Introduction
Macrophages in the body are known to exhibit the
effect of immune surveillance on tumor cells (TCs) (1), although it is now considered that
this effect occurs only in the early stages of tumor growth. A
large number of immune inflammatory cells, including macrophages,
do not provide immune surveillance effects in the aggressive stage.
They are therefore known as tumor-associated immune cells, and the
macrophage, as a notable member of the inflammatory cells, is known
as a tumor-associated macrophage (TAM) (2,3).
These cells are widely observed in numerous malignant tumors
(4–9). TAMs are recruited into the tumor by
inflammatory chemokines and play a key role in tumor proliferation
and progression (10–14). With respect to neural tumors,
Charles et al described in detail various cell/molecular
phenotypes of TAMs and other inflammatory cells in the brain tumor
microenvironment and their promoting effects of TC proliferation,
invasion and metastasis, although the study did not mention the
issue of canceration of the TAMs themselves (15). Bouvet et al used a liver
metastasis model of spleen-inoculated colon cancer cells, and
demonstrated the synergistic effects of spleen cells in the process
of colon cancer cell metastasis and colonization (16). It is well known that the spleen is
an innate organ enriched with immune cells, and that spleen cells
with original immune surveillance play a similar role to that of
TAMs, described above. However, whether spleen cells would
themselves develop canceration has again not been elucidated. We
were inspired by the study of Bouvet et al, who demonstrated
that the self-built double-color fluorescent tracer tumor model was
beneficial in the discovery of spleen cell-assisted colon cancer
metastasis and colonization, and clearly identified a correlation
between the TCs and the host. Therefore, we used a similar method,
establishing a double-color fluorescent tracer tumor model of red
fluorescent protein as a tracer for TCs and green fluorescent
protein for host cells (RFP/GFP) (17), and demonstrated that there were
always a number of continuous passaging GFP+ cells in
the transplanted tumor tissues. These tumor tissue cell suspensions
were then cultured in vitro, where the existence of
continuous passaging GFP+ cells was also noted. The
cells were identified as TAM cancerous cells originating from the
transplanted tumor tissues through a series of macrophage-related
and cancer phenotype tests, including cancer genetics and cell
biology tests.
Materials and methods
Establishment of tumor model
Lentiviral vectors (Genechem Chemical Technology
Co., Ltd., Shanghai, China) were used to transfect the RFP gene
into human SU3 glioma stem/progenitor cells (self-built in the lab)
(18), and SU3 cells were obtained
with high expression of RFP (SU3-RFP) screened by puromycin.
SU3-RFP cells were cultured in high-glucose Dulbecco’s modified
Eagle’s medium (DMEM; Gibco-Invitrogen, Life Technologies,
Carlsbad, CA, USA) containing 10% fetal bovine serum (Hyclone,
Logan, UT, USA), or in DMEM/F12 (Gibco-Invitrogen) containing 20
ng/ml recombinant human basic fibroblast growth factor (PeproTech,
Princeton, NJ, USA) and recombinant human epidermal growth factor
(PeproTech). SU3-RFP (150 μl) containing 1×106 cells was
injected with a micro-syringe directly into the abdominal cavity of
the NC-C57BL/6J-GFP nude mice, which were ~6 weeks old and
expressing GFP (19). The mice
were then bred in an IVC isolation device (Fengshi Laboratory
Animal Equipment Co., Ltd., Suzhou, China) according to specific
pathogen-free level management requirements. Approximately one
month later, when the abdominal circumference of the mice was
observed to have increased from the in vivo imaging system
(Carestream Health, Rochester, NY, USA), the mice were sacrificed,
the ascites were obtained, and an abdominal anatomical procedure
was performed to obtain the solid invasive-growing tumors.
Cryosectioning was also performed on the peritoneal tumors for
observation with a laser scanning confocal microscope (Carl Zeiss,
Oberkochen, Germany). This study was carried out in strict
accordance with the recommendations in the Guide for the Care and
Use of Laboratory Animals of the National Institutes of Health. The
animal use protocol was reviewed and approved by the Institutional
Animal Care and Use Committee (IACUC) of Soochow University.
Proliferative host cells cloned from
tumor model
Ascites were obtained from the tumor-bearing mice
and red blood cells were removed. Solid tumor tissue that had
invaded into the liver and gastrointestinal wall was obtained and
digested with trypsin into a single cell suspension, then the above
targets were subcultured and amplified in DMEM (Gibco) containing
10% fetal bovine serum (Hyclone). Then flow cytometry (Beckman
Coulter, Miami, FL, USA) was used to separate GFP+ cells
for continuous cultivation. The limiting dilution method and the
capillary method were performed for the monoclonal cell lines. Once
the amplification identified cells of single-cell origin, the cells
were frozen in liquid nitrogen for future use. Among these, the
cell lines originating from the solid tumor on the gastrointestinal
wall underwent further study and were named SU3-induced host celiac
tumor cells (SU3-ihCTCs).
Detection of characteristics of
SU3-ihCTCs grown in vitro
DMEM medium containing 10% fetal bovine serum was
used for the cultivation of SU3-ihCTCs, then the cell growth was
observed with an inverted fluorescence microscope (Carl Zeiss).
After developing the SU3-ihCTCs on slides, hematoxylin and eosin
(H&E) staining was performed and the cell morphology was
observed. A total of 1×103 cells (100 μl) were added
onto a 96-well plate, and the
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
method was used to draw the cell growth curve. To determine the
colony formation rate the cells in the logarithmic growth phase
were digested with 0.25% trypsin, seeded in six-well plates with
100 cells per well and incubated overnight at 37°C and with 5%
CO2, then the number of adherent cells was calculated.
After culturing for a further 6–8 days, cells were fixed with
methanol for 10 min and stained with crystal violet for 20 min,
then a microscope was used to determine colony counts (a colony was
defined as ≥50 cells grown together) for the final calculation of
the clone formation rate: clone formation rate = number of
colonies/number of seeded cells × 100. This was performed three
times for each well.
Molecular genetic testing of
SU3-ihCTCs
The cellular DNA content of SU3-ihCTCs in the
logarithmic growth phase was detected by flow cytometry. Following
the method of Seabright (20),
cell chromosome G-banding analysis was performed. A DNeasy blood
and tissue kit (Qiagen GmbH, Hilden, Germany) was used to extract
the cell or tissue DNA, and the cell species was identified using
the method reported by Parodi et al (21). The primers used for polymerase
chain reaction (PCR) amplification of the human-specific h-cox1
gene were 5′-TTCGGCGCATGAGCTGGAGTCC and 5′-TAT GCGGGGAAACGCCATATCG,
with a PCR product of 228 bp. The primers for the mouse-specific
m-cox1 gene were 5′-ATTACAGCCGTACTGCTCCTAT and 5′-CCCAAAGAA
TCAGAACAGATGC, with an amplified product of 150 bp. Western blot
analysis was performed to detect GFP expression. RIPA cell
disruption buffer (Millipore, Billerica, USA) was added to the
collected SU3-ihCTCs and SU3-RFP cells and mouse spleen tissue to
extract the total protein, and proteins were quantified using the
bicinchoninic acid method. Total protein (50 μg) was isolated by
12% SDS-PAGE electrophoresis and transferred to a PVDF membrane,
then reacted with rabbit anti-GFP antibody (1:2,000; Abcam, Hong
Kong, China) and mouse anti-GAPDH antibody (1:1,000; Sigma, St.
Louis, MO, USA), respectively. Following reaction with the
corresponding horseradish peroxidase-conjugated secondary antibody,
chemiluminescence detection and X-ray film developing were
performed. Immunocytochemistry was performed to detect the
expression of macrophage-specific marker protein CD68 with rat
anti-mouse CD68 monoclonal antibody (1:200; Abcam).
Tumorigenicity testing of SU3-ihCTCs and
RAW264.7 cells
SU3-ihCTCs and the murine macrophage cell line
RAW264.7 (Chinese Academy of Seed Cell Bank, Shanghai, China) were
inoculated into the abdominal cavity and right forelimb armpit of
nude mice at a concentration of 1×107 cells/150 μl, and
the tumorigenicity was observed. RAW264.7 belongs to the mouse
macrophage cell line established by Raschke et al (22), with established tumorigenicity, and
acted as a control of macrophage canceration in this
experiment.
Results
Transfecting effects of SU3-RFP
The RFP gene was stably transfected into SU3 cells
through lentiviral vectors for both differentiated adherent-growing
cells in serum culture conditions and suspended-growing stem
progenitor cells in growth-factor-containing culture conditions. A
red color was observed under the fluorescence microscope due to RFP
expression (Fig. 1).
Double-color fluorescent tracer effects
of RFP/GFP tumor model
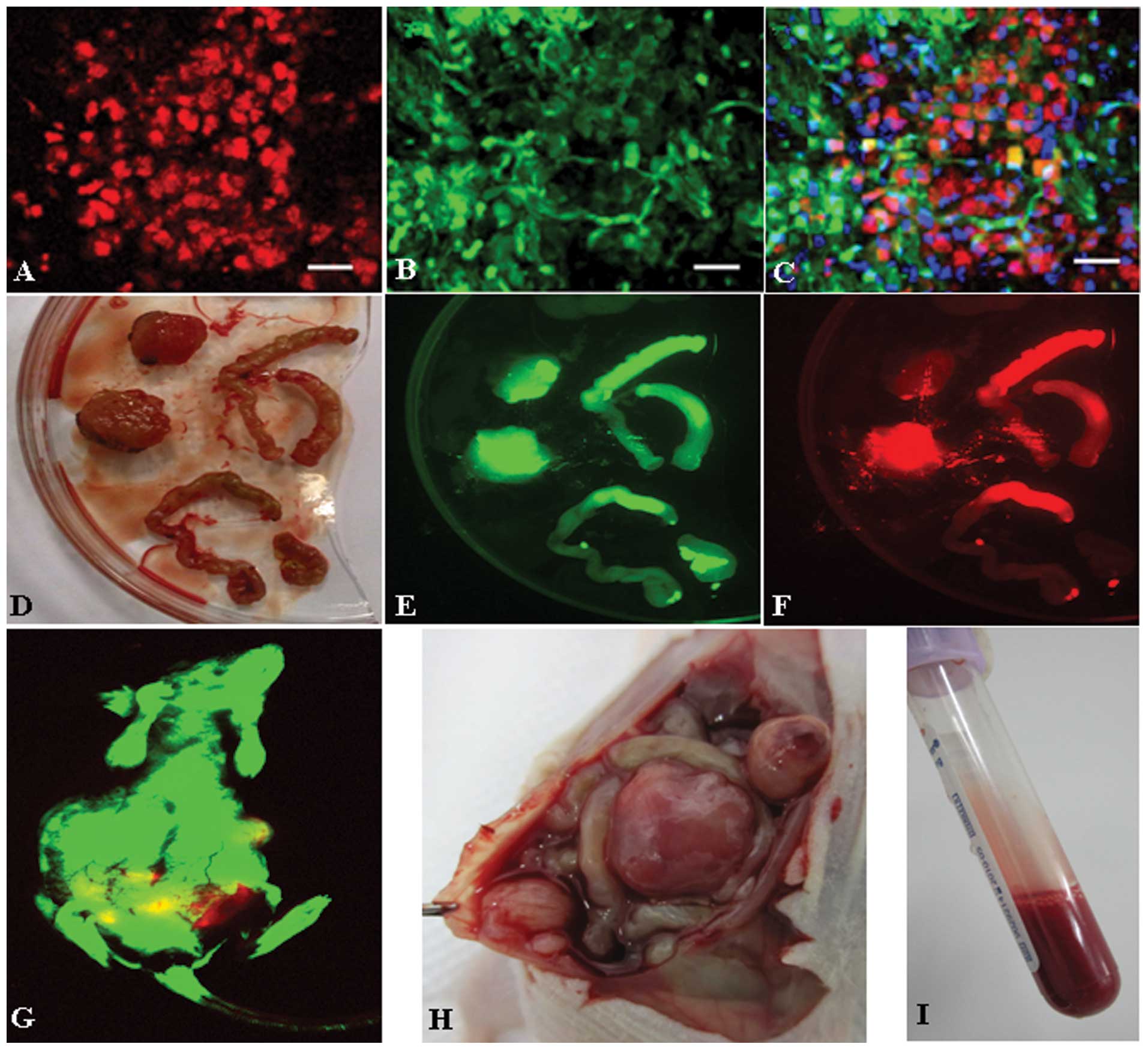
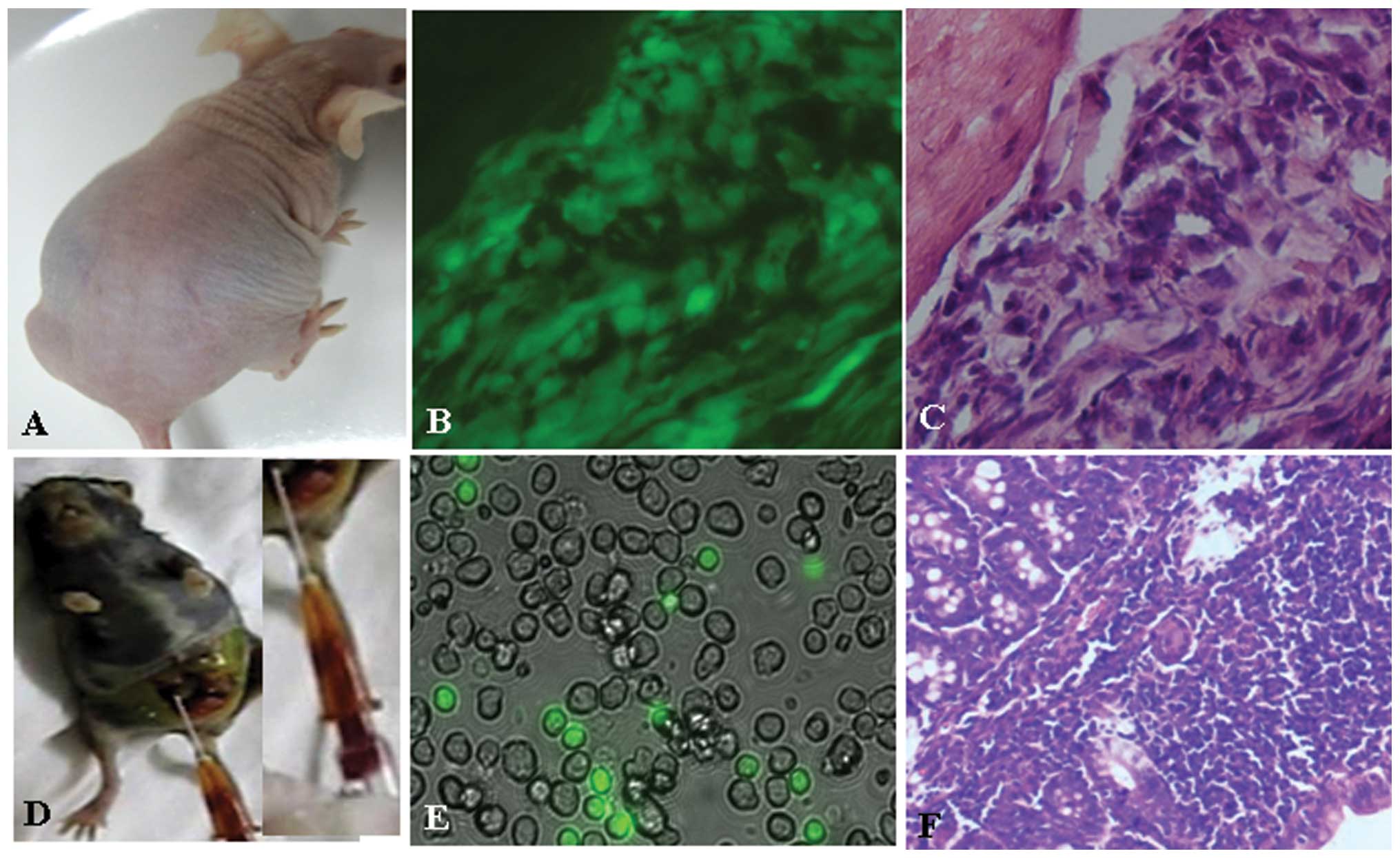
SU3-RFP cells gave rise to solid tumors (Fig. 2D and H) and malignant ascites
(Fig. 2I) in the abdominal cavity
of GFP nude mice. In the living tumor-bearing mice (Fig. 2G), in tumor nodules obtained
following sacrifice (Fig. 2D–F)
and in frozen sections of tumor tissues (Fig. 2A–C), observations revealed that the
tumor tissues were composed of the red SU3-RFP fluorescent
progenitor cells and the green fluorescent host cells.
Collection of GFP+ cells in
transplanted tumors
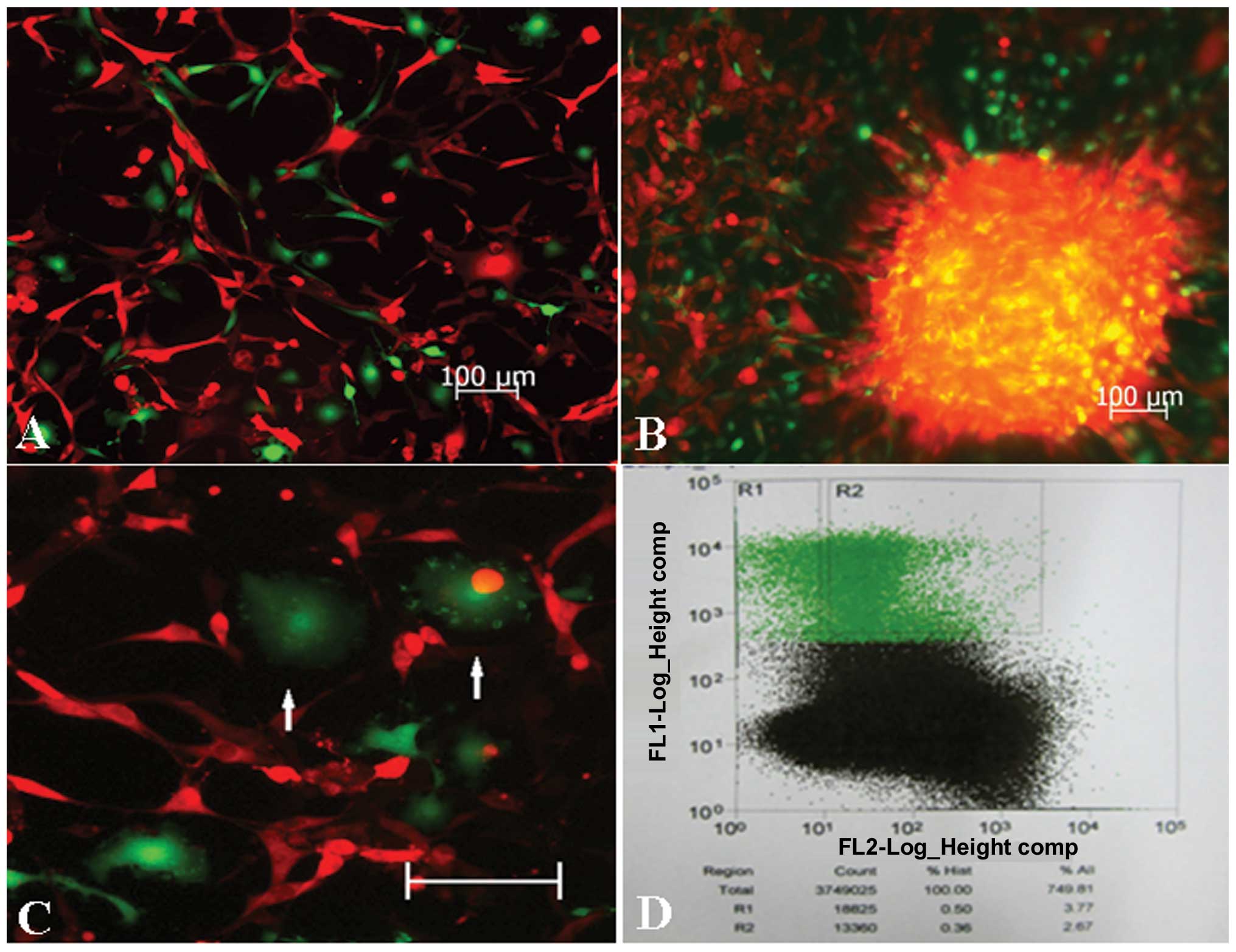
The bloody ascites of tumor-bearing mice or tumor
nodule tissues were cultured in serum-containing medium, and, while
few demonstrated proliferative activity, green fluorescent cells
were observed under the fluorescence microscope (Fig. 3A and B). In these green cells,
macrophages with a fluctuating membrane edge could be observed
(Fig. 3C), among which the
majority revealed red fluorescence. Following the digestion of the
tumor tissues, GFP+ host cells accounted for 3.77% of
the mixed cells in flow cytometry (Fig. 3D). The GFP+ cells were
then separated, enriched with flow cytometry, and the cells could
be subcultured continuously.
In vitro characteristics of
SU3-ihCTCs
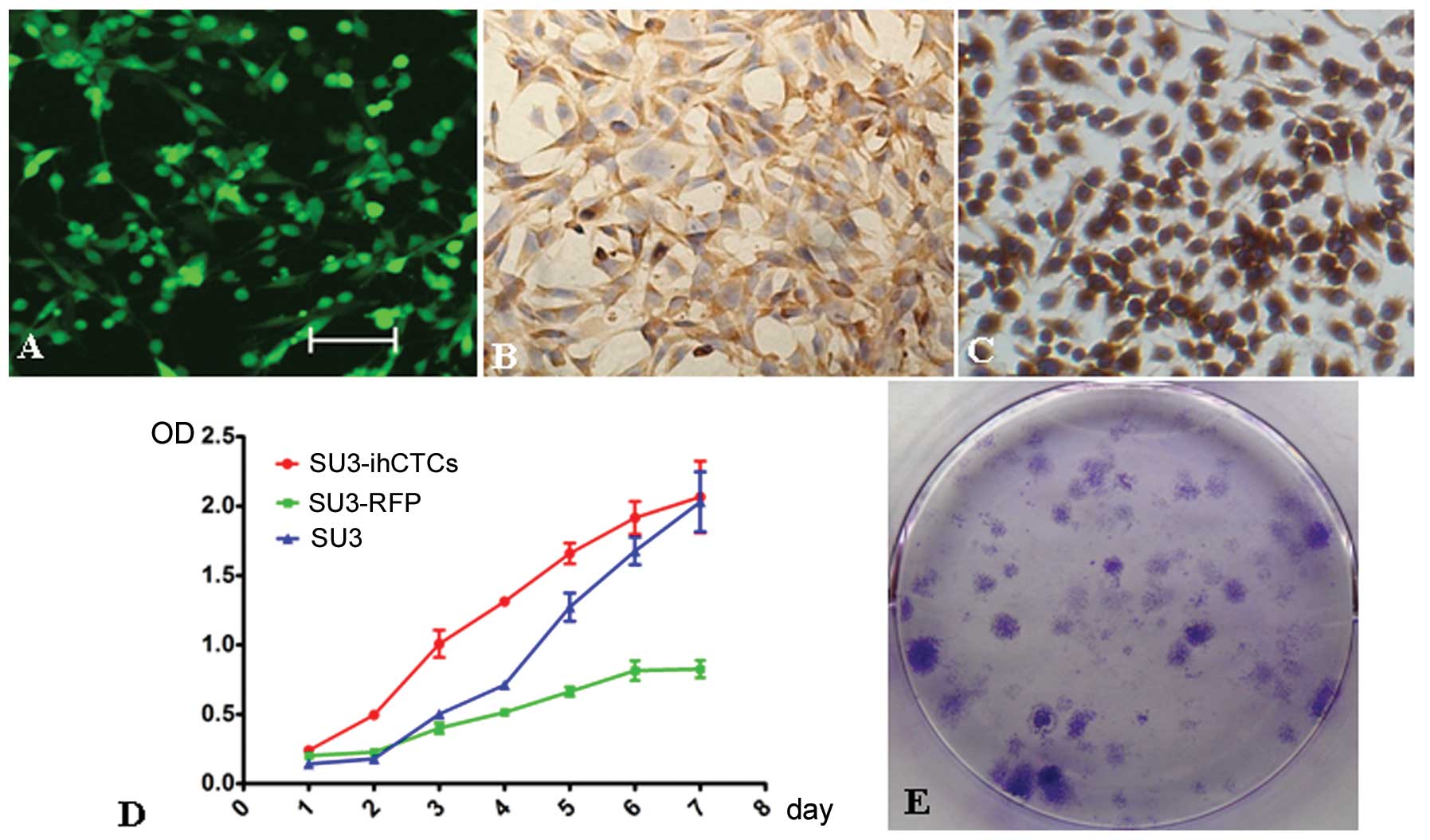
Monocloning was performed on the above collected
GFP+ cells, and unlimited proliferative capacity was
confirmed under in vitro cultivation. The progenitor cells
exhibited green fluorescence during subcultivation. SU3-ihCTCs were
pleomorphic, and cell-cell contact inhibition disappeared (Fig. 4A). The growth incubation period and
doubling time of SU3-ihCTCs were shorter than those of SU3 and
SU3-RFP cells; the cells quickly entered the logarithmic growth
phase, exhibiting rapid proliferation in the logarithmic growth
phase and a short cell growth cycle (Fig. 4D). The clone formation rates of
SU3-ihCTCs and SU3 cells were 57.0±4.4 and 7.3±1.0%, respectively,
revealing the capacity of proliferation and formation of a single
SU3-ihCTC to be stronger than that of SU3 cells, with low
population dependence (Fig. 4E).
SU3-ihCTCs highly expressed macrophage-specific marker protein CD68
in the same way as mouse macrophage cell line RAW264.7 (Fig. 4B and C).
Genetic characteristics of SU3-ihCTCs of
murine origin and heteroploidy
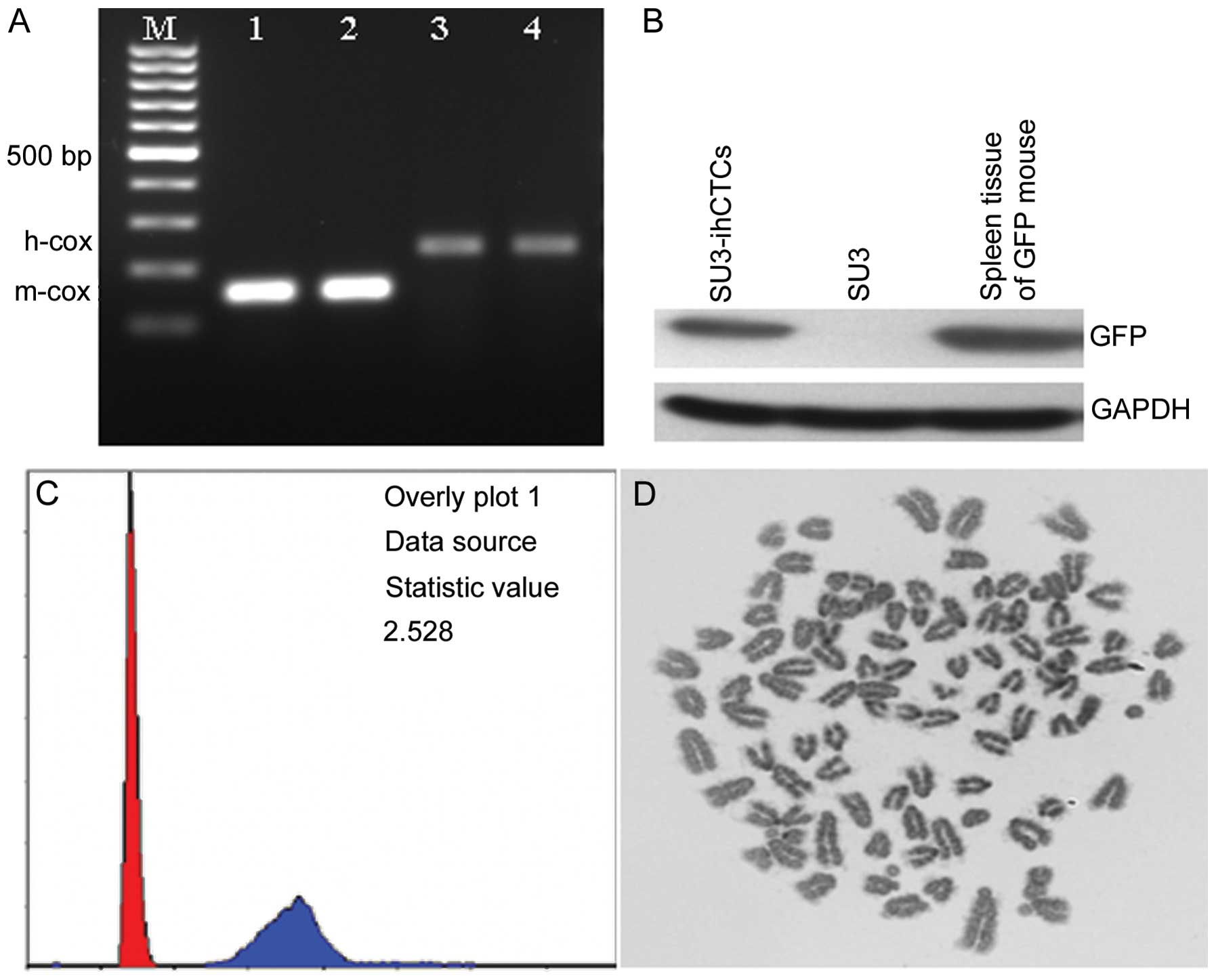
High cellular DNA synthesis and chromosome changes
are major features of TCs. Measured with flow cytometry, the DNA
synthesis capacity of SU3-ihCTCs was 2.528 times higher than that
of normal diploid mouse lymphocytes, from which it can be assumed
that the chromosome number of SU3-ihCTCs was 2.528 times higher
than that of normal mouse lymphocytes, and thus that SU3-ihCTCs
exceed pentaploidy (2.528×2; Fig.
5C). G-banding calculation was used to calculate the number of
chromosomes per cell. In the experiment, the cell chromosome
splitting phase was randomly extracted, and the chromosome number
was counted to be 92.70±7.15 (n=10) (Fig. 5D). PCR amplification was performed
on mouse- and human-specific primers, respectively. Only the mouse
cox1 gene was amplified from SU3-ihCTCs (150 bp), and GFP
expression was detected using western blot analysis, while the
cells revealed features including telocentric chromosomes (Fig. 5C). The above results proved that
SU3-ihCTCs were host-derived TCs.
High tumorigenicity of SU3-ihCTCs
In intraperitoneally or subcutaneously inoculated
nude mice, the tumorigenicity rate of SU3-ihCTCs was 100% (5/5);
ascites and tumor nodules were also formed when SU3-ihCTCs were
inoculated into the abdominal cavity (Fig. 6A). Since SU3-ihCTCs were
deliberately inoculated in no-fluorescence nude mice, the grown
TCs, either ascites or solid tumors, expressed GFP (Fig. 6B); therefore, it could be
determined that the TCs were daughter cells of SU3-ihCTCs. The
peritoneal solid tumors exhibited clear characteristics of
pleomorphism following H&E staining, with a dense arrangement
(Fig. 6C). RAW264.7 cells were
also inoculated into the abdominal cavity and subcutaneous part of
the GFP nude mice, with tumorigenicity rates of 5/5 and 6/6
(Fig. 6D). As the cells were not
transfected with fluorescence protein, there was a clear
distinction between the GFP− RAW264.7 daughter cells and
the GFP+ host cells in cultures of the bloody ascites
(Fig. 6E). The peritoneal tumor
nodules appeared mainly as densely arranged small round cells, and
fat vacuoles were also observed (Fig.
6F).
Discussion
During the progressive stage of malignant tumors,
the location of macrophages which play a key role in the innate
immune response system has aroused widespread debate in the field
of oncology (11–14). Charles et al (15) observed that macrophages not only
exist in malignant glioma, but also participate in the formation of
tumor vessels. This idea contradicts the widespread ‘tumor immune
escape’ theory. For tumor immune escape, the TCs escape from the
attack of immune cells, and continue to grow. This growth is caused
by TCs themselves, and does not involve immune cells. In addition,
this theory suggests that the immune cells attacking tumors are
differentiated functional cells. Once they enter the senescence or
apoptosis stage, the TC attacking ability is lost. The escape of
TCs from immune cell attack is due to the insufficient number of
immune cells. In contrast, our hypothesis is that immune cells
without attacking function do not fade or die, but continue to
proliferate under the induction of TCs. At the same time, certain
immune cells transform into cancer cells with unlimited
proliferative capacity in the same way as TCs. This has not been
noted in the study of Charles et al (15) or any other publication. It is a
novel discovery in a transplanted tumor model using an RFP/GFP
double-color fluorescent tracer (Fig.
2).
In this RFP/GFP double-color fluorescent tracer
model, the transplanted tumor tissue is cultivated, and one
immortalized cell line expressing GFP, named SU3-ihCTC, is cloned
(Fig. 4). It is difficult to
distinguish between the tumor and host in the traditional
non-fluorescence tracer model. At the same time, only the mouse
cox1 gene is amplified from SU3-ihCTCs. The DNA synthesis ability
detected by flow cytometry and the chromosome G-banding result
demonstrate that these cells are heteroploid TCs (Fig. 5) with high tumorigenicity (Fig. 6). Therefore, SU3-ihCTCs are
cancerous host cells. Considering the evidence-based medicine
level, this can be regarded as level-one evidence. However, the
type of host cells from which these cells originate still needs to
be confirmed. As observed from the transplanted tumor tissue
structure in the double-color fluorescent tracer model (Fig. 2), cells expressing GFP belong to
the stromal cell group, which are divided into two categories
(cells having parasitic TC tissue, and bone marrow-derived or
external circulating cells recruited by TCs) (23). As SU3-ihCTCs are from
abdomen-transplanted tumors, they are likely to be peritoneal
macrophage-derived cells. Considering that the cells express
macrophage marker protein CD68, it is likely that SU3-ihCTCs are
macrophage-derived cancer cells. This has been identified from the
level of cellular immune marker protein (24). According to a study by Mossor
(25), TAMs are divided into two
types: typical type (M1) and alternative type (M2). M2 macrophages
highly express CD163 and CD204, and their number is positively
correlated with the degree of tumor malignancy, playing a role in
promoting tumor growth. M1 macrophages are correlated with the
immune inflammatory response, demonstrating an anticancer effect.
Mora and Regnier-Vigouroux (26)
identified that, under induction of lipopolysaccharide, M1
macrophages secrete tumor necrosis factor and induce the apoptosis
of M2 cells. Wu et al (27)
and Waldron et al (28)
have preliminarily illustrated the molecular mechanism of the
transformation of M1 cells to M2 cells and proposed that the
transformation is associated with the activation of the STAT3 and
PI3K-Akt-mTOR signal transduction pathways. All of these should be
confirmed in future studies, in particular the identification of
the molecular phenotype and molecular regulation mechanism of
SU3-ihCTC and M2 macrophages.
Studies of malignant transformation of stromal cells
in human cancer xenografts in nude mice date back to the 1980s
(29–37). Goldenberg and Pavia (29) reported that, following in
vitro short-term subculture, a malignant transformation of host
stromal cells is observed in transplanted human cancer in nude
mice, but no similar phenomena have been observed in established
cell lines. It is speculated that this malignant transformation is
induced by a stromal component and not the TCs for inoculation in
human cancer tissue. In the present study, the SU3 cells have no
stromal component. They are cloned from human malignant glioma
tissue with high CD133/nestin expression. Following transplantation
into the brain, abdominal cavity and subcutaneous tissue in nude
mice, malignant-transformed mouse-derived stromal cells are
obtained (SU3-ihBTC, SU3-ihCTC and SU3-ihSTC monoclonal cells,
respectively). Therefore, we question the proposal of Goldenberg
and Pavia. A significant difference is that the SU3 cells used in
our study were stem progenitor cells of malignant glioma, and not
traditional TCs cultured in vitro, as used in the study of
Goldenberg and Pavia. We therefore speculate that in transplanted
tumors in nude mice, the initiation factor inducing transformation
of murine stromal cells reside in tumor stem progenitor cells for
inoculation, and not the original tumor stroma. Of course, this
speculation still needs to be confirmed by inoculation of different
types of cancer stem progenitor cells to nude mice, which will be
performed in the future.
It is naturally essential to clarify the promoter
factor inducing the malignant transformation of stromal cells in
tumors. Several factors have been considered in previous studies,
including horizontal transmission of malignancy by cell fusion
(38–42), exosomes (or microvesicles)
(43,44) or even sera (45,46).
However, more significant is the fact that, if the change in
stromal cells in the development of tumors in patients is
consistent with the transplanted tumor model, the results of this
study have added a new basis to the theory that malignant tumors
have heterogeneity (47), with
practical value in research on resistance to radiation and
chemotherapy of heterogeneous tumors (48). Whether the TCs are of monoclonal or
polyclonal origin is still unknown. Goldenberg and Pavia (29) support the latter. We are of the
opinion that the initiating cells of the transplanted tumor have
gone through the whole process from normal cell to cancer cell, and
there is no final conclusion on the origin of the TCs. Recently, Xu
et al (49) and Hou et
al (50) applied a single-cell
exome sequencing method and established that renal cell carcinoma
and leukemia cells are monoclonal-originated. This will be of great
assistance in our future research on this topic.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81172400, 81071766,
81272799, 81472739 and 81302196) and the Jiangsu Provincial Natural
Science Foundation (grant no. BK2011341 and BK2010227).
References
|
1
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fukuda K, Kobayashi A and Watabe K: The
role of tumor-associated macrophage in tumor progression. Front
Biosci (Schol Ed). 4:787–798. 2012. View
Article : Google Scholar
|
|
3
|
Galdiero MR, Garlanda C, Jaillon S, Marone
G and Mantovani A: Tumor associated macrophages and neutrophils in
tumor progression. J Cell Physiol. 228:1404–1412. 2013. View Article : Google Scholar
|
|
4
|
Lin EY, Li JF, Gnatovskiy L, et al:
Macrophages regulate the angiogenic switch in a mouse model of
breast cancer. Cancer Res. 66:11238–11246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurahara H, Takao S, Maemura K, et al:
M2-polarized tumor-associated macrophage infiltration of regional
lymph nodes is associated with nodal lymphangiogenesis and occult
nodal involvement in pN0 pancreatic cancer. Pancreas. 42:155–159.
2013. View Article : Google Scholar
|
|
6
|
Behnes CL, Bremmer F, Hemmerlein B, et al:
Tumor-associated macrophages are involved in tumor progression in
papillary renal cell carcinoma. Virchows Arch. 464:191–196. 2014.
View Article : Google Scholar
|
|
7
|
Shirabe K, Mano Y, Muto J, et al: Role of
tumor-associated macrophages in the progression of hepatocellular
carcinoma. Surg Today. 42:1–7. 2012. View Article : Google Scholar
|
|
8
|
Obeid E, Nanda R, Fu YX and Olopade OI:
The role of tumor-associated macrophages in breast cancer
progression (review). Int J Oncol. 43:5–12. 2013.PubMed/NCBI
|
|
9
|
Lee CH, Liu SY, Chou KC, et al:
Tumor-associated macrophages promote oral cancer progression
through activation of the Axl signaling pathway. Ann Surg Oncol.
21:1031–1037. 2014. View Article : Google Scholar
|
|
10
|
Balkwill FR: The chemokine system and
cancer. J Pathol. 226:148–157. 2012. View Article : Google Scholar
|
|
11
|
Pollard JW: Tumour-educated macrophages
promote tumour progression and metastasis. Nat Rev Cancer. 4:71–78.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Allavena P, Sica A, Solinas G, Porta C and
Mantovani A: The inflammatory micro-environment in tumor
progression: the role of tumor-associated macrophages. Crit Rev
Oncol Hematol. 66:1–9. 2008. View Article : Google Scholar
|
|
13
|
Solinas G, Germano G, Mantovani A and
Allavena P: Tumor-associated macrophages (TAM) as major players of
the cancer-related inflammation. J Leukoc Biol. 86:1065–1073. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pello OM and Andrés V: Role of c-MYC in
tumor-associated macrophages and cancer progression.
Oncoimmunology. 2:e229842013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Charles NA, Holland EC, Gilbertson R,
Glass R and Kettenmann H: The brain tumor microenvironment. Glia.
59:1169–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bouvet M, Tsuji K, Yang M, et al: In vivo
color-coded imaging of the interaction of colon cancer cells and
splenocytes in the formation of liver metastases. Cancer Res.
66:11293–11297. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong J, Dai XL, Lu ZH, et al: Incubation
and application of transgenic green fluorescence nude mice in
visualization studies on glioma tissue remodeling. Chin Med J
(Engl). 125:4349–4354. 2012.
|
|
18
|
Wan Y, Fei XF, Wang ZM, et al: Expression
of miRNA-125b in the new highly invasive glioma stem cell and
progenitor cell line SU3. Chin J Cancer. 31:207–214. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu ZH, Lv K, Zhang JS, et al:
Establishment of a green fluorescent protein tracing murine model
focused on the functions of host components in necrosis repair and
the niche of subcutaneously implanted glioma. Oncol Rep.
31:657–664. 2014.
|
|
20
|
Seabright M: A rapid banding technique for
human chromosomes. Lancet. 2:971–972. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parodi B, Aresu O, Bini D, et al: Species
identification and confirmation of human and animal cell lines: a
PCR-based method. Biotechniques. 32:432–434. 436438–440.
2002.PubMed/NCBI
|
|
22
|
Raschke WC, Baird S, Ralph P and Nakoinz
I: Functional macrophage cell lines transformed by Asbelson
leukemia virus. Cell. 15:261–267. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kerkar SP and Restifo NP: Cellular
constituents of immune escape within the tumor microenvironment.
Cancer Res. 72:3125–3130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shabo I and Svanvik J: Expression of
macrophage antigens by tumor cells. Adv Exp Med Biol. 714:141–150.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mossor DM: The many faces of macrophage
activation. J Leukoc Biol. 73:209–212. 2003. View Article : Google Scholar
|
|
26
|
Mora R and Regnier-Vigouroux A:
Autophagy-driven cell fate decision maker: activated microglia
induce specific death of glioma cells by a blockade of basal
autophagic flux and secondary apoptosis/necrosis. Autophagy.
5:419–421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu A, Wei J, Kong LY, et al: Glioma cancer
stem cells induce immunosuppressive macrophages/microglia. Neuro
Oncol. 12:1113–1125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Waldron JS, Yang I, Han S, et al:
Implications for immunotherapy of tumor-mediated T-cell apoptosis
associated with loss of the tumor suppressor PTEN in glioblastoma.
J Clin Neurosci. 17:1543–1547. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goldenberg DM and Pavia RA: Malignant
potential of murine stromal cells after transplantation of human
tumors into nude mice. Science. 212:65–67. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sparrow S, Jones M, Billington S and Stace
B: The in vivo malignant transformation of mouse fibroblasts in the
presence of human tumour xenografts. Br J Cancer. 53:793–797. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gupta V, Rajaraman S, Gadson P and
Costanzi JJ: Primary transfection as a mechanism for transformation
of host cells by human tumor cells implanted in nude mice. Cancer
Res. 47:5194–5201. 1987.PubMed/NCBI
|
|
32
|
Russell PJ, Brown J, Grimmond S, et al:
Tumour-induced host stromal-cell transformation: induction of mouse
spindle-cell fibrosarcoma not mediated by gene transfer. Int J
Cancer. 46:299–309. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gupta V, Rajaraman S and Eberle R:
Spontaneous induction of malignancy in mouse cells by a human small
cell lung cancer implanted in nude mice. Carcinogenesis.
11:713–722. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bryzgalov IP, Iudicheva TV, Galetskiĭ SA,
Solov’ev IuN and Revazova ES: Induction of stromal cell
transformation in xenografts of human colonic cancer. Biull Eksp
Biol Med. 113:399–402. 1992.(In Russian). View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cate CC, Belloni DR and Marin-Padilla M:
Acquisition and enhanced expression of the metastatic phenotype
following transfections of genomic mouse tumor DNA containing human
SCLC gene sequences. Clin Exp Metastasis. 13:203–217. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ozen M, Multani AS, Kuniyasu H, et al:
Specific histologic and cytogenetic evidence for in vivo malignant
transformation of murine host cells by three human prostate cancer
cell lines. Oncol Res. 9:433–438. 1997.PubMed/NCBI
|
|
37
|
Pathak S, Nemeth MA, Multani AS, et al:
Can cancer cells transform normal host cells into malignant cells?
Br J Cancer. 76:1134–1138. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sinkovics JG: Horizontal gene transfers
and cell fusions in microbiology, immunology and oncology (Review).
Int J Oncol. 35:441–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goldenberg DM, Gold DV, Loo M, et al:
Horizontal transmission of malignancy: in-vivo fusion of human
lymphomas with hamster stroma produces tumors retaining human genes
and lymphoid pathology. PLoS One. 8:e553242013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Goldenberg DM, Zagzag D, Heselmeyer-Haddad
KM, et al: Horizontal transmission and retention of malignancy, as
well as functional human genes, after spontaneous fusion of human
glioblastoma and hamster host cells in vivo. Int J Cancer.
131:49–58. 2012. View Article : Google Scholar :
|
|
41
|
Goldenberg DM: Horizontal transmission of
malignancy by cell-cell fusion. Expert Opin Biol Ther.
12:S133–S139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rappa G, Mercapide J and Lorico A:
Spontaneous formation of tumorigenic hybrids between breast cancer
and multipotent stromal cells is a source of tumor heterogeneity.
Am J Pathol. 180:2504–2515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Holmgren L, Bergsmedh A and Spetz AL:
Horizontal transfer of DNA by the uptake of apoptotic bodies. Vox
Sang. 83:305–306. 2002. View Article : Google Scholar
|
|
44
|
Yu F, Hsieh WS, Petersson F, et al:
Malignant cells derived from 3T3 fibroblast feeder layer in cell
culture for nasopharyngeal carcinoma. Exp Cell Res. 322:193–201.
2014. View Article : Google Scholar
|
|
45
|
García-Olmo DC, Domínguez C, García-Arranz
M, et al: Cell-free nucleic acids circulating in the plasma of
colorectal cancer patients induce the oncogenic transformation of
susceptible cultured cells. Cancer Res. 70:560–567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
García-Olmo DC, Picazo MG and García-Olmo
D: Transformation of non-tumor host cells during tumor progression:
theories and evidence. Expert Opin Biol Ther. 12(Suppl 1):
S199–S207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Durrett R, Foo J, Leder K, Mayberry J and
Michor F: Intratumor heterogeneity in evolutionary models of tumor
progression. Genetics. 188:461–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee AJ and Swanton C: Tumour heterogeneity
and drug resistance: personalising cancer medicine through
functional genomics. Biochem Pharmacol. 83:1013–1020. 2012.
View Article : Google Scholar
|
|
49
|
Xu X, Hou Y, Yin X, et al: Single-cell
exome sequencing reveals single-nucleotide mutation characteristics
of a kidney tumor. Cell. 148:886–895. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hou Y, Song L, Zhu P, et al: Single-cell
exome sequencing and monoclonal evolution of JAK2-negative
myeloproliferative neoplasm. Cell. 148:873–885. 2012. View Article : Google Scholar : PubMed/NCBI
|