Introduction
Breast cancer is the most common malignancy in
females and a critical cause of cancer-associated mortality, second
only to lung carcinoma (1).
Metastasis, predominantly in the lung, is a leading cause of breast
cancer mortality (2,3). Breast cancer metastasis is regulated
not only by intrinsic genetic changes in malignant cells, but also
by the tumor microenvironment, and emerging data have suggested a
role for systemic inflammation in cancer metastasis (3).
Lipopolysaccharide (LPS) is a major structural
component of the outer membrane of Gram-negative bacteria and is a
potent inducer of inflammation through the production of various
cytokines, growth factors and inflammatory mediators (4–7).
Previous studies have shown that LPS may induce systemic
inflammation and increase hepatic recruitment of cancer cells in
mice (5,8,9). In
addition, LPS-induced inflammation has been shown to increase the
growth of experimental metastases in a murine tumor model and
increase angiogenesis in vivo and in vitro (9). These findings were associated with
increased vascular endothelial growth factor (VEGF) expression,
vascular permeability and tumor cell invasion/migration (6,10,11).
Numerous studies have indicated that activation of Toll-like
receptor 4 signaling and the nuclear factor-κB pathway are involved
in increases of LPS-induced metastasis in each process, including
tumor cell adhesion and invasion (12,13).
LPS was previously reported to upregulate the expression of
metadherin, which has been shown to induce lung metastasis of 4T1
mammary tumor cells, and has been associated with tumor
angiogenesis, including the expression of VEGF and increased
microvessel density (14,15). These studies suggested that LPS may
promote angiogenesis and metastasis. However, the underlying
mechanisms remain elusive. The present study used breast cancer
cells to generate a metastasis model in LPS-induced mice in order
to investigate the underlying mechanisms by which LPS promotes
metastasis. Prostaglandin E2 (PGE2) is a metabolite of arachidonic
acid derived via the cyclooxygenase pathway. In previous studies,
PGE2 was found to play a major role in promoting tumor cell growth
and correlate with malignant cell invasion (16). The present study therefore also
evaluated the role of PGE2 in breast cancer metastasis.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM),
penicillin-streptomycin and 0.25% trypsin-0.02% EDTA were obtained
from HyClone Laboratories, Inc. (Logan, UT, USA); fetal bovine
serum (FBS) was purchased from Gibco Life Technologies (Carlsbad,
CA, USA); Matrigel™ was from BD Biosciences (San Jose, CA, USA);
VEGF was from PeproTech, Inc. (Rocky Hill, NJ, USA); and
prostaglandin E2 (PGE2), LPS and celecoxib were all purchased from
Sigma-Aldrich (St. Louis, MO, USA). AH6809 was obtained from Cayman
Chemical Company (Ann Arbor, MI, USA); and the prostaglandin E
receptor (EP) agonists (EP1 receptor agonist ONO-DI-004, EP2
receptor agonist ONO-AE1-259-01, EP3 receptor agonist ONO-AE-248
and EP4 receptor agonist ONO-AE1-329) were obtained from Ono
Pharmaceutical Co, Ltd. (Osaka, Japan). Monoclonal
anti-cyclooxygenase-2 (COX-2; 1:100) and monoclonal β-actin (1:100)
antibodies were purchased from Biosynthesis Biotechnology Co., Ltd.
(Beijing, China) and monoclonal anti-VEGF antibody (100 ng/ml;
1:100; R&D Systems Inc., Minneapolis, MN, USA); and rat
anti-mouse CD31 (1:100) antibody was from Santa Cruz Biotechnology
(Dallas, TX, USA).
Animals
A total of 50 female BALB/c mice, aged 6–8 weeks
(20–22 g), were obtained from the Animal Center of Shandong
University (Jinan, China) and equally assigned to five groups:
Phosphate-buffered saline (PBS) group, LPS group, PBS+4T1 group (PT
group), LPS+4T1 group (LT group) and LPS+4T1+celecoxib group (LTC
group). The mice were housed in five cages with a 12/12 h
light-dark cycle at 21–22°C. Food and water were available ad
libitum. Mice were anesthetized with an injection of a 1:1
mixture of Hypnorm (fentanyl citrate, fuanisone; VetaPharma Ltd,
Leeds, UK) and Midazolam (dormicum; Roche Diagnostics, Basel,
Switzerland) prior to all procedures and sacrificed by a lethal
dose of CO2, followed by cervical dislocation. Following
sacrification, serum was collected from the mice and stored at
−20°C until further use. The lungs were harvested and stored at
−80°C or fixed with 4% formalin. All of the animal experiments were
approved by the Institutional Animal Care and Use Committee at the
animal facility of Shandong University (Jinan, China).
LPS treatment
The mice in the LPS, LT and LTC groups received
intraperitoneal injection of 5 mg/kg LPS for three consecutive
days. This concentration of LPS was sufficiently low to ensure
survival of the mice. All of the mice were sacrificed four weeks
after the final injection of LPS. The lungs were harvested and
systemic inflammation was detected using hematoxylin and eosin
(H&E) staining (Biosynthesis Biotechnology Co., Ltd). Lungs
from the various groups of mice were dissected, fixed in 4%
paraformaldehyde (Beijing Biosynthesis Biotechnology Co., Ltd.,
Beijing, China) for 48h, embedded in paraffin, cut into
3-4-μm sections and stained with H&E.
CD31 immunofluorescence analysis
Mouse lung tissues were cut into 5-μm slices
using a YD-355 tissue slicing machine (Jihua Yidi Medical Appliance
Co., Ltd, Jinhua, China) and fixed in 3% paraformaldehyde
overnight, followed by treatment with proteinase K (20
μg/ml). Rat anti-mouse CD31 antibodies were used as primary
antibodies (1:50) and goat anti-rat Alex-555 Red (Invitrogen Life
Technologies) antibodies were used as the secondary antibodies
(1:100). Slides were examined under a confocal microscope (LSM510;
Carl Zeiss AG, Oberkochen, Germany). By scanning 10 sections (4–5
μm distances) of each sample, three dimensional images of
each tissue sample were assembled.
Experimental lung metastasis model
4T1 murine breast cancer cells, purchased from the
China Cell Culture Centre (Shanghai, China), were resuspended in
FBS-free DMEM (1×106 cells/ml), and 100 μl 4T1
cells were intravenously injected into each mouse of the PT, LT and
LTC groups through the tail vein. Following injection, the mice in
the LTC group received treatment with celecoxib (5 mg/kg/d by
intraperitoneal injection, once per day) and the other two groups
were treated with PBS for 4 weeks until sacrification. Lung
metastatic tumors were counted and a pathological examination was
performed.
Isolation of endothelial cells
Murine pulmonary endothelial cells (MPVECs) were
isolated from the lungs of normal 3–4 week old female BALB/c mice.
Once harvested, the lungs were rinsed in PBS and the visceral
pleura were stripped. The lungs were minced into 1.0×1.0×1.0
mm3 tissue blocks and cultured in FBS for 30 min. The
tissue blocks were then carefully placed in gelatin-coated flasks
(Biosynthesis Biotechnology Co., Ltd.) and cultured at 37°C in an
atmosphere containing 5% CO2 for 1 h. The flasks were
kept inverted to ensure that the tissue blocks were well adhered to
the bottom of the culture flasks. Subsequently, the flasks were
reverted and DMEM containing 10% FBS was added. The lung tissue
blocks were carefully removed after 60 h and the culture medium was
replaced every other day. Cellular morphology was fusiform or a
monolayered ‘cobblestone’ appearance, which is considered typical
for endothelial cells (14).
Endothelial cell culture
MPVECs were grown in 0.1% gelatin-coated culture
flasks with DMEM supplemented with 10% FBS and 100 U/ml
penicillin-streptomycin at 37°C in a humidified atmosphere
containing 5% CO2. The cells were passaged at a 1:3
ratio using 0.25% trypsin-EDTA once grown to 90% confluence.
Experiments were performed using subcultured cells between the
third and sixth in vitro passages. MPVECs were
serum-deprived for 2 h in FBS-free DMEM prior to treatment with
various doses of PGE2 (0, 10, 100 and 200 nM), AH6809 (10
μM) and EP receptor agonists (10 μM) for 24 h. The
culture supernatants were collected following centrifugation at
447.6 × g for 15 min and stored at −20°C until further use.
Cell proliferation assay
The proliferation rate of 4T1 cells was determined
using an MTT assay. The 4T1 cells were seeded in 96-well plates at
a density of 1×105 cells/ml and treated with LPS once
grown to 90% confluence. Following incubation with various
concentrations of LPS (0, 10, 100 μg/ml) for 24 h or with
100 μg/ml LPS for various durations (0, 24, 48, 96 and 120
h), 20 μl MTT (5 mg/ml; HyClone, Logan, UT, USA) was added
to the wells and the plates were incubated at 37°C for an
additional 4 h. The reaction was terminated by lysing the cells
with 200 μl dimethyl sulfoxide (HyClone) for 20 min, with
agitation. The optical density (OD) of the cells was measured at
590 nm (ELx800; BioTek Instruments, Inc., Winooski, VT, USA) and
the proliferation rate of the 4T1 cells was calculated. All
experiments were repeated three times.
Measurement of cytokines by ELISA
VEGF and PGE2 levels in the animal serum samples,
and supernatants of the treated monolayer MPVECs were quantified
using VEGF and PGE2 ELISA kits (R&D Systems, Inc.). All
experiments were performed in accordance with the manufacturer’s
instructions. Standards and samples, in duplicate, were added to
96-well plates pre-coated with monoclonal antibody and incubated at
room temperature for 2 h. The plates were then washed three times,
100 ng/ml biotinylated antibody was added and the plates were
incubated for another 2 h at room temperature, followed by
incubation with streptavidin-horseradish peroxidase for 20 min.
Bound horseradish peroxidase was detected using
tetramethylbenzidine, following the final wash with PBS. The
reaction was terminated with stop solution and the OD was measured
using a Dynatech MR5000 microplate reader at 450 nm in order to
quantify cytokine production.
Western blot analysis
Western blotting was performed to examine the
protein expression levels of COX-2 and VEGF in treated MPVECs, as
well as in the lungs of treated mice. The cells were treated with
various concentrations of PGE2 in six-well plates, washed with PBS
and lysed on ice with 80 μl/well lysis buffer (HyClone) for
30 min. The lysates were clarified by centrifugation at 15,000 × g
for 30 min at 4°C. The mouse lungs were lysed with
radioimmunoprecipitation assay buffer (HyClone) and the protein
samples from each group were collected and quantified using a
Bicinchoninic Acid Quantification kit (HyClone), according to the
manufacturer’s instructions. Equal amounts of protein (40
μg) were added to the loading buffer (HyClone), boiled and
separated by 10% SDS-PAGE (BD Biosciences), prior to being
electrophoretically transferred to polyvinylidene fluoride
membranes. Following blocking with 5% skimmed milk, the membranes
were incubated with anti-COX-2 antibody (1:1,000 dilution) or
anti-VEGF antibody (1:300 dilution) overnight at 4°C, followed by
incubation with anti-rabbit secondary antibody for 1 h at room
temperature. The blots were then exposed to enhanced
chemiluminescence (ECL) film. The ECL kit, reagents and apparatus
were purchased from Biosynthesis Biotechnology Co., Ltd.
Chemiluminescence was detected using EpiChemi3 Darkroom imaging
system and LabWorks densitometry software, version 4.6 (both from
UVP Bioimaging, Upland, CA, USA). Data were corrected for
background signal and normalized to positive controls using RayBio
Analysis Tool software (UVP Bioimaging).
MPVEC endothelial tube formation
assay
A tube formation assay was used to determine the
effects of celecoxib, PGE2 and VEGF on angiogenesis of MPVECs.
Matrigel™ (80 μl) was used to coat 24-well plates, which
were then incubated at 37°C for 30 min, for gelatinization.
Serum-deprived MPVECs were resuspended in FBS-free DMEM
(1×106 cells/ml) and 600 μl cell suspension was
seeded into each well. Following 1 h incubation, the MPVECs were
treated with VEGF (100 nM) and PGE2 (100 nM), alone or subsequently
with celecoxib (10 μM). Culture plates were then incubated
for 8 h and images of the tubules and branch points were captured.
The number of tubular structures was counted at 10x magnification
in five randomly selected fields using a Zeiss Confocal LSM510
microscope (Carl Zeiss AG). The experiments were repeated three
times.
Statistical analysis
Values are expressed as the mean ± standard
deviation. One way analysis of variance was followed by the
Bonferroni test for post hoc analysis to evaluate
statistical significance. Data were analyzed using JMP version 6
(SAS Institute, Cary, NC, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
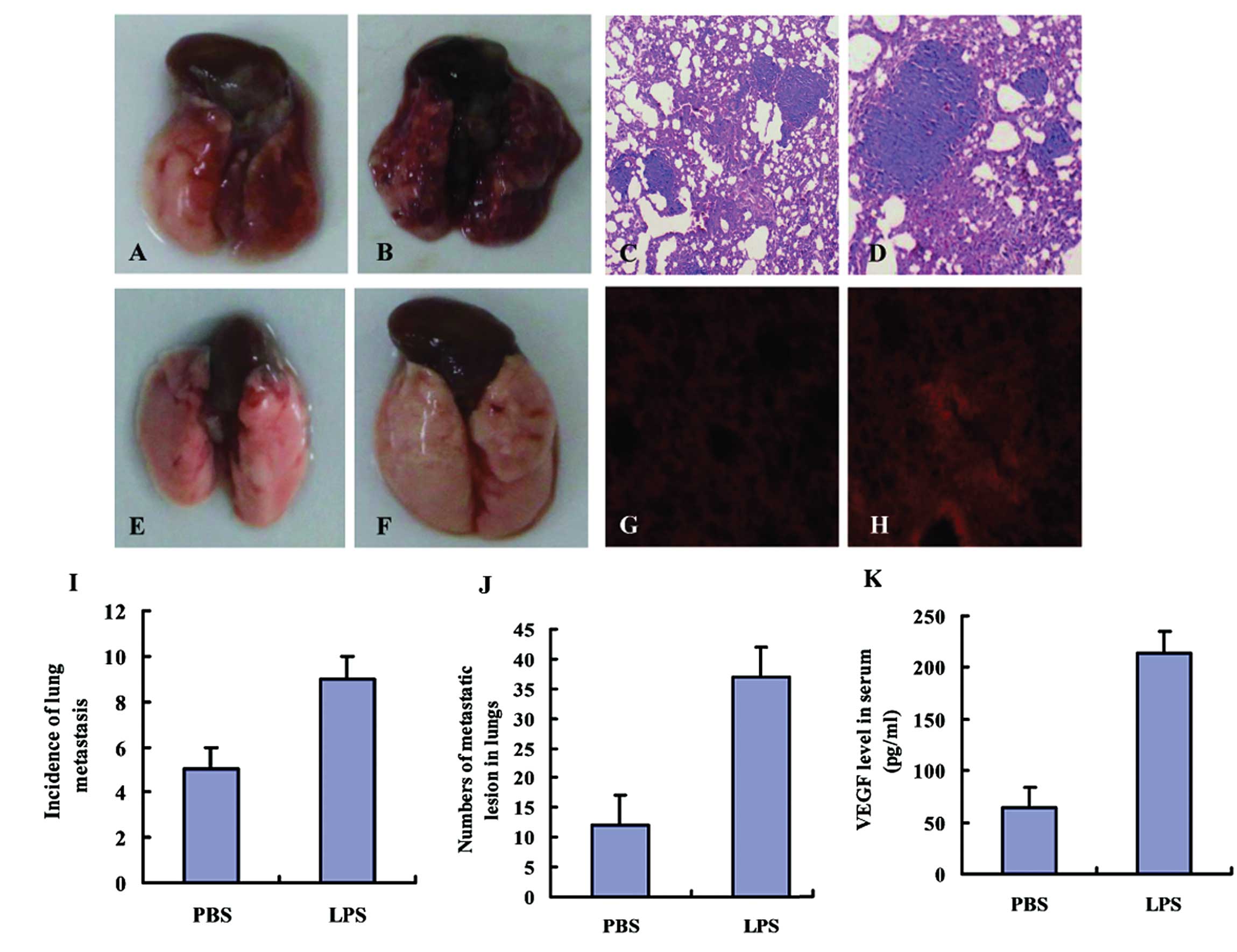
LPS promotes lung metastasis of 4T1
cells
To investigate the effects of LPS on lung
metastasis, 4T1 cells were intravenously injected into LPS-treated
and PBS-treated mice through the tail vein. The mice were
sacrificed after 14 days and a significant increase in the
incidence of lung metastasis was observed in the mice of the LT
group (9/11), as compared with those in the PT group (5/11). Lung
metastatic lesions were visualized by H&E staining. The number
and size of metastatic lesions in LPS-treated mice was
significantly greater as compared with that in PBS-treated mice
(Fig. 1A–D).
LPS induces the production of VEGF and
angiogenesis in lungs
The lungs of the LPS-treated mice were edematous and
enlarged as compared with the lungs of those treated with PBS
(Fig. 1E and F). The blood vessels
in the lungs of LPS-treated mice appeared as primitive and dilated
sinusoidal vascular structures, which consisted of disorganized,
tortuous and interconnected vascular plexuses (Fig. 1G and H), as determined by CD31
immunofluorescence. Quantification analysis indicated that the
total vessel density in the lungs was markedly increased in the
LPS-treated mice, as compared with that in the lungs of PBS-treated
mice (data not shown). The concentration of VEGF in the serum of
the mice was measured by ELISA and was significantly higher in the
LPS-treated group compared with that in the PBS-treated group
(214.3 pg/ml vs. 64.1 pg/ml). Differences between the LPS and PBS
groups were statistically significant (P<0.05; Fig. 1I-K).
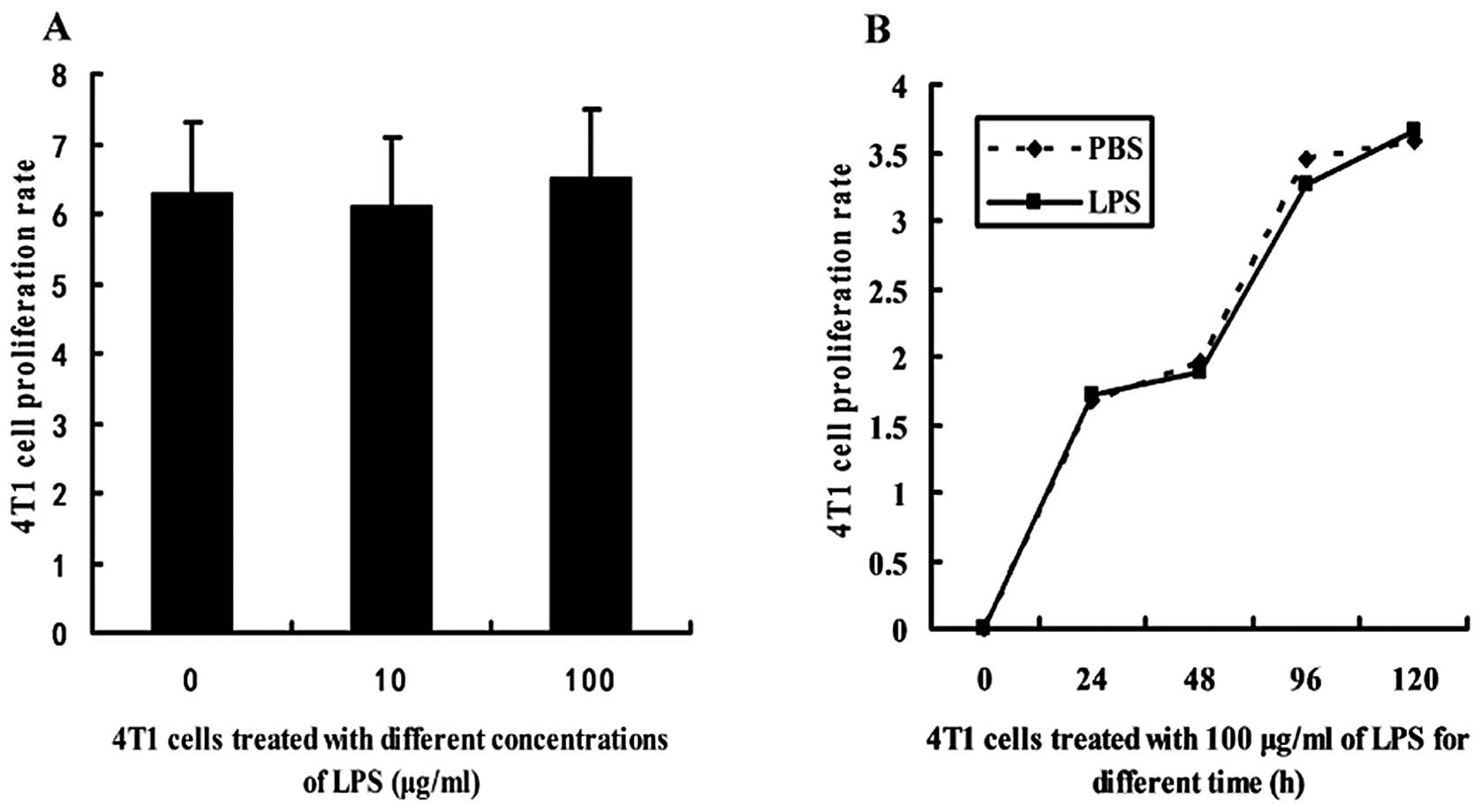
LPS does not influence the proliferation
of 4T1 cells
4T1 cells were treated with LPS and the
proliferation rate was determined using an MTT assay. There was no
significant difference between the proliferative rate of 4T1 cells
treated with various concentrations of LPS (10 and 100
μg/ml) and that of the PBS-treated cells (Fig. 2A). In addition, the proliferative
rate of the 4T1 cells treated with 100 μg/ml of LPS for
various durations (0, 24, 48, 96 and 120 h) was not significantly
altered (Fig. 2B).
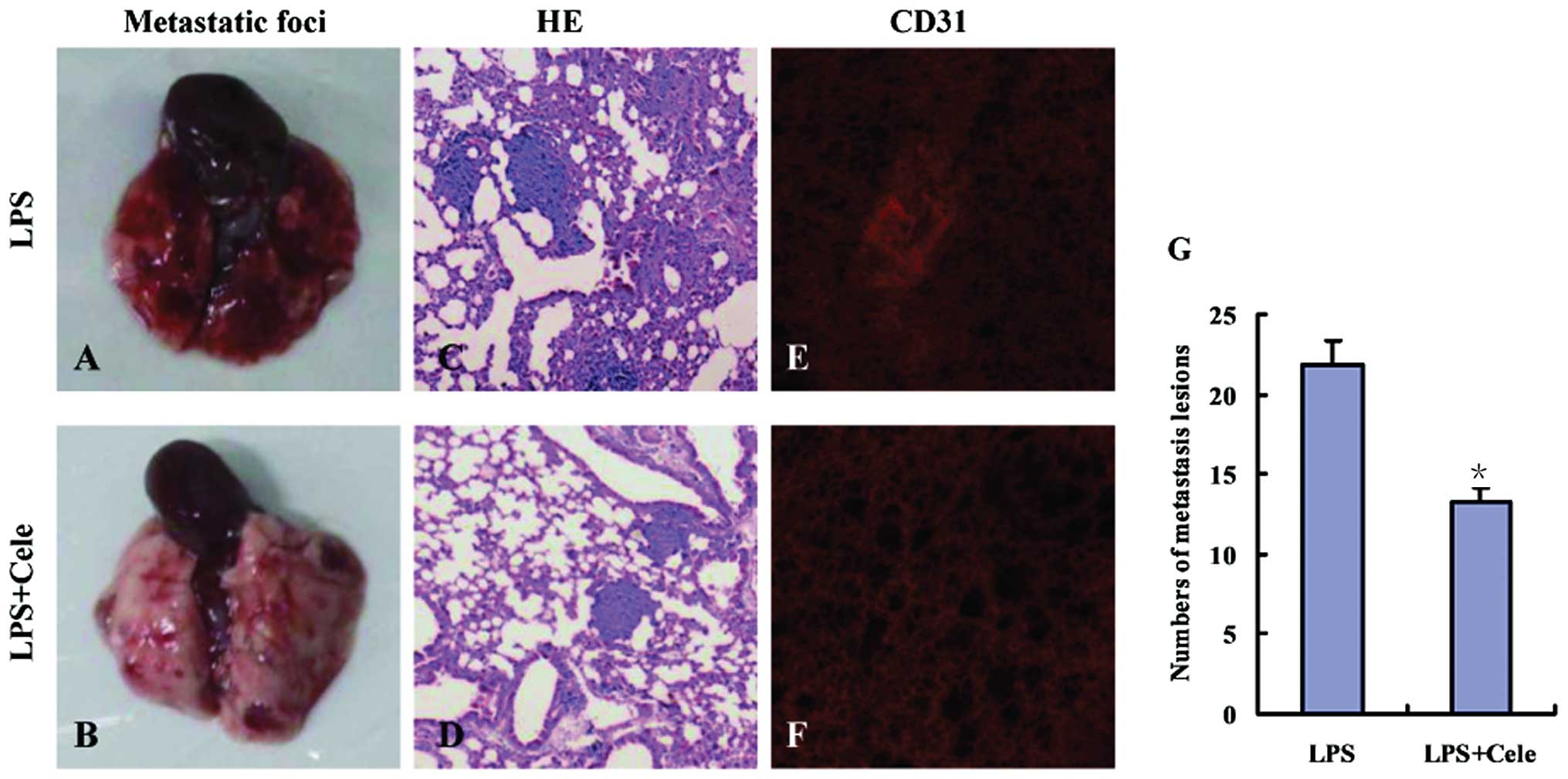
Celecoxib reduces metastasis and
angiogenesis in LPS-treated mice
Anti-COX-2 treatment of the mice in the LT group was
performed by intraperitoneal injection of celecoxib (5 mg/kg/d,
once per day), from the final injection of LPS until the mice were
sacrificed. Celecoxib significantly reduced lung metastasis in the
tumor-bearing mice (Fig. 3). Laser
scanning confocal microscopy detected reduced numbers of
CD31-positive cells (red fluorescence) in the lungs of
celecoxib-treated LTC mice as compared with those in the mice in
the LT group (Fig. 3E and F).
Celecoxib reduces LPS-induced production
of VEGF
To verify the effects of PGE2 in systemic
inflammation-induced metastasis, the circulating levels of PGE2 and
VEGF in the LPS- and celecoxib-treated mice were examined by ELISA.
The concentration of PGE2 in the serum of LPS-treated mice was
significantly decreased in response to treatment with celecoxib, a
selective COX-2 inhibitor. These results were concordant with the
levels of VEGF in the serum of LPS- and celecoxib-treated mice
(Fig. 4A). The release of VEGF by
MPVECs into monolayer culture medium was evaluated by ELISA in
order to determine whether PGE2 affected the production of VEGF
in vitro. Following incubation of the cells with various
concentrations of PGE2 (0, 10, 100, 200 nM) for 24 h, the
production of VEGF was significantly increased in a
concentration-dependent manner (Fig.
4C). Furthermore, the production of VEGF was increased almost
linearly from the first (4 h) to the last time-point assessed (48
h) in response to culturing with PGE2 (100 nM) (Fig. 4D). Western blot analysis showed
that protein expression levels of VEGF in the PGE2-treated cells
were increased in a concentration-dependent manner in PGE2-treated
MPVECs as compared with those in the control cells (Fig. 4E).
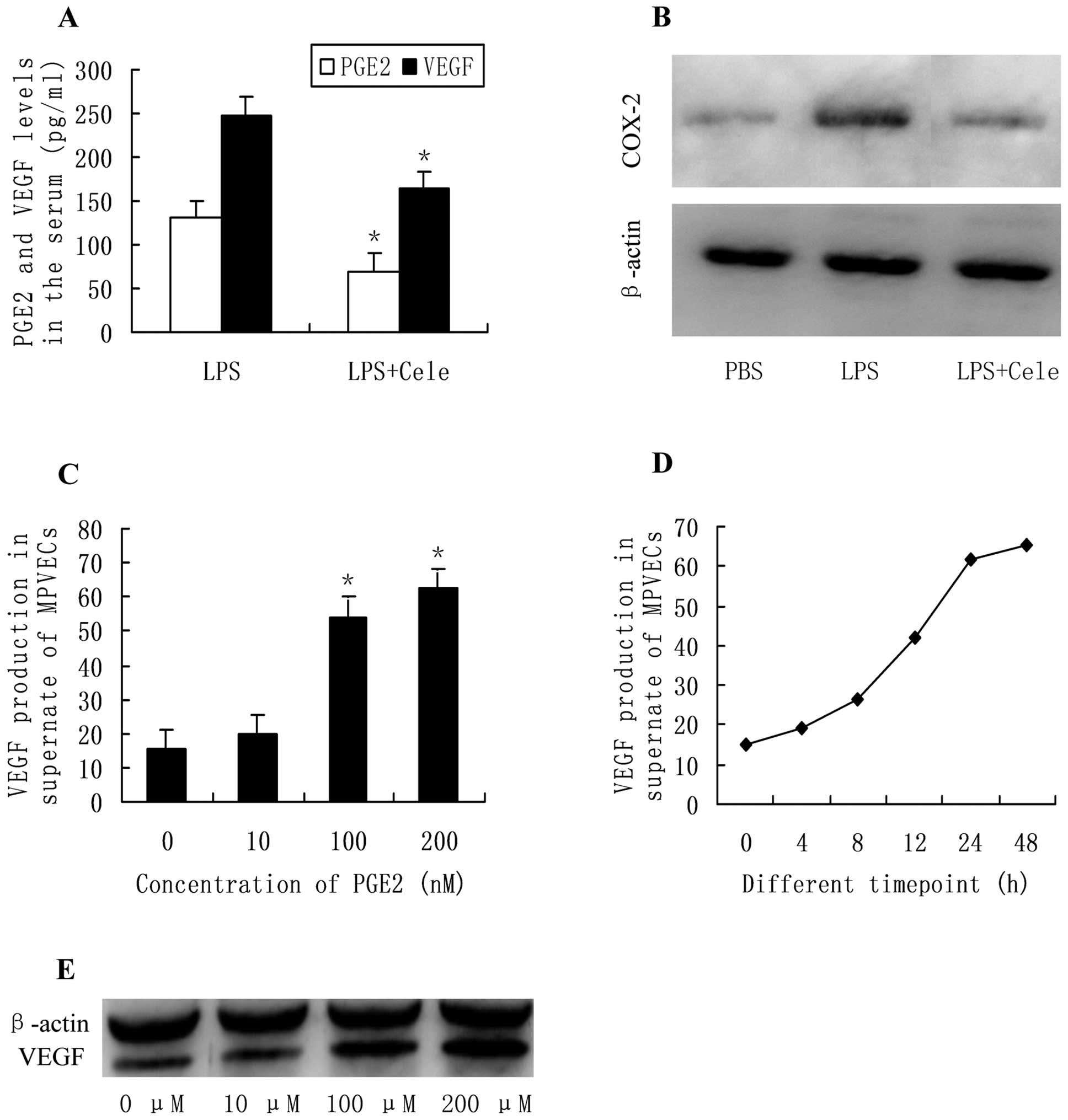 | Figure 4(A) PGE2 and VEGF concentration
(pg/ml) in the serum of lipopolysaccharide (LPS)-treated
tumor-bearing mice and celecoxib-treated mice
(*P<0.05); (B) COX-2 protein expression levels in the
lungs, quantified by western blotting; (C) VEGF production in the
supernatant of MPVECs treated with various concentrations of PGE2
(0, 10, 100 and 200 nM) for 24 h (*P<0.05); (D) VEGF
production in the supernatant of MPVECs treated with PGE2 (100 nM)
for various durations (0, 4, 8, 12, 24 and 48 h); (E) VEGF protein
expression levels in MPVECs treated with PGE2, as determined by
western blotting. PBS, phosphate-buffered saline; PGE2,
prostaglandin E2; COX-2, cyclooxygenase-2; MPVECs, murine pulmonary
endothelial cells; VEGF, vascular endothelial growth factor. |
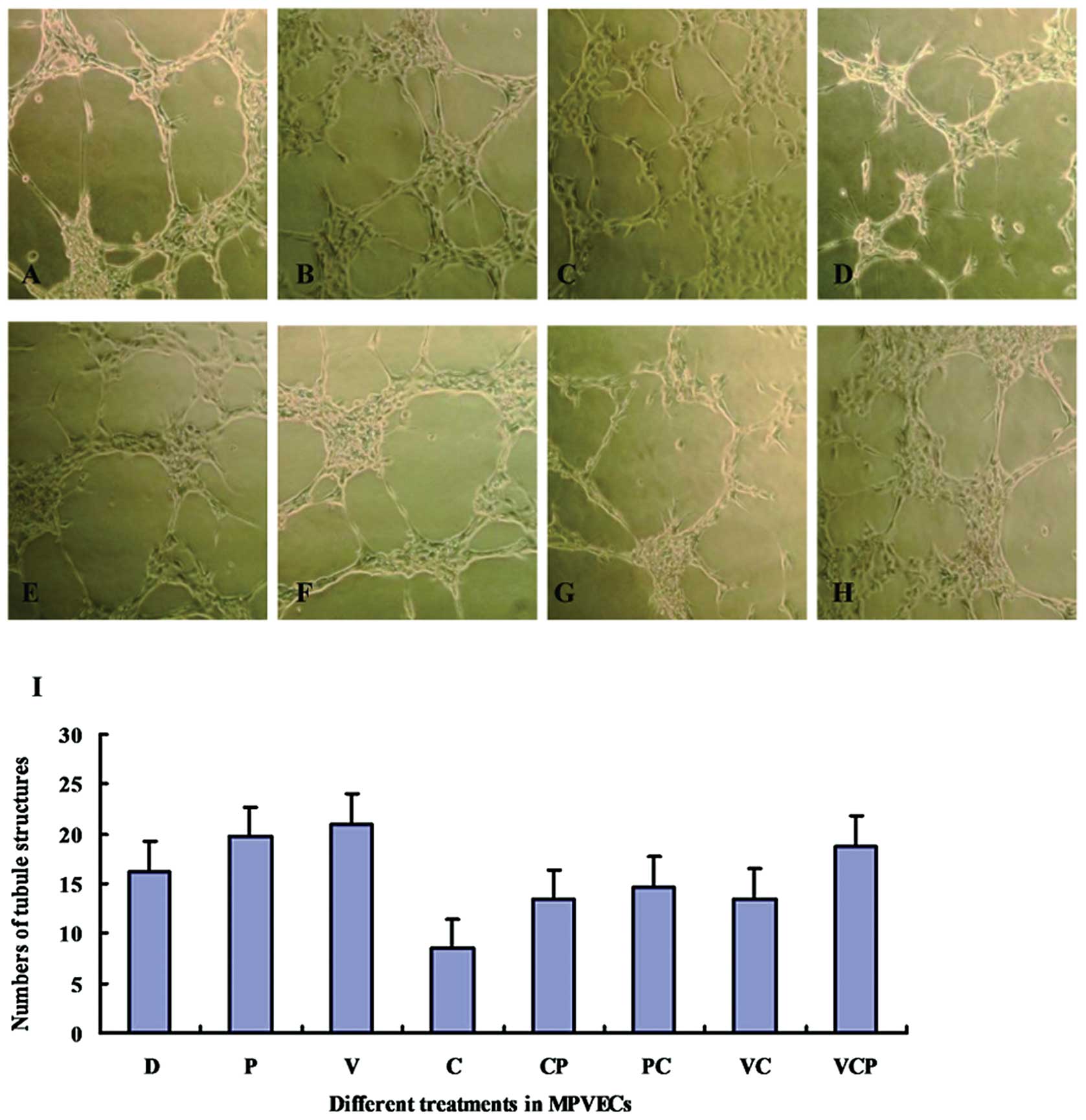
Treatment with celecoxib inhibits
VEGF-induced angiogenesis in vitro
Treatment with PGE2 or VEGF alone promoted tube
formation by MPVECs on Matrigel™, whereas treatment with celecoxib
reduced the number of branch points and the total tube length.
Treatment with PGE2 also attenuated the celecoxib-induced
inhibition of tube formation. However, simultaneous treatment with
celecoxib reduced tube formation in the VEGF-treated MPVECs, as
compared with that in the control group (Fig. 5).
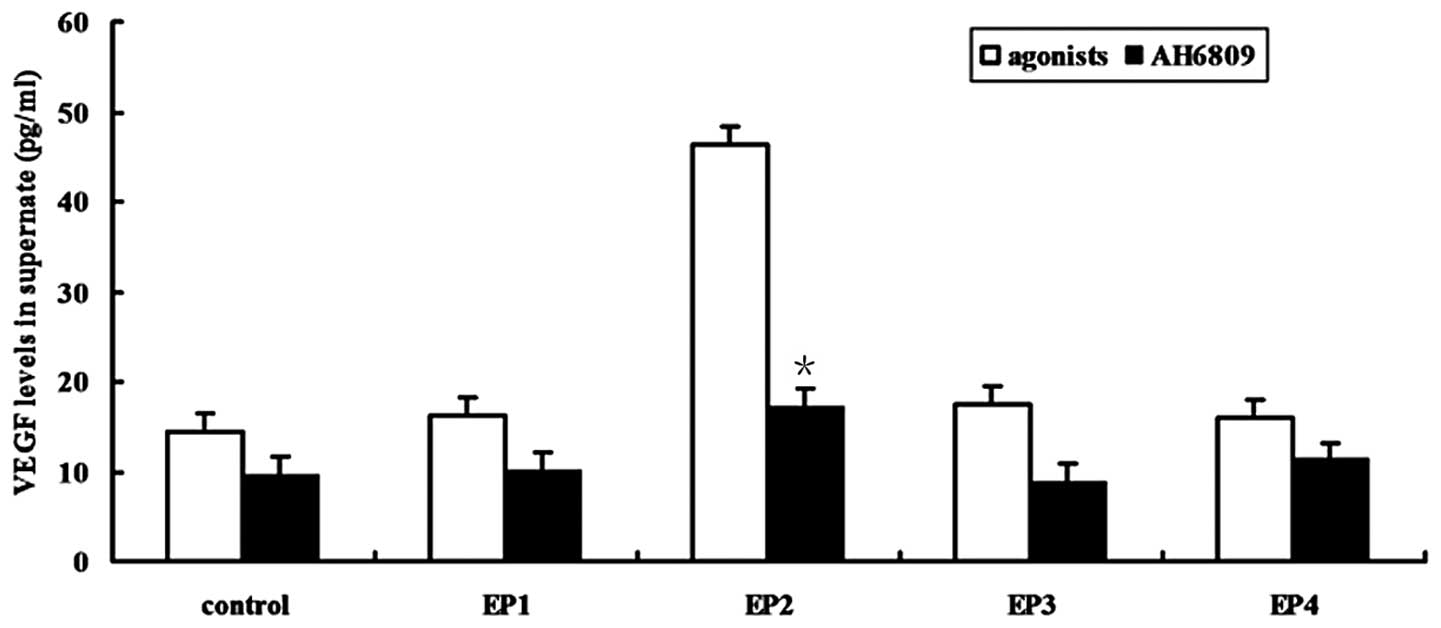
LPS induces the production of VEGF via
the PGE2-EP2 pathway
Four specific EP receptor agonists were used to
stimulate MPVECs and the resulting release of VEGF into the
monolayer culture medium was quantified by ELISA. Following
incubation for 24 h, the EP2 receptor agonist ONO-AE1-259-01 (10
μM) significantly increased the production of VEGF, which
was significantly alleviated to levels similar to those in the
control group in response to treatment with the EP2 receptor
antagonist AH6809 (10 μM). However, EP1 receptor agonist
ONO-DI-004, EP3 receptor agonist ONO-AE-248 and EP4 receptor
agonist ONO-AE1-329 had no significant effect on the production of
VEGF (P>0.05; Fig. 6).
Discussion
The results of the present study demonstrated that
the incidence of lung metastasis and the number of metastatic
lesions was significantly increased in the lungs of LPS-treated
mice as compared with those in PBS-treated mice. It may therefore
be suggested that inflammation induced by LPS increases metastasis.
Compared with the PBS-treated mice, the vessel density in the lungs
of the LPS-treated mice was markedly increased, thus suggesting
that an inflammatory milieu alongside angiogenesis may be
responsible for attracting the 4T1 metastatic cells to the lung
microenvironment. Inflammation may instigate cancer initiation and
progression through increasing levels of pro-inflammatory
mediators, including cytokines, chemokines and transcription
factors that mediate tumor cell proliferation, metastasis, invasion
and angiogenesis (17–19). To investigate the possible
mechanisms by which LPS enhances lung metastasis, the proliferative
activity of 4T1 cells was determined, with or without LPS
treatment. No significant difference was observed in the rate of
cellular proliferation following treatment with LPS at various
concentrations and time-points. This excluded the possibility of
LPS inducing metastasis through altering the proliferation of
cancer cells.
The present study showed that the concentration of
VEGF in the serum was markedly increased in LPS-treated mice as
compared with that in PBS-treated mice. This suggested that
LPS-induced angiogenesis and metastasis in the lungs may be
associated with a high expression of VEGF in the inflammatory mouse
model. VEGF was initially identified as a vascular permeability
factor in the supernatant of a tumor cell line (20,21)
and was subsequently reported as a growth factor with mitogenic
effects on vascular endothelial cells (22,23).
VEGF and its receptors are involved in tumor formation and distant
metastasis (24). VEGF promotes
endothelial cell survival and regulates migration, permeability and
proliferation of blood vessels (25,26),
which is critical in tumor angiogenesis and metastasis (27).
PGE2 is a metabolite of arachidonic acid derived
from the COX pathway (28,29). Sakurai et al (30) previously reported that activation
of COX-2 stimulated angiogenesis in the ovarian corpusluteum, and
the effect of PGE2 was shown to overcome the inhibition of
angiogenesis by COX-2 inhibitors (16,31).
Furthermore, VEGF has been shown to enhance the production of PGE2
by stimulating COX-2 and membrane-associated PGE synthase-1
expression, and PGE2 concurrently induced VEGF expression in rat
ovarian luteal cells (32).
Nakanishi et al (27)
reported that PGE2 stimulated the release of VEGF from human lung
fibroblasts. It was therefore hypothesized that PGE2 may exert a
similar effect and serve as a mediator in the release of VEGF by
pulmonary endothelial cells. The present study showed that the
differences in the serous levels of VEGF between the PBS, LPS and
celecoxib-treated groups were concordant with PGE2 levels and COX-2
expression. In vitro, PGE2 treatment increased the
production of VEGF in MPVECs in a concentration-dependent manner.
Therefore, the results of the present study verified that PGE2
promotes the expression of VEGF in lung endothelial cells.
Angiogenesis is a process whereby new blood vessels
grow from existing vessels (33,34),
which is an important function of endothelial cells. PGE2 has
previously been reported to mediate tumor angiogenesis through
numerous mechanisms, including increasing the production of VEGF
(35,36). In the present study, a tube
formation assay was conducted with MPVECs in order to demonstrate
the role of PGE2 in tumor angiogenesis. Treatment with PGE2 or VEGF
alone promoted tube formation, and these effects were decreased by
celecoxib treatment. Furthermore, PGE2 counteracted the
celecoxib-induced inhibition of VEGF-stimulated tube formation.
These results suggested that VEGF promotes angiogenesis by MPVECs
via the PGE2 pathway.
PGE2 exerts its functions through four G
protein-coupled receptors with distinct signaling properties
(37). EP1 is associated with Gq
protein, and upon stimulation, causes an increase in intracellular
Ca2+ and subsequent protein kinase C activation
(38,39). The EP2 and EP4 receptors couple to
Gs and protein kinase A/adenyl cyclase and mediate elevations in
intracellular cyclic adenosine monophosphate, whereas the EP3
receptor couples to Gi and decreases concentrations of cyclic
adenosine monophosphate (40–42).
To identify the specific EP receptor involved in the release of
VEGF, the present study treated MPVECs with four EP receptor
agonists. Only ONO-AE1-259-01, a specific EP2 receptor agonist,
showed an effect similar to that of PGE2, and the specific EP2
receptor antagonist AH6809 counteracted this effect. This indicated
that the EP2 receptor participates in regulating the release of
VEGF by the PGE2 pathway.
In conclusion, LPS promoted lung metastasis of
breast cancer by inducing elevated VEGF production, increased
vessel density and angiogenesis of MPVECs via the PGE2 pathway.
Therefore, treatment with a PGE2 inhibitor or prostaglandin-EP2
receptor antagonist may provide a novel approach for the prevention
and treatment of tumor metastasis.
Acknowledgments
The present study was supported by the General
Programs of Natural Science Foundation of China (grant no.
81272351), the General Programs of Natural Science Foundation of
Shandong Province (grant no. ZR2012HM020) and the Key Development
Program for Basic Research of Shandong Province (grant no.
2012G0021826).
Abbreviations:
|
LPS
|
lipopolysaccharide
|
|
PGE2
|
prostaglandin E2
|
|
VEGF
|
vascular endothelial growth factor
|
|
H&E
|
hematoxylin and eosin
|
|
MPVECs
|
mouse pulmonary endothelial cells
|
|
PBS
|
phosphate-buffered solution
|
|
OD
|
optical density
|
References
|
1
|
Liu H, Kato Y, Erzinger SA, Kiriakova GM,
Qian Y, Palmieri D, Steeg PS and Price JE: The role of MMP-1 in
breast cancer growth and metastasis to the brain in a xenograft
model. BMC Cancer. 12:5832012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee YT: Breast carcinoma: pattern of
metastasis at autopsy. J Surg Oncol. 23:175–180. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DeSantis C, Siegel R, Bandi P and Jemal A:
Breast cancer statistics, 2011. CA Cancer J Clin. 61:409–418. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Harmey JH, Bucana CD, Lu W, Byrne AM,
McDonnell S, Lynch C, Bouchier-Hayes D and Dong Z:
Lipopolysaccharide-induced metastatic growth is associated with
increased angiogenesis, vascular permeability and tumor cell
invasion. Int J Cancer. 101:415–422. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ikebe M, Kitaura Y, Nakamura M, Tanaka H,
Yamasaki A, Nagai S, Wada J, Yanai K, Koga K, Sato N, Kubo M,
Tanaka M, Onishi H and Katano M: Lipopolysaccharide (LPS) increases
the invasive ability of pancreatic cancer cells through the
TLR4/MyD88 signaling pathway. J Surg Oncol. 100:725–731. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He W, Liu Q, Wang L, Chen W, Li N and Cao
X: TLR4 signaling promotes immune escape of human lung cancer cells
by inducing immunosuppressive cytokines and apoptosis resistance.
Mol Immunol. 44:2850–2859. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gassmann P, Hemping-Bovenkerk A, Mees ST
and Haier J: Metastatic tumor cell arrest in the liver-lumen
occlusion and specific adhesion are not exclusive. Int J Colorectal
Dis. 24:851–858. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He Z, Zhu Y and Jiang H: Inhibiting
toll-like receptor 4 signaling ameliorates pulmonary fibrosis
during acute lung injury induced by lipopolysaccharide: an
experimental study. Respir Res. 10:1262009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang EL, Qian ZR, Nakasono M, Tanahashi T,
Yoshimoto K, Bando Y, Kudo E, Shimada M and Santo T: High
expression of Toll-like receptor 4/myeloid differentiation factor
88 signals correlates with poor prognosis in colorectal cancer. Br
J Cancer. 102:908–915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan L, Cai Q and Xu Y: The ubiquitin-CXCR4
axis plays an important role in acute lung infection-enhanced lung
tumor metastasis. Clin Cancer Res. 19:4706–4716. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Killeen SD, Wang JH, Andrews EJ and
Redmond HP: Bacterial endotoxin enhances colorectal cancer cell
adhesion and invasion through TLR-4 and NF-kappaB-dependent
activation of the urokinase plasminogen activator system. Br J
Cancer. 100:1589–1602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Liang J and Li G:
Lipopolysaccharide promotes adhesion and invasion of hepatoma cell
lines HepG2 and HepG2.2.15. Mol Biol Rep. 37:2235–2239. 2010.
View Article : Google Scholar
|
|
13
|
Brown DM and Ruoslahti E: Metadherin, a
cell surface protein in breast tumors that mediates lung
metastasis. Cancer Cell. 5:365–374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Y, Kong X, Li X, Yan S, Yuan C, Hu W
and Yang Q: Metadherin mediates lipopolysaccharide-induced
migration and invasion of breast cancer cells. PLoS One.
6:e293632011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sethi G, Shanmugam MK, Ramachandran L,
Kumar AP and Tergaonkar V: Multifaceted link between cancer and
inflammation. Biosci Rep. 32:1–15. 2012. View Article : Google Scholar
|
|
16
|
Senger DR, Galli SJ, Dvorak AM, Perruzzi
CA, Harvey VS and Dvorak HF: Tumor cells secrete a vascular
permeability factor that promotes accumulation of ascites fluid.
Science. 219:983–985. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Doan HQ, Bowen KA, Jackson LA and Evers
BM: Toll-like receptor 4 activation increases Akt phosphorylation
in colon cancer cells. Anticancer Res. 29:2473–2478.
2009.PubMed/NCBI
|
|
18
|
Wang L, Liu Q, Sun Q, Zhang C, Chen T and
Cao X: TLR4 signaling in cancer cells promotes chemoattraction of
immature dendritic cells via autocrine CCL20. Biochem Biophys Res
Commun. 366:852–856. 2008. View Article : Google Scholar
|
|
19
|
Ferrara N and Henzel WJ: Pituitary
follicular cells secrete a novel heparin-binding growth factor
specific for vascular endothelial cells. Biochem Biophys Res
Commun. 161:851–858. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsuji T, Aoshiba K, Yokohori N and Nagai
A: A systemically administered EP2 receptor agonist stimulates
pulmonary angiogenesis in a murine model of emphysema.
Prostaglandins Other Lipid Mediat. 90:85–88. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sato Y, Kanno S, Oda N, Abe M, Ito M,
Shitara K and Shibuya M: Properties of two VEGF receptors, Flt-1
and KDR, in signal transduction. Ann NY Acad Sci. 902:201–207.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lohela M, Bry M, Tammela T and Alitalo K:
VEGFs and receptors involved in angiogenesis versus
lymphangiogenesis. Curr Opin Cell Biol. 21:154–165. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perrot-Applanat M and Di Benedetto M:
Autocrine functions of VEGF in breast tumor cells: adhesion,
survival, migration and invasion. Cell Adh Migr. 6:547–553. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koch AE: Angiogenesis as a target in
rheumatoid arthritis. Ann Rheum Dis. 62:ii60–ii67. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baeriswyl V and Christofori G: The
angiogenic switch in carcinogenesis. Semin Cancer Biol. 19:329–337.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carmeliet P: VEGF as a key mediator of
angiogenesis in cancer. Oncology. 69(Suppl 3): 4–10. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakanishi M, Sato T, Li Y, Nelson AJ,
Farid M, Michalski J, Kanaji N, Wang X, Basma H, Patil A, Goraya J,
Liu X, Togo S, L Toews M, Holz O, Muller KC, Magnussen H and
Rennard SI: Prostaglandin E2 stimulates the production of vascular
endothelial growth factor through the E-prostanoid-2 receptor in
cultured human lung fibroblasts. Am J Respir Cell Mol Biol.
46:217–223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang S, Wettlaufer SH, Hogaboam C,
Aronoff DM and Peters-Golden M: Prostaglandin E2
inhibits collagen expression and proliferation in patient-derived
normal lung fibroblasts via E prostanoid 2 receptor and cAMP
signaling. Am J Physiol Lung Cell Mol Physiol. 292:L405–L413. 2007.
View Article : Google Scholar
|
|
29
|
Sakurai T, Tamura K, Okamoto S, Hara T and
Kogo H: Possible role of cyclooxygenase II in the acquisition of
ovarian luteal function in rodents. Biol Reprod. 69:835–842. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sakurai T, Tamura K and Kogo H:
Stimulatory effects of eicosanoids on ovarian angiogenesis in early
luteal phase in cyclooxygenase-2 inhibitor-treated rats. Eur J
Pharmacol. 516:158–164. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bos CL, Richel DJ, Ritsema T,
Peppelenbosch MP and Versteeg HH: Prostanoids and prostanoid
receptors in signal transduction. Int J Biochem Cell Biol.
36:1187–1205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sakurai T, Tamura K and Kogo H: Vascular
endothelial growth factor increases messenger RNAs encoding
cyclooxygenase-II and membrane-associated prostaglandin E synthase
in rat luteal cells. J Endocrinol. 183:527–533. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Folkman J and D’Amore PA: Blood vessel
formation: what is its molecular basis? Cell. 87:1153–1155. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Keith RL, Geraci MW, Nana-Sinkam SP,
Breyer RM, Hudish TM, Meyer AM, Malkinson AM and Dwyer-Nield LD:
Prostaglandin E2 receptor subtype 2 (EP2) null mice are protected
against murine lung tumorigenesis. Anticancer Res. 26:2857–2861.
2006.PubMed/NCBI
|
|
35
|
Chang SH, Liu CH, Conway R, Han DK,
Nithipatikom K, Trifan OC, Lane TF and Hla T: Role of prostaglandin
E2-dependent angiogenic switch in cyclooxygenase 2-induced breast
cancer progression. Proc Natl Acad Sci USA. 101:591–596. 2004.
View Article : Google Scholar :
|
|
36
|
Gately S and Li WW: Multiple roles of
COX-2 in tumor angiogenesis: a target for antiangiogenic therapy.
Semin Oncol. 31:2–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma X, Kundu N, Ioffe OB, et al:
Prostaglandin E receptor EP1 suppresses breast cancer metastasis
and is linked to survival differences and cancer disparities. Mol
Cancer Res. 8:1310–1318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Narumiya S, Sugimoto Y and Ushikubi F:
Prostanoid receptors: structures, properties and functions. Physiol
Rev. 79:1193–1226. 1999.PubMed/NCBI
|
|
39
|
Yang T and Du Y: Distinct roles of central
and peripheral prostaglandin E2 and EP subtypes in blood pressure
regulation. Am J Hypertens. 25:1042–1049. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sugimoto Y and Narumiya S: Prostaglandin E
receptors. J Biol Chem. 282:11613–11617. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Breyer RM, Bagdassarian CK, Myers SA and
Breyer MD: Prostanoid receptors: subtypes and signaling. Annu Rev
Pharmacol Toxicol. 41:661–690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chell S, Kaidi A, Williams AC and
Paraskeva C: Mediators of PGE2 synthesis and signalling downstream
of COX-2 represent potential targets for the prevention/treatment
of colorectal cancer. Biochim Biophys Acta. 1766:104–119.
2006.PubMed/NCBI
|