicated antibodies.
The membranes were incubated with primary antibodies at room
temperature for 4 h or at 4°C overnight and horseradish
peroxidase-conjugated anti-mouse secondary antibody (1:5,000) at
room temperature for 1 h. Signals were visualized using Enhanced
Chemiluminescence (ECL) western blotting substrate (Thermo Fisher
Scientific). Monoclonal mouse anti-human CYP3A4/5 antibodies were
diluted at a ratio of 1:1,000 in Tris-buffered saline containing 5%
bovine serum albumin and 0.1% sodium azide.. Each blot was stripped
using Stripping Buffer (0.5 mM NaCl/0.2 mM acetic acid) and
reprobed. Monoclonal mouse anti-α-tubulin (1:3,000) was used as an
internal control.
Luciferase reporter assay
Luciferase reporter assays were performed using a
Dual-Luciferase® Reporter Assay system (Promega
Corporation). Specifically, pGL3-CYP3A4-promotor Luciferase
reporter vectors were co-transfected with or without CYP3A5 siRNA,
as indicated, using Lipofectamine 2000 in 24-well plates, where
HepG2 cells were at 70% confluence. Following treatment, cells were
washed and lysed with 80 μl lysis buffer from the
Dual-Luciferase®Reporter kit. Following three freeze and
thaw cycles, cell lysates were centrifuged at 4°C and 9,300 × g for
10 min and 10 μl was mixed with 100 μl of buffer LARII by pipetting
in luminometer tubes. Firefly fluorescence was then read on a
Turner DesignsTD-20/20 luminometer (Promega Corporation)
immediately. The fluorescence of Renilla luciferase was
measured in a second reading following the addition of 100 μl Stop
& Glo®Reagent and vortexed for 5 sec at ~100
rpm.
High performance liquid
chromatography-mass spectometry (HPLC-MS) detection of CYP3A4
substrate metabolites
Enzyme catalytic activity experiments were performed
using microsomes extracted from treated HepG2 cells, as previously
described (24). Microsomes were
suspended in PBS (0.1 mM, pH 7.4). Microsomal protein (50 μg) in
250 μl total incubation volume was achieved in all samples
following addition of substrates. Samples were pre-incubated for 5
min in a 37°C shaker, and enzyme reactions were initiated by
applying nicotinamide adenine dinucleotide phosphate (NADPH)
regenerating buffer to a working concentration of 1 mM
NADP+, 7.5 mM isocitric acid, 10 mM magnesium chloride
and 0.2 units of isocitric dehydrogenase. Total organic solvent did
not exceed 1% v/v. The linear range of each substrate was
determined. Quinidine was chosen as a selective probe for CYP3A4
and amlodipine, widely used clinically as an antihypertensive, was
used as a representative CYP3A4-metabolized drug. A starting
concentration of 1 mM quinidine and amlodipine was used for the 20
min incubation. Reactions were terminated with 100 μl acetonitrile
containing 0.1 μM tolbutamide as an internal standard. Samples were
then analyzed by HPLC- MS, as previously described (25).
Statistical analysis
The data were collected by at least three
independent experiments. Averages and standard deviations were
calculated using GraphPad Prism version 5.0 (GraphPad Software,
Inc., La Jolla, CA, USA) software. Student’s t test was used for
evaluating differences and a P<0.05 was considered to indicate a
statistically significant difference.
Results
Overexpression of CYP3A5 does not affect
the expression of CYP3A4
Increasing evidence has demonstrated that CYP3A5 is
important in the pathogenesis of hypertension (14), not only via the control of water
retention (26,27), but also through the metabolism of
certain antihypertensive drugs. However, conflicting data have been
reported regarding the influence of CYP3A5 on the response of blood
pressure to amlodipine (21,28).
It was hypothesized that there may be additional factors involved
in moderating the metabolism of drugs other than the innate
catalytic ability of CYP3A5.
The effect of CYP3A5 on CYP3A4 expression was
investigated in a HepG2 cell line, which was induced to overexpress
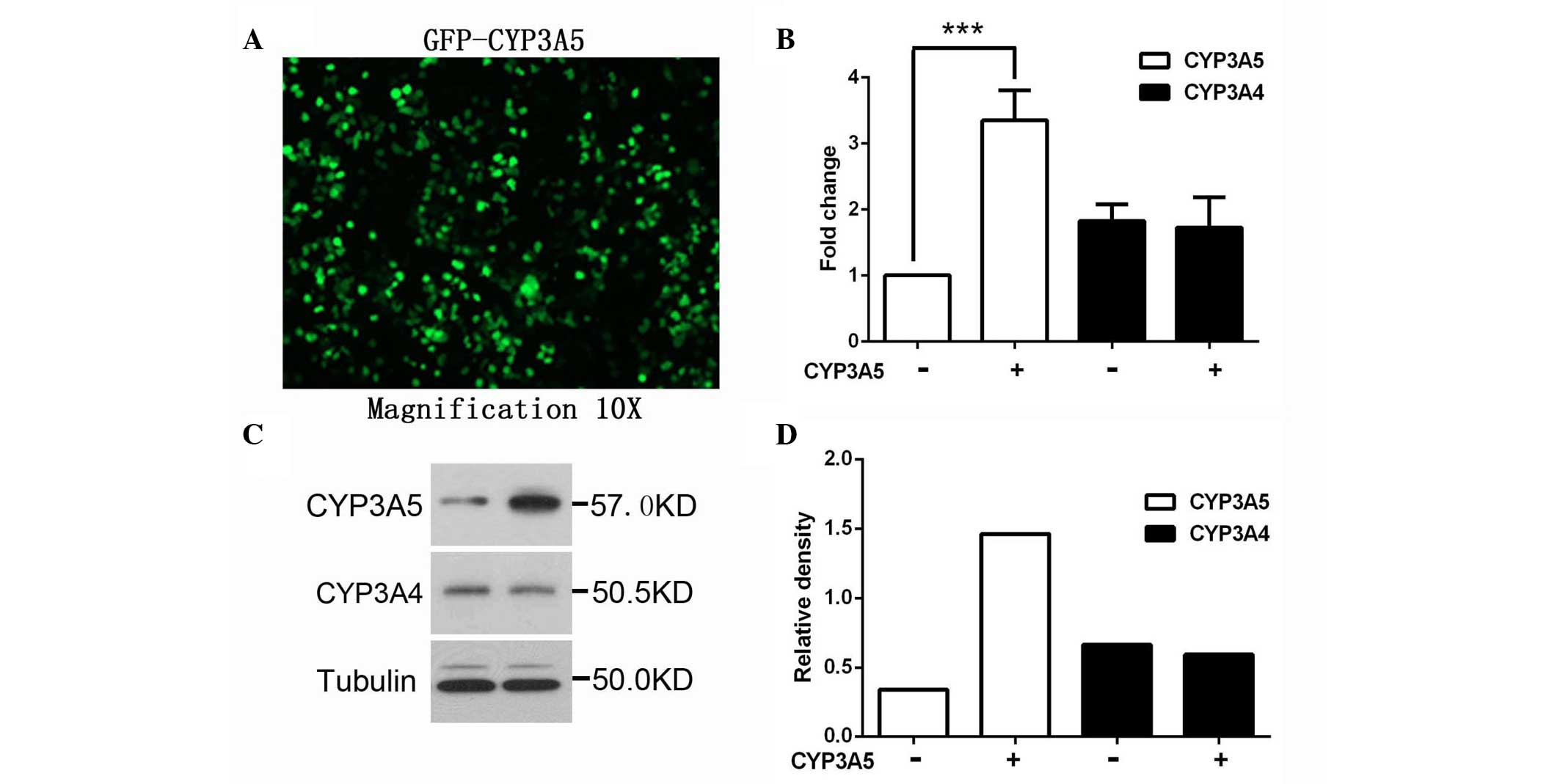
CYP3A5 using GFP-CYP3A5. The expression level of CYP3A5 was
confirmed by western blotting and RT-qPCR. As shown in Fig. 1, RNA and protein levels of CYP3A4
were not affected by CYP3A5 overexpression, suggesting that CYP3A4
is not direct regulated by CYP3A5.
Overexpression of CYP3A5 reduces
induction of CYP3A4 expression by DEX
The effect of clinical conditions, such as the
presence of substrates or inducers was then investigated. DEX was
selected due to its wide range of clinical applications. HepG2-wild
type (WT) and HepG2-CYP3A5+ cells were seeded onto
6-well plates at 5×105 cells/well. Following overnight
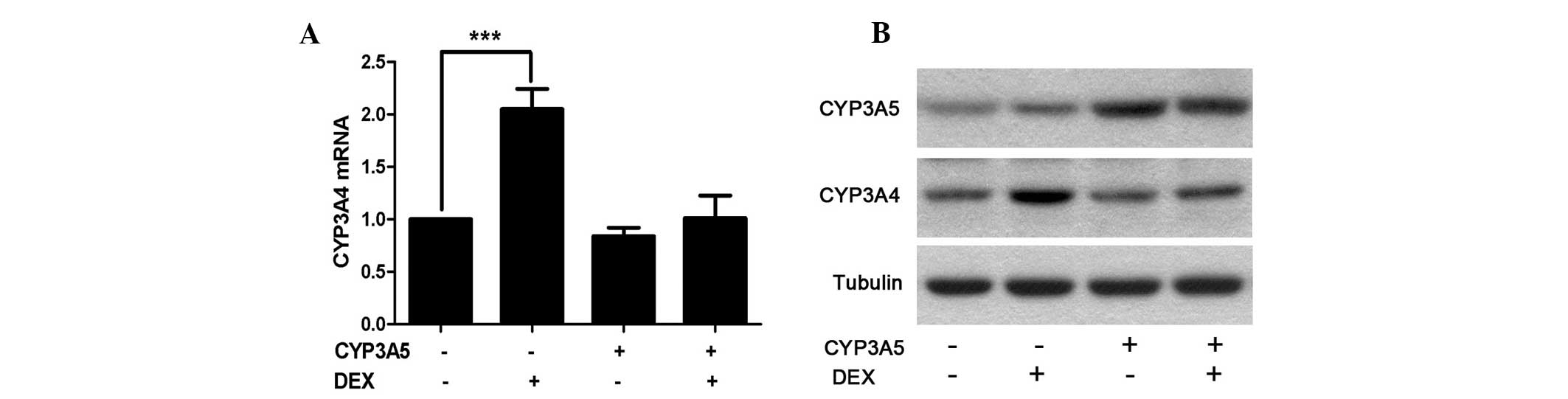
adhesion, 100 nM DEX was applied for a further 48 h incubation.
Cells were then harvested for RNA and protein analysis. As
hypothesized, CYP3A4 RNA as well as protein levels were
significantly elevated following DEX induction in the WT groups
compared with the control group (Fig.
2). However, this induction was suppressed in the cells
overexpressing CYP3A5. This indicates that CYP3A5 affects CYP3A4
indirectly under certain conditions.
Overexpression of CYP3A5 attenuates
CYP3A4 promoter activity in the presence of DEX
To further explore the effect of overexpression of
CYP3A5 on DEX induction of CYP3A4 expression, the 5′-flanking
region of CYP3A4 was cloned as described. Three short interfering
RNAs (siRNAs) were designed and synthesized at GenePharma Ltd.
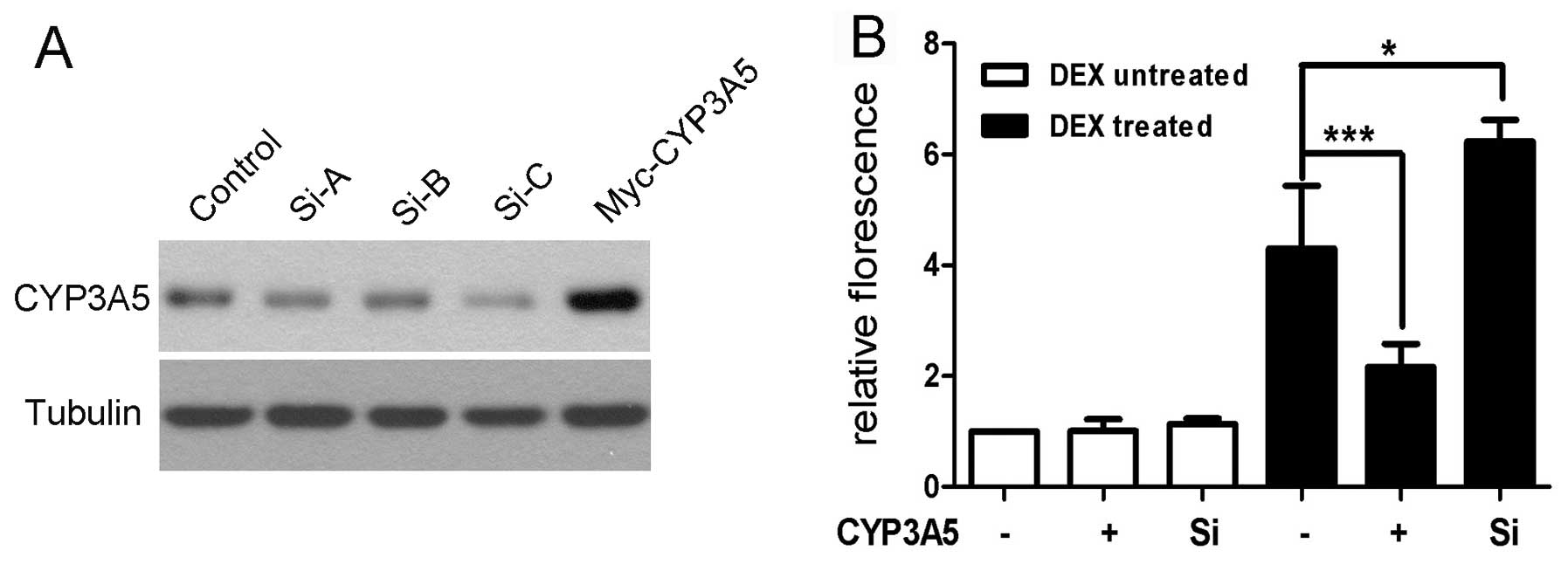
(Shanghai, China), and transfected into HepG2 cells. The effects of
silencing CYP3A5 were examined with western blotting and the most
potent siRNA (Si-C) was selected for subsequent experiments
(Fig. 3). HepG2-WT and
HepG2-CYP3A5+ cells were seeded onto 24-well plates at a
density of 4×104 cells/well. The purified
pGL3-CYP3A4-promoter plasmid was co-transfected with Si-C or
control RNA following cell adhesion. Fresh medium containing 100 nM
DEX was exchanged at 6 h post-transfection. Samples were harvested,
and Luciferase activity was measured, following 48 h incubation as
described. In concordance with the CYP3A4 expression data shown
above, the CYP3A4 promoter activity was unaffected by
overexpressing or silencing of CYP3A5. DEX was shown to stimulate
activation of the CYP3A4 promoter, which was also consistent with
previous reports. Notably, overexpression of CYP3A5 suppressed
CYP3A4 promoter activity compared with control in the presence of
DEX. This suggests that an abundance of CYP3A5 may affect CYP3A4
expression at the transcriptional level under certain
conditions.
Overexpression of CYP3A5 decreases the
activity of CYP3A4 in the presence of DEX
To further investigate the impact of the suppression
of CYP3A4 function in the presence of DEX, CYP3A4 activity in cells
that had undergone the same treatment was measured. In order to
remove the influence of CYP3A5, quinidine was used as a selective
CYP3A4 probe based on the availability of clinical DDI data and the
structural characteristics of the probe substrate (20). The 3-hydroxylated quinidine
generated in each group was normalized to non-treated controls, in
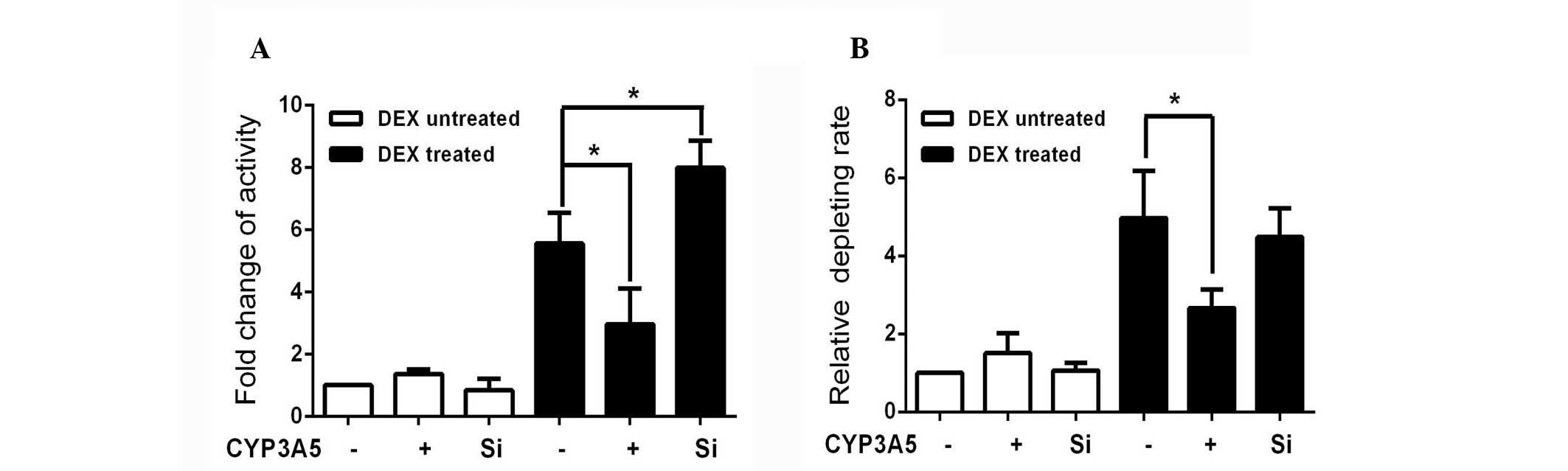
order to assess the alteration in CYP3A4 activity. As in the
luciferase experiments, the enzyme activity was also reduced
compared with DEX-treated controls (Fig. 4A). Greater activity was observed in
the CYP3A5-silenced group. CYP3A4 levels appeared to be inversely
associated with the expression of CYP3A5 in the presence of
DEX.
The impact of this interaction on potential clinical
events was examined using amlodipine, a calcium channel blocker,
widely used as an antihypertensive drug. The un-transformed
amlodipine was measured by HPLC-MS/MS and normalized to the levels
in the DEX-untreated control (Fig.
4B). The rate of amlodipine metabolism increased in cells
overexpressing CYP3A5, although it was not statistically
significant. The clearance of amlodipine increased following DEX
induction, and this induction was reduced by CYP3A5 overexpression.
This may be attributed to the fact that the metabolism of
amlodipine by CYP3A4 is four times higher than that of CYP3A5.
Following DEX induction, the rate of metabolism of amlodipine in
the CYP3A5-silenced group was not significantly changed compared
with the DEX-treated control group. This result indicates that
CYP3A4 is the primary enzyme involved in the metabolism of
amlodipine. However, CYP3A5 may influence CYP3A4 metabolism of this
and other substrates following exposure to DEX.
Discussion
Primary hepatocytes are a valuable in vitro
model with which to identify compounds that are potentially toxic
to humans. However, there are a number of disadvantages to the use
of primary hepatocytes, including a shortage of available human
liver material, limited proliferation ability and loss of metabolic
activity after a limited number of passages, which have constrained
its application. A number of groups have explored the possibility
of obtaining hepatocytes from differentiated adult or embryonic
stem cells, or immortalized human hepatocytes, but this technique
remains inconvenient. HepG2 cells express the predominant liver
functional CYP isoforms associated with drug metabolism. CYP3A7 is
the dominant isoform in these cells compared with CYP3A4 in the
human adult liver. Thus, HepG2 cells possess a phenotype most
similar to that of the fetal liver (29,30).
Although less suitable for predicting effects on metabolism in
humans, HepG2 cells provide a system that is easy to handle and
reproducible, and thus suitable for the investigation of gene
regulation. Quinidine was reported to be predominantly metabolized
by CYP3A4 (25), which is
consistent with the observation in the present study that
overexpression of CYP3A5 did not increase the rate of quinidine
metabolism significantly in the absence of other compounds. The
results confirm that quinidine is a reliable marker of CYP3A4
activity.
Environmental factors, such as cigarette smoking and
food intake induce the expression of CYPs and increase clearance of
phenacetin and theophylline (31).
Hepatic diseases, such as hepatitis B virus infection and
cirrhosis, and age, gender, hormones, inflammation and pregnancy
alter the expression pattern of CYP enzymes (32). The complexity of the
transcriptional regulation of CYP3A4 has been attributed to the
response of CYP3A4 to such factors. Transcription factors, such as
pregnane X receptor (PXR; −362/+53) (33), constitutive androstane receptor
(CAR) (34), nuclear factor I
(−243/−220) (29), differentially
expressed in chondrocyte 1 (35)
and hepatocyte nuclear factor 4a (HNF4a) (36) have been reported to account for a
component of CYP3A4 inter-individual variability. Constitutive
liver enhancer module of CYP3A4 (CLEM4) (37) and CCAAT-enhancer-binding proteins
(C/EBP) response elements (38)
were also found within its proximal promoter. However a significant
degree of CYP3A4 variation remains unexplained. Epigenetic
regulation of CYP3A4 has also been explored recently. The 12 kb
CYP3A4 regulatory region shows highly variable CpG methylation in
the adult liver, which corresponds to important CYP3A4
transcription factor binding sites, including xenobiotic responsive
enhancer module, CLEM4, C/EBP and HNF4a (39). In addition, a high degree of
methylation was observed in the fetal liver, which is consistent
with the minimal expression of CYP3A4 at this stage. The results
from the promoter activity experiments demonstrated that CYP3A5
does not affect CYP3A4 transcription directly. However, in the
presence of DEX, the promoter activity appears to be inversely
correlated with the expression of CYP3A5 (Fig. 3). This data may indicate that
excessive CYP3A5 prevents DEX from binding to its response
elements. However, which transcriptional factors are involved
remains to be elucidated.
CYP3A5 protein expression was found to be highly
variable in a manner that was generally independent of age but
dependent on race (4). The
expression of the CYP3A5*3 polymorphism results in a
truncated mRNA (40). This
polymorphism is observed at a similar frequency in Chinese and
Japanese populations, but three times higher in Caucasian
populations (41,42), which implies that more Asian
subjects are extensive CYP3A5 metabolizers. CYP3A5 expression
appears to be inducible via the glucocorticoid receptor, PXR and
CAR-β, as is the case for CYP3A4. However, the 5′-flanking regions
of CYP3A5 shares only 60% sequence similarity with that of CYP3A4.
The low homology may be one of the factors that differentiates
their regulation (43). This may
explain the different effects of DEX on the induction of the
expression of each of these genes (Fig. 2). CYP3A5 has been associated with
disease, however these associations are independent to its drug
metabolizing function. CYP3A5*1 homozygotes may have
higher systolic blood pressure (14). Certain combined CYP3A4/CYP3A5
haplotypes exhibit differential susceptibility to prostate cancer
(44). Females positive for
CYP3A5*1 appear to reach puberty earlier, which may
affect their risk of developing breast cancer (45). The results from the current study
demonstrating an interaction between CYP3A5 and CYP3A4 extends the
potential impact of CYP3A5 polymorphisms and variations in
expression, since the metabolic capacity of CYP3A4 appears to be
higher than of CYP3A5 and CYP3A7 (46) for the majority of substrates.
DEX is a potent synthetic member of the
glucocorticoid class of steroid drugs that are widely used as
anti-inflammatory and immunosuppressant treatments. DEX is
preferentially metabolized by CYP3A4 into 6β-OH-DEX in the human
adult liver. Thus DEX has been used in a number of studies as a
probe for CYP3A4 activity. However, DEX is also a potent inducer of
CYP3A4 (47). Evidence reported by
Pascussi et al (48)
demonstrated that the mechanism underlying DEX induction of CYP3A4
is concentration-related, as a low dose (10–100 nM) of DEX induced
CYP3A4 via the glucocorticoid receptor, whilst a high concentration
(10 μM) activates CYP3A4 through the PXR pathway. In the present
study, DEX strongly induced CYP3A4 in HepG2 cells, whilst the
induction by CYP3A5 was limited (Fig.
2). This is consistent with previous reports (30,46).
Since DEX is also a common substrate of CYP3A5, although at a
relatively lower metabolic rate, it is postulated that the
overexpression of CYP3A5 accelerates the metabolism of DEX, and
thus reduces the level of expression of CYP3A4 that it can induce.
Thus, DEX may be a bridge linking CYP3A4 and CYP3A5 function. A
similar observation was made that DEX increased erythromycin breath
test (ERBT) only in CYP3A5*1 non-carriers as they may be
more susceptible to the inductive effects of DEX due to lower basal
CYP3A activity (49).
Adverse drug interactions are an important cause of
morbidity, hospitalization, and mortality. Drug interactions are be
the fourth leading cause of death in hospitalized patients in the
US (50). The greatest risk of
drug interactions occurs due to effects on the cytochrome system.
CYP3A4, the most prevalent cytochrome P450, accounts for 30–50% of
drugs metabolized by type I enzymes. Previous DDI studies have
focused on drug metabolism by single specific enzymes, such as
CYP3A4 or CYP3A5. However, conflicting results have been reported
using this model. For instance, CYP3A5*3 carriers
require a lower dose of substrate drugs, such as cyclosporine and
tacrolimus (51). However,
CYP3A5*3 showed no association with the response of
blood pressure to amlodipine in African-Americans with early
hypertensive renal disease (21).
The current study demonstrated that the contribution of CYP3A5 may
be an important source of inter-individual variability in response
to drugs. Furthermore, the identification of this novel interaction
may provide further insights when predicting drug metabolism and
designing individualized treatment regimes, particularly when a
patient with multiple co-morbidities is prescribed more than two
drugs.
Acknowledgements
This study was supported by the National Basic
Research Program of China (grant no. 2011CB512001), The National
Key Technology R&D Program (grant no. 2012BAI37B05), the
National Natural Science Foundation of China (grant no. 81273594)
and the Fundamental Research Funds for the Central Universities of
Central South University (grant no. 2012zzts036).
References
|
1
|
Kruijtzer CM, Beijnen JH and Schellens JH:
Improvement of oral drug treatment by temporary inhibition of drug
transporters and/or cytochrome P450 in the gastrointestinal tract
and liver: an overview. Oncologist. 7:516–530. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Casarett LJ and Doull J: Toxicology: The
Basic Science of Poisons. Macmillan Publishing Company; London, UK:
1975
|
|
3
|
Anzenbacher P and Anzenbacherová E:
Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci.
58:737–747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stevens JC, Hines RN, Gu C, et al:
Developmental expression of the major human hepatic CYP3A enzymes.
J Pharmacol Exp Ther. 307:573–582. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo G, Cunningham M, Kim S, et al: CYP3A4
induction by drugs: correlation between a pregnane X receptor
reporter gene assay and CYP3A4 expression in human hepatocytes.
Drug Metab Dispos. 30:795–804. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Katoh M, Nakajima M, Yamazaki H and Yokoi
T: Inhibitory effects of CYP3A4 substrates and their metabolites on
P-glycoprotein-mediated transport. Eur J Pharm Sci. 12:505–513.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kivistö KT, Niemi M and Fromm MF:
Functional interaction of intestinal CYP3A4 and P-glycoprotein.
Fundam Clin Pharmacol. 18:621–626. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galetin A, Burt H, Gibbons L and Houston
JB: Prediction of time-dependent CYP3A4 drug-drug interactions:
impact of enzyme degradation, parallel elimination pathways, and
intestinal inhibition. Drug Metab Dispos. 34:166–175. 2006.
View Article : Google Scholar
|
|
9
|
Wang RW, Newton DJ, Liu N, Atkins WM and
Lu AY: Human cytochrome P-450 3A4: in vitro drug-drug interaction
patterns are substrate-dependent. Drug Metab Dispos. 28:360–366.
2000.PubMed/NCBI
|
|
10
|
Walsky RL, Obach RS, Hyland R, et al:
Selective mechanism-based inactivation of CYP3A4 by CYP3cide
(PF-04981517) and its utility as an in vitro tool for delineating
the relative roles of CYP3A4 versus CYP3A5 in the metabolism of
drugs. Drug Metab Dispos. 40:1686–1697. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yuan R, Madani S, Wei XX, Reynolds K and
Huang SM: Evaluation of cytochrome P450 probe substrates commonly
used by the pharmaceutical industry to study in vitro drug
interactions. Drug Metab Dispos. 30:1311–1319. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang W, Lin YS, McConn DJ II, et al:
Evidence of significant contribution from CYP3A5 to hepatic drug
metabolism. Drug Metab Dispos. 32:1434–1445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai Y, Hebert MF, Isoherranen N, et al:
Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in
vitro. Drug Metab Dispos. 34:836–847. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kivistö KT, Niemi M, Schaeffeler E, et al:
CYP3A5 genotype is associated with diagnosis of hypertension in
elderly patients: data from DEBATE study. Am J Pharmacogenomics.
5:191–195. 2005. View Article : Google Scholar
|
|
15
|
Dai Y, Iwanaga K, Lin YS, et al: In vitro
metabolism of cyclosporine A by human kidney CYP3A5. Biochem
Pharmacol. 68:1889–1902. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuehl P, Zhang J, Lin Y, et al: Sequence
diversity in CYP3A promoters and characterization of the genetic
basis of polymorphic CYP3A5 expression. Nat Genet. 27:383–391.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang YP, Zuo XC, Huang ZJ, et al: CYP3A5
polymorphism, amlodipine and hypertension. J Hum Hypertens.
28:145–149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Y, Li Y, Tang J, et al: Influence of
CYP3A4, CYP3A5 and MDR-1 polymorphisms on tacrolimus
pharmacokinetics and early renal dysfunction in liver transplant
recipients. Gene. 512:226–231. 2013. View Article : Google Scholar
|
|
19
|
Zuo XC, Ng CM, Barrett JS, et al: Effects
of CYP3A4 and CYP3A5 polymorphisms on tacrolimus pharmacokinetics
in Chinese adult renal transplant recipients: a population
pharmacokinetic analysis. Pharmacogenet Genomics. 23:251–261. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suzuki Y, Itoh H, Fujioka T, et al:
Association of plasma concentration of 4β-hydroxycholesterol with
CYP3A5 polymorphism and plasma concentration of indoxyl sulfate in
stable kidney transplant recipients. Drug Metab Dispos. 42:105–110.
2014. View Article : Google Scholar
|
|
21
|
Bhatnagar V, Garcia EP, O’Connor DT, et
al: CYP3A4 and CYP3A5 polymorphisms and blood pressure response to
amlodipine among African-American men and women with early
hypertensive renal disease. Am J Nephrol. 31:95–103. 2010.
View Article : Google Scholar :
|
|
22
|
Kim KA, Park PW, Lee OJ, et al: Effect of
CYP3A5*3 genotype on the pharmacokinetics and
pharmacodynamics of amlodipine in healthy Korean subjects. Clin
Pharmacol Ther. 80:646–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Overbergh L, Valckx D, Waer M and Mathieu
C: Quantification of murine cytokine mRNAs using real time
quantitative reverse transcriptase PCR. Cytokine. 11:305–312. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paine MF, Khalighi M, Fisher JM, et al:
Characterization of interintestinal and intraintestinal variations
in human CYP3A-dependent metabolism. J Pharmacol Exp Ther.
283:1552–1562. 1997.
|
|
25
|
Galetin A, Brown C, Hallifax D, Ito K and
Houston JB: Utility of recombinant enzyme kinetics in prediction of
human clearance: impact of variability, CYP3A5, and CYP2C19 on
CYP3A4 probe substrates. Drug Metab Dispos. 32:1411–1420. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ghosh SS, Basu AK, Ghosh S, et al: Renal
and hepatic family 3A cytochromes P450 (CYP3A) in spontaneously
hypertensive rats. Biochem Pharmacol. 50:49–54. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang L, Miyaki K, Wang W and Muramatsu M:
CYP3A5 polymorphism and sensitivity of blood pressure to dietary
salt in Japanese men. J Hum Hypertens. 24:345–350. 2010. View Article : Google Scholar
|
|
28
|
Ho H, Pinto A, Hall SD, et al: Association
between the CYP3A5 genotype and blood pressure. Hypertension.
45:294–298. 2005. View Article : Google Scholar
|
|
29
|
Riffel AK, Schuenemann E and Vyhlidal CA:
Regulation of the CYP3A4 and CYP3A7 promoters by members of the
nuclear factor I transcription factor family. Mol Pharmacol.
76:1104–1114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maruyama M, Matsunaga T, Harada E and
Ohmori S: Comparison of basal gene expression and induction of
CYP3As in HepG2 and human fetal liver cells. Biol Pharm Bull.
30:2091–2097. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sarkar MA and Jackson BJ: Theophylline
N-demethylations as probes for P4501A1 and P4501A2. Drug Metab
Dispos. 22:827–834. 1994.PubMed/NCBI
|
|
32
|
Badyal DK and Dadhich AP: Cytochrome P450
and drug interactions. Ind J Pharmacol. 33:248–259. 2001.
|
|
33
|
Moore LB, Parks DJ, Jones SA, et al:
Orphan nuclear receptors constitutive androstane receptor and
pregnane X receptor share xenobiotic and steroid ligands. J Biol
Chem. 275:15122–15127. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Drocourt L, Ourlin JC, Pascussi JM, Maurel
P and Vilarem MJ: Expression of CYP3A4, CYP2B6, and CYP2C9 is
regulated by the vitamin D receptor pathway in primary human
hepatocytes. J Biol Chem. 277:25125–25132. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mao Z, Luan X, Cao G, et al: DEC1 binding
to the proximal promoter of CYP3A4 ascribes to the downregulation
of CYP3A4 expression by IL-6 in primary human hepatocytes. Biochem
Pharmacol. 84:701–711. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tirona RG, Lee W, Leake BF, et al: The
orphan nuclear receptor HNF4alpha determines PXR-and CAR-mediated
xenobiotic induction of CYP3A4. Nat Med. 9:220–224. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsumura K, Saito T, Takahashi Y, et al:
Identification of a novel polymorphic enhancer of the human CYP3A4
gene. Mol Pharmacol. 65:326–334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jover R, Bort R, Gómez-Lechón MJ and
Castell JV: Down-regulation of human CYP3A4 by the inflammatory
signal interleukin-6: molecular mechanism and transcription factors
involved. FASEB J. 16:1799–1801. 2002.PubMed/NCBI
|
|
39
|
Kacevska M, Ivanov M, Wyss A, et al: DNA
methylation dynamics in the hepatic CYP3A4 gene promoter.
Biochimie. 94:2338–2344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Busi F and Cresteil T: CYP3A5 mRNA
degradation by nonsense mediated mRNA decay. Mol Pharmacol.
68:808–815. 2005.PubMed/NCBI
|
|
41
|
Hu YF, He J, Chen GL, et al:
CYP3A5*3 and CYP3A4*18 single nucleotide
polymorphisms in a Chinese population. Clin Chim Acta. 353:187–192.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chou FC, Tzeng SJ and Huang JD: Genetic
polymorphism of cytochrome P450 3A5 in Chinese. Drug Metab Dispos.
29:1205–1209. 2001.PubMed/NCBI
|
|
43
|
Jounaïdi Y, Guzelian PS, Maurel P and
Vilarem MJ: Sequence of the 5′-flanking region of CYP3A5:
comparative analysis with CYP3A4 and CYP3A7. Biochem Biophys Res
Commun. 205:1741–1747. 1994. View Article : Google Scholar
|
|
44
|
Zeigler-Johnson C, Friebel T, Walker AH,
et al: CYP3A4, CYP3A5, and CYP3A43 genotypes and haplotypes in the
etiology and severity of prostate cancer. Cancer Res. 64:8461–8467.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bajpai P, Tripathi AK and Agrawal D:
Genetic polymorphism of CYP3A5 in Indian chronic myeloid leukemia
patients. Mol Cell Biochem. 336:49–54. 2010. View Article : Google Scholar
|
|
46
|
Krusekopf S, Roots I and Kleeberg U:
Differential drug-induced mRNA expression of human CYP3A4 compared
to CYP3A5, CYP3A7 and CYP3A43. Eur J Pharmacol. 466:7–12. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lu C and Li AP: Species comparison in P450
induction: effects of dexamethasone, omeprazole, and rifampin on
P450 isoforms 1A and 3A in primary cultured hepatocytes from man,
Sprague-Dawley rat, minipig, and beagle dog. Chem Biol Interact.
134:271–281. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pascussi JM, Drocourt L, Gerbal-Chaloin S,
et al: Dual effect of dexamethasone on CYP3A4 gene expression in
human hepatocytes. Sequential role of glucocorticoid receptor and
pregnane X receptor. Eur J Biochem. 268:6346–6358. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roberts PJ, Rollins KD, Kashuba AD, et al:
The influence of CYP3A5 genotype on dexamethasone induction of
CYP3A activity in African Americans. Drug Metab Dispos.
36:1465–1469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pirmohamed M, James S, Meakin S, et al:
Adverse drug reactions as cause of admission to hospital:
prospective analysis of 18,820 patients. BMJ. 329:15–19. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao Y, Song M, Guan D, et al: Genetic
polymorphisms of CYP3A5 genes and concentration of the cyclosporine
and tacrolimus. Transplant Proc. 37:178–181. 2005. View Article : Google Scholar : PubMed/NCBI
|