Introduction
Leukemia is a malignant clonal hematopoietic stem
cell disease; leukemia cells are arrested at various cell
development stages due to enhanced self-renewal, out of control
proliferation, differentiation disorder and blocked apoptosis
(1). In the bone marrow and other
hematopoietic tissues, leukemia cells proliferate rapidly,
inhibiting normal hematopoiesis and infiltrating other tissues and
organs. In malignant tumor mortality rankings, leukemia-associated
mortality is sixth most common in males, eighth in females, and
first in individuals under the age of 35 (2). The primary treatment for leukemia is
chemotherapy; however, treatment failure and drug-resistance of
cancer cells often occur. Adriamycin (ADR) is a commonly used
chemotherapeutic agent used in the treatment of leukemia. ADR
inhibits cancer cell cycle progression by inducing DNA damage and
disordering DNA replication. ADR inserts between the base pairs of
DNA in the anthraquinone plane to form specific structures, termed
interstrand crosslinks (ICLs) (3).
DNA ICLs can block DNA replication. Therefore, the DNA damage
repair mechanism may be an important molecular basis of tumor cell
drug-resistance to ADR.
The Fanconi anemia (FA) pathway is an important DNA
repair pathway, which can effectively repair DNA interstrand
crosslinks (4). Fifteen different
complementation groups for FA have been defined (5). The FA pathway consists of three major
steps: FA core complex formation, FANCD2 ubiquitination and DNA
repair in the downstream FA pathway (6). The FA core complex is composed of
eight FA proteins. The primary function of the FA core complex is
to monoubiquitinate FANCD2, a key step in the FA pathway.
Monoubiquitined FANCD2 is able to activate nucleases in the
downstream FA pathway and subsequently repair DNA damage, such as
ICLs. Chen et al (7) found
that overexpression of FANCF in drug-sensitive cells partially
reproduced the drug-resistant cell phenotype. The ubiquitination of
FANCD2 has an important role in drug resistance in multiple myeloma
(8). FANCD2 monoubiquitination and
FANCD2 nuclear foci formation are increased in TMZ/BCNU-resistant
glioma cells (9). In addition,
FANCD2 ubiquitination and FANCF demethylation have been shown to be
associated with cisplatinum resistance of ovarian carcinoma cells
(10). The aim of the current
study was to explore the roles of FANCD2 and FANCF, members of the
FA pathway, in ADR-resistant leukemia cells.
Materials and methods
Cell lines and drugs
The K562 and K562/R human leukemia cell lines were
obtained from the Cell Center of Xiangya School of Medicine
(Changsha, China). Cell lines were grown in RPMI-1640 medium
(Hyclone, Logan, UT, USA) supplemented with 10% heat-inactivated
fetal bovine serum (Evergreen, Suzhou, China). K562/R ADR-resistant
cells were grown in culture medium containing 1 μM ADR
(Zhejiang Haizheng Pharmaceutical, Taizhou, China) to maintain
drug-resistance.
Methyl-thiazoltetrazolium (MTT)
cytotoxicity assay
Cells were seeded at a density of 5,000 cells/well
in 96-well plates (Costar, Corning, NY, USA). To establish a dose
response to ADR, cells were incubated with ADR in 2-fold serial
dilutions ranging from 1 to 64 μmol/l. The blank group did
not contain cells. Each group included three parallel samples. The
total medium volume of each well was 200 μl. Two days after
drug addition, 20 μl MTT solution [5.0 g/l in
phosphate-buffered saline (PBS; ZSGB-BIO, Beijing, China)] was
added. After 4 h, 100 μl dimethylsulfoxide (Sigma-Aldrich,
St. Louis, MO, USA) was added per well and the cells were allowed
to stand for 30 min prior to detection.
Cell cycle analysis
Propidium iodide (PI; Sigma-Aldrich) staining was
used to measure the effects of ADR on cell-cycle progression. K562
and K562/R cells were treated with 2 or 20 μmol/l ADR for 2
h and cultured in drug-free medium; cells were collected at 24 and
48 h. Subsequently, cells were fixed with 70% ethanol at 4°C
overnight, resuspended in 500 μl PBS containing 500
μg/ml PI and 10 mg/ml RNase A (Invitrogen Life Technologies,
Carlsbad, CA, USA), and incubated at 37°C for 30 min in the dark.
The cell cycle was analyzed by flow cytometry (MofloXDP; Beckman
Coulter, Brea, CA, USA) using FlowJo 4.4.4 software (Tree Star,
Inc., Ashland, OR, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
TotalRNA Extractor (Shenggong), ReverTra Ace qPCR
RT-kit (Toyobo Co., Ltd., Osaka, Japan) and SYBR Green®
Realtime PCR Master mix (Toyobo Co., Ltd.) were used for total RNA
(1 μg) extraction, cDNA synthesis and PCR analysis,
respectively. qPCR of FANCF (forward, 5′-CTCCTGTCTATCTGGGTCTGCT-3′
and reverse, 5′-GAGGCTTTGAAACCTATTGTGC-3′) and FANCD2 (forward,
5′-AGCAATGTATGCCGCTCCT3′ and reverse, 5′-TTGGGTGAGTCTCGTGTCC-3′)
was run for 40 cycles. Each cycle consisted of denaturation (30 sec
at 95°C; first cycle, 2 min 30 sec), annealing (30 sec at 58°C) and
extension (30 sec at 72°C; last cycle 7 min 30 sec). The gene
expression level was normalized using the endogenous control gene
GAPDH. qPCR reactions were performed using Mastercycler ep realplex
(Eppendorf, Hamburg, Germany).
Western blot analysis
FANCD2 and FANCF proteins were detected by western
blot analysis. Cells were lysed using a commercial lysis buffer kit
(Beyotime Institute of Biotechnology, Haimen, China) according to
the manufacturer’s instructions. Following quantification of
proteins using a BCA kit (Beyotime, Shanghai, China), equal
quantities of protein lysates were mixed with SDS-PAGE protein
loading buffer, boiled for 5 min, and then run on 10% SDS-PAGE at
100 V for 180 min and transferred onto a polyvinylidine fluoride
membrane (Beyotime Insitute of Biotechnology). The primary
antibodies used for western blotting were mouse monoclonal
immunoglobulin G1 (IgG1) anti-human FANCF
(sc-271952; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit
polyclonal IgG anti-human FANCD2 (sc-28194; Santa Cruz
Biotechnology) and β-actin (Sigma-Aldrich). The secondary
antibodies were polyclonal horseradish peroxidase-conjugated
AffiniPure goat anti-rabbit IgG, Fc fragment specific
(111-035-008), polyclonal horseradish peroxidase-conjugated
AffiniPure goat anti-rabbit IgG (H+L) (111-035-003) and polyclonal
rhodamine (TRITC) AffiniPure Goat Anti-Mouse IgG (H+L)
(115-025-062) (Jackson ImmunoResearch Laboratories, Inc., West
Grove, PA, USA). Results were visualized using a DAB Horseradish
Peroxidase Color Development kit (Beyotime Institute of
Biotechnology) for chemiluminescent detection.
Alkaline comet assay
The alkaline comet assay was used to detect
ADR-induced DNA cross-links in drug-sensitive (K562) and
drug-resistant (K562/R) leukemia cells. A total of 1×105
cells were treated with 80 μM ADR or vehicle control for 24
h, washed in PBS and resuspended in ice-cold PBS. A volume of ~10
μl of the resuspended cells was mixed with 65 μl of
low melting point agarose at 4°C. The slides were placed in the
dark at 4°C until gelling occurred and then immersed in prechilled
lysis buffer (Jingcai, Xi’an, China) at 4°C. After a 60-min
incubation, the buffer was aspirated and replaced with pre-chilled
alkaline solution for 90–120 min at 4°C. After lysis and unwinding,
the slides were placed in a horizontal electrophoresis tank filled
with freshly prepared alkaline electrophoresis buffer (Jingcai).
The electrophoresis was run for 30 min at 25 V and 300 mA. After
electrophoresis, the slides were transferred to neutralization
buffer (Jingcai) for 10 min. The neutralization buffer was
aspirated and replaced three times. Thereafter, the slides were
allowed to air dry and 50 μl/well PI (5 μg/ml) was
added to each slide for 5 min in the dark at room temperature, for
DNA staining. DNA migration was observed using a fluorescence
microscope (AE31; Motic, Hong Kong, China). For each sample, 200
randomly selected cells were analyzed using CASPLab version 1.2.2
(Comet Assay Software Project, Wrocław, Poland).
Statistical analysis
Statistical analysis was performed using Student’s
t-test or one-way analysis of variance with SPSS version 21.0 (IBM,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
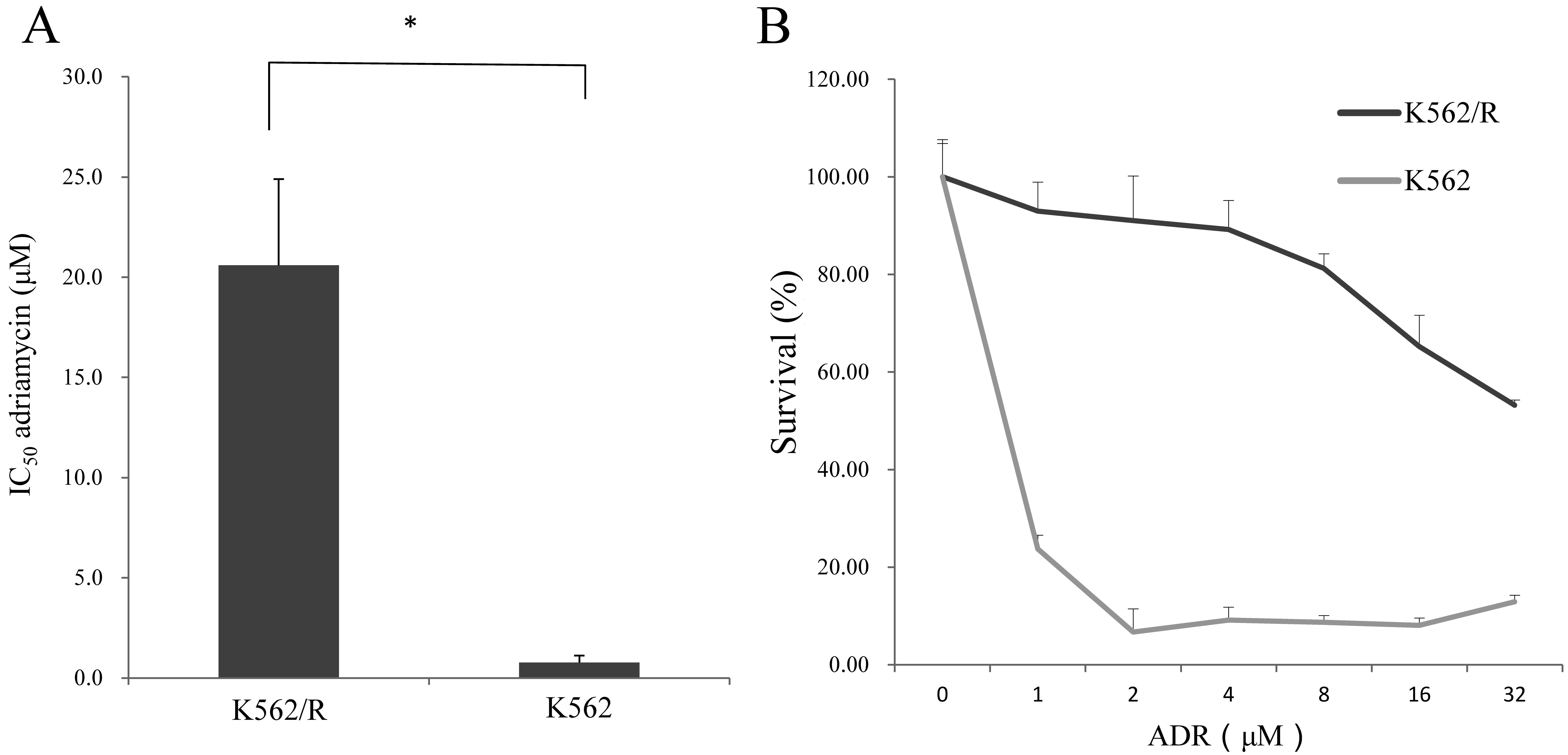
Drug-resistant cells have a higher
survival rate after ADR treatment
An MTT assay was used to compare ADR sensitivity in
the drug-sensitive (K562) and drug-resistant (K562/R) leukemia cell
lines. The IC50 of K562/R cells was 20.6±4.30 μM
and the IC50 of K562 cells was 0.78±0.05 μM
(Fig. 1A). ADR-resistant K562/R
cells exhibited increased survival (Fig. 1B) compared with that of their
drug-sensitive parental K562 cells.
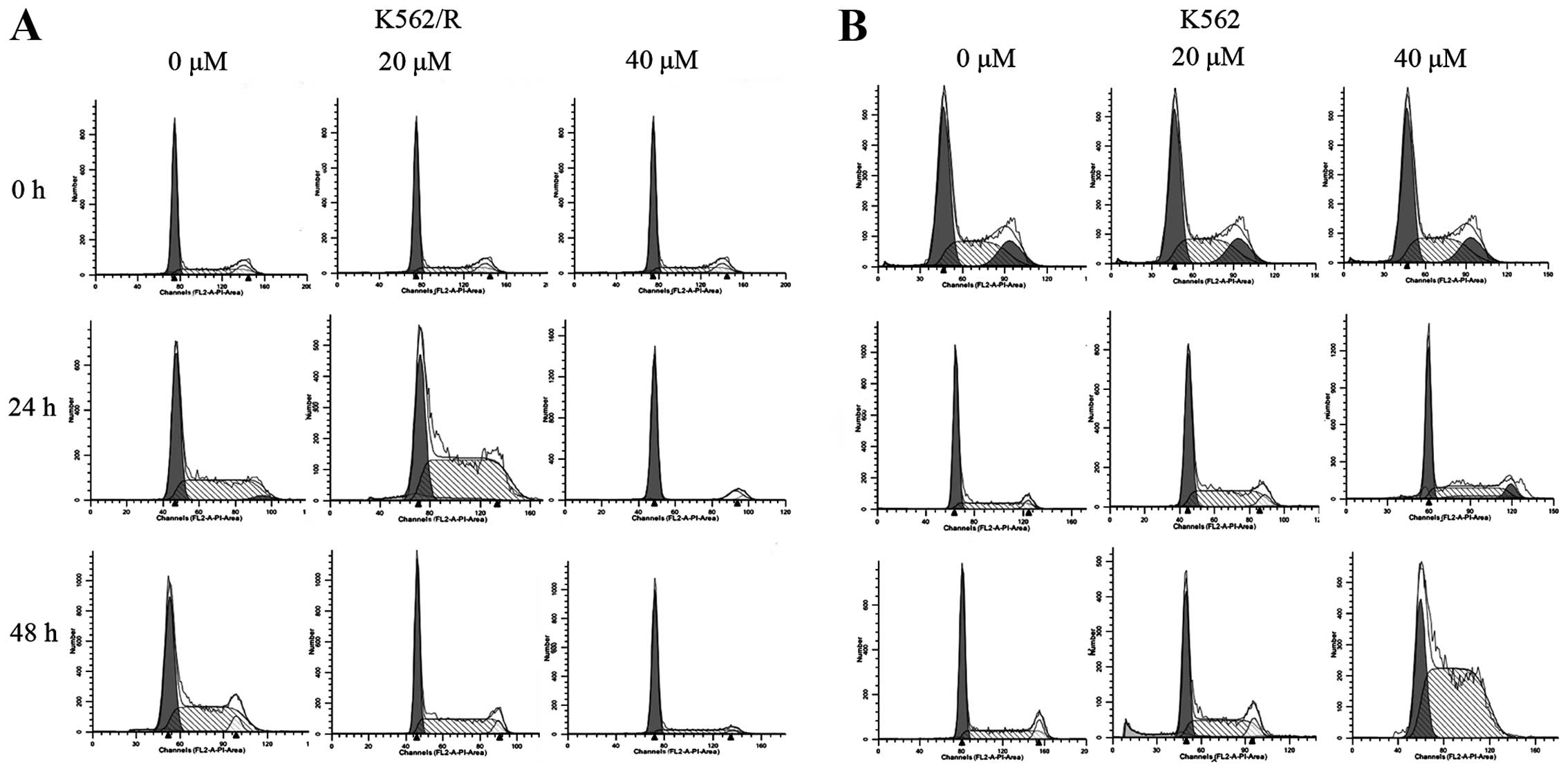
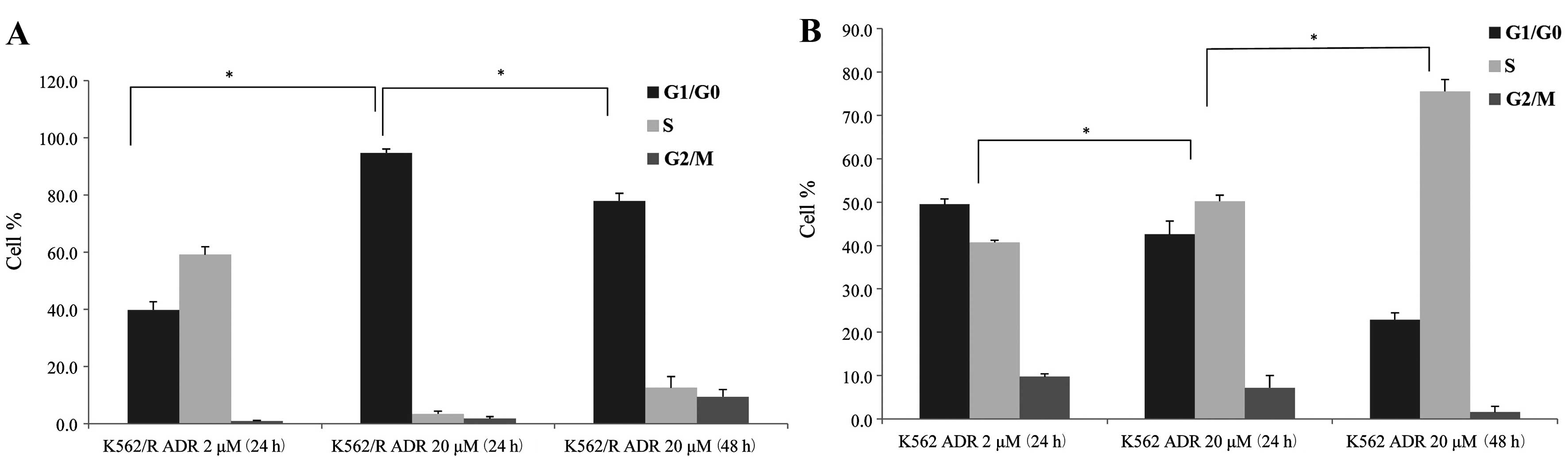
Effect of ADR on cell-cycle progression
in K562 and K562/R cells
To measure dose-response and time-response effects
of ADR on cell-cycle progression, K562 and K562/R cells were
treated with 2 or 20 μM ADR for 2 h, cultured in drug-free
medium, and collected at 24 and 48 h. After 24 h, high-dose (20
μM) and low-dose (2 μM) ADR treatment resulted in an
accumulation of 40.7±0.55 and 50.2±1.35% K562 cells in the S phase,
respectively (Figs. 2 and 3). K562/R cells were arrested in the G1
phase (94.7±1.36%) after high-dose ADR treatment (20 μM)
(Figs. 2 and 3). However, low-dose ADR treatment (2
μM) induced K562/R cell arrest in the S phase (59.19±2.71%).
Following ADR (20 μM) treatment for 24 or 48 h, K562 cells
were arrested in the S phase 50.2±1.35% and 75.6±7.72%,
respectively (Fig. 2 and 3). ADR treatment over a longer period
resulted in a greater number of K562 cells arresting in the S
phase. K562/R cells were arrested in the G1 phase after ADR (20
μM) treatment for 24 (94.7±1.36%) or 48 h (77.9±2.69%);
however, K562/R cells that were treated for 48 h had fewer cells in
the G1 phase (Figs. 2
and 3). As time passed, the
G1 phase-arresting effect of ADR on K562/R cells became
weaker, however, the effect on cell cycle arrest of K562 cells
became stronger.
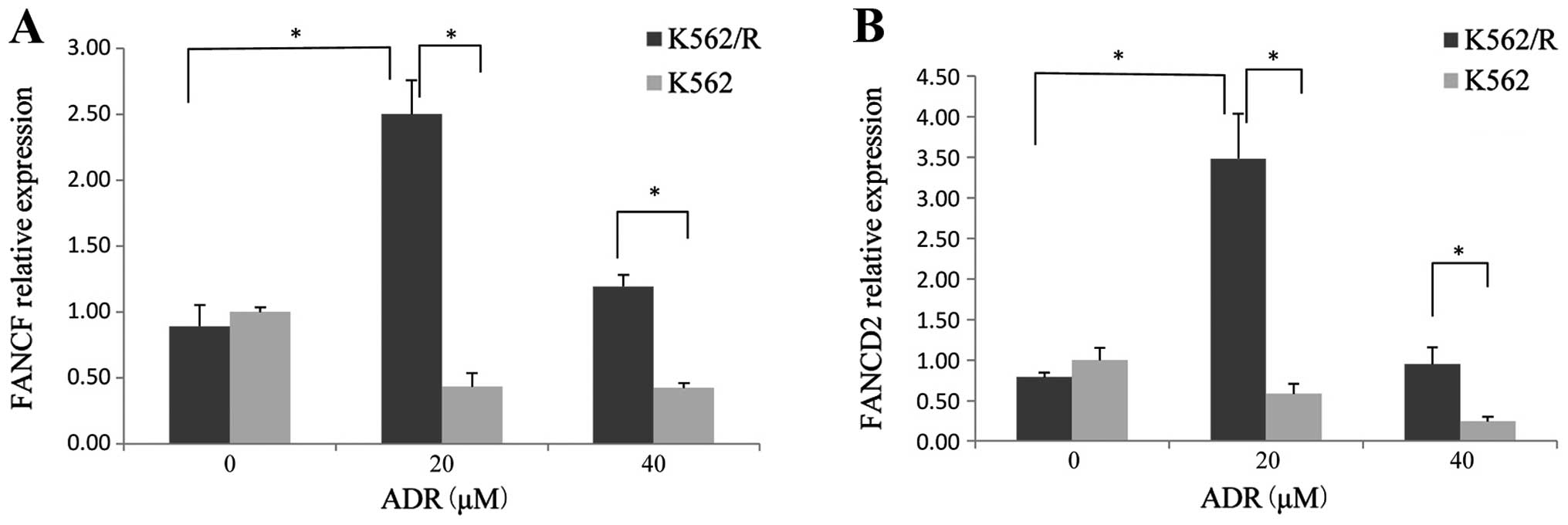
ADR-resistant leukemia cells exhibit
enhanced expression of FANCD2 and FANCF
qPCR showed that K562/R cells had higher levels of
FANCF and FANCD2 expression after ADR (20 μM) treatment for
24 h compared with the untreated K562/R cells. ADR (20 and 40
μM) treatment resulted in greater FANCF and FANCD2
expression in K562/R cells than K562 cells (Fig. 4). The results revealed that the
expression of the two components of the FA pathway were upregulated
in drug-resistant K562/R cells compared with drug-sensitive K562
cells.
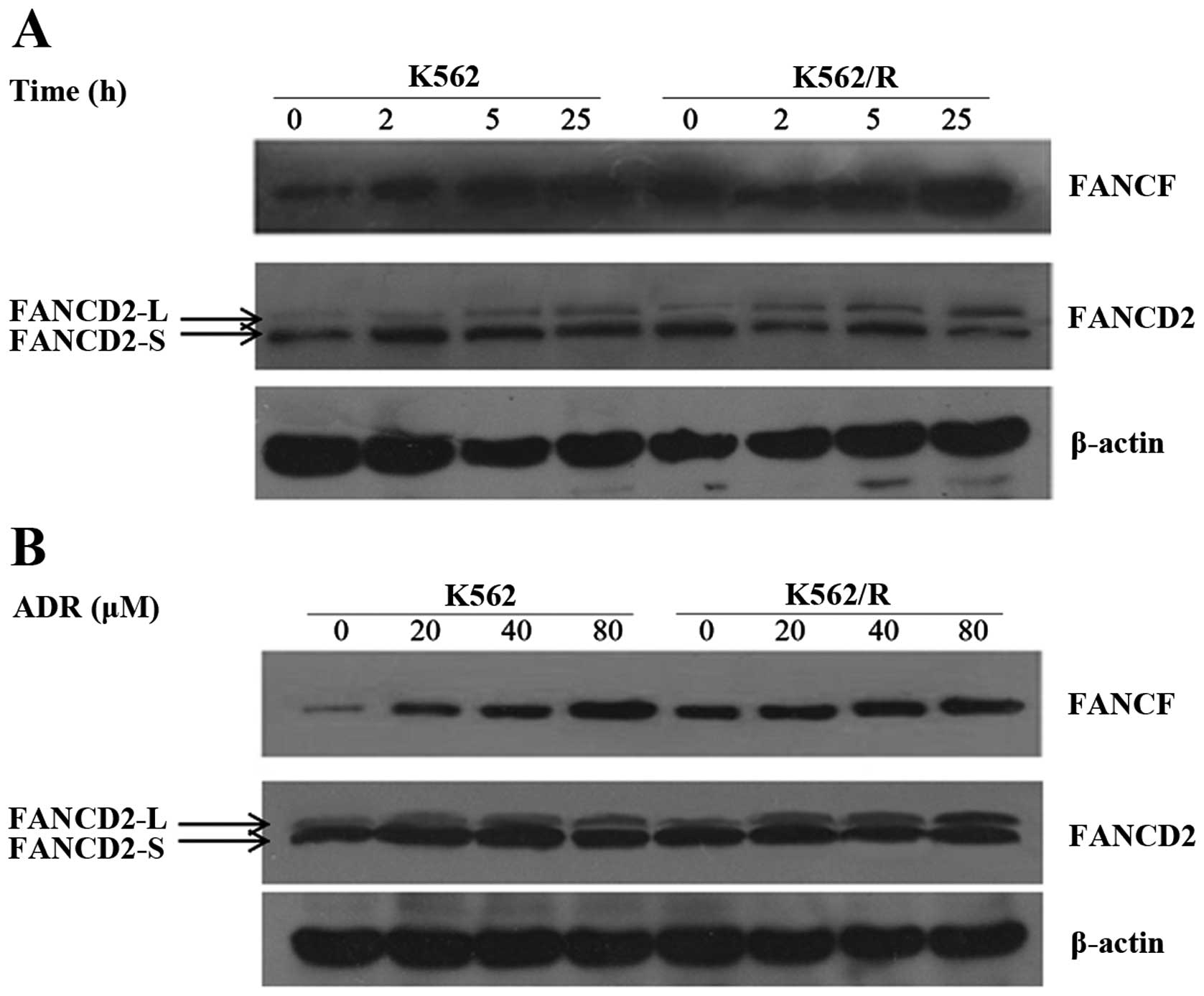
FANCD2 ubiquitination and FANCF protein
expression are enhanced in ADR-resistant leukemia cells
FANCD2 protein has two isomers, namely FANCD2-S and
FANCD2-L. FANCD2-S is non-ubiquitinated FANCD2 protein ndFANCD2-L
is ubiquitinated FANCD2 protein. In the FA pathway, ubiquitinated
FANCD2 proteins are functional; thus, primarily the FANCD2
ubiquitination level was compared with the FANCD2-L/S (the ratio of
ubiquitinated FANCD2 protein to non-ubiquitinated FANCD2 protein).
FANCD2 is one of the most important proteins in the FA pathway, and
FANCF is important for forming the FA core complex. Therefore,
FANCD2-L/S and FANCF were monitored at various time points after
multi-concentration drug treatment in the two cell lines. The
results revealed that the levels of FANCD2-L/S and FANCF were
significantly increased within 2 h of ADR treatment, and the
maximum increase of FANCD2-L/S and FANCF occurred within 25 h after
ADR treatment (Fig. 5).
Additionally, the results revealed that the level of FANCD2-L/S and
FANCF was clearly increased after 20 μM ADR treatment for 25
h and the maximum increase of FANCD2-L/S and FANCF occurred after
80 μM ADR treatment for 25 h (Fig. 5). The level of FANCD2-L/S and FANCF
in K562 and K562/R cell lines increased in a time-dependent manner
and dose-dependent manner, but higher expression levels of
FANCD2-L/S and FANCF were observed at all times and doses in
drug-resistant cells compared with drug-sensitive cells. In
addition, the results suggest that increased FANCD2-L may be due to
the enhanced expression of FANCF protein. A stable FA complex
functions as the ubiquitin ligase for FANCD2. When the expression
of FANCF was increased, the expression of FANCF and
monoubiquitinated FANCD2 protein were significantly increased in
ADR-resistant leukemia cells. Therefore it was concluded that FANCF
and ubiquitinated FANCD2 proteins were involved in ADR-resistant
leukemia cells.
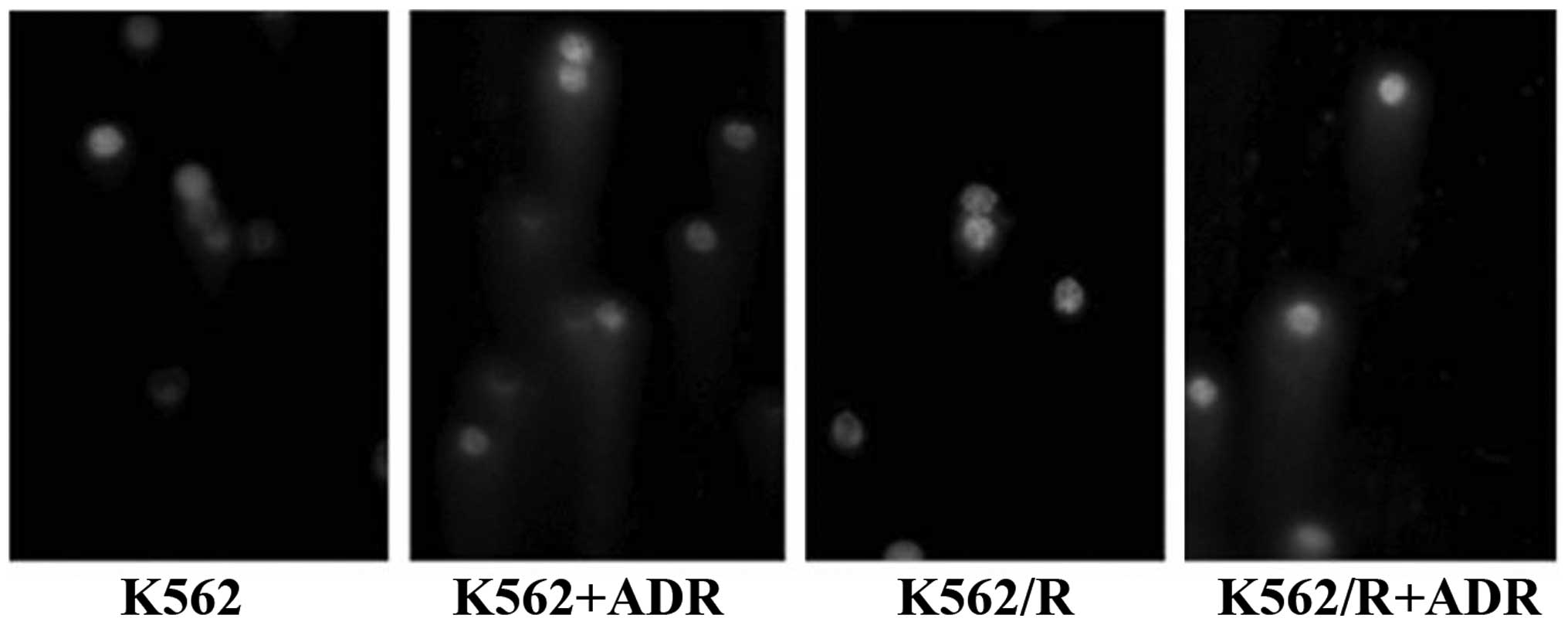
ADR-resistant cells experience less DNA
damage compared with drug-sensitive cells
Photomicrographs of the ADR-induced DNA damage are
shown in Fig. 6. The ADR-induced
DNA ICL formation effect was obvious in K562 and K562/R cells after
ADR treatment. The ICLs were significantly increased in K562 and
K562/R cells after ADR treatment compared with those of the group
without ADR treatment. However, drug-resistant cells had fewer ICLs
than drug-sensitive cells, indicating that ADR-resistant cells
exhibited less DNA damage compared with drug-sensitive cells.
Discussion
Currently the curative effect of ADR for leukemia
treatment remains limited. Resistance to ADR is often observed in
cancer cells. DNA repair pathway activation is one of the
mechanisms for resistance to ADR in tumor cells. A number of
chemotherapeutic agents cause DNA damage, and then inhibit tumor
cell replication and induce cell apoptosis to achieve their
therapeutic effect. The FA pathway is a DNA repair pathway, which
repairs DNA in one of three ways: Base excision repair, homologous
recombination repair or translesion DNA synthesis. It has been
shown that the FA pathway is involved in resistance to a variety of
drugs in tumor cells. A total of 15 FA proteins have already been
identified, including FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE,
FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCP and RAD51C.
The formation of the FA core complex and FANCD2 monubiquitylation
are two key steps in the FA pathway. The FA core complex has E3
ligase activity and is composed of eight FA proteins (A, B, C, E,
F, G, L and M), FAAP100 protein and FAAP24 protein (11). FANCF protein is an important
adapter, which connects the FANCA/FANCG and FANCC/FANCE subunits in
the FA core complex (12). It
interacts directly with FANCG protein and aids in stabilizing a
complex involving FANCA and FANCC proteins using its C-terminal. In
addition, FANCF interacts with the FANCC-FANCE sub-complex through
the N-terminal region (13).
Therefore, FANCF can stabilise the whole FA core complex. When DNA
is damaged, the stable FA core complex uses its E3 ubiquitin ligase
activity to assist in the ubiquitination of FANCD2. After FANCD2
ubiquitination, the downstream DNA repair enzymes can be recruited
to repair DNA damage. Therefore FANCF and FANCD2 were selected to
investigate the association between the FA pathway and
drug-resistant leukemia.
ADR is an anthracycline cytotoxic drug, it is a
non-specific cell cycle drug; however, S-phase tumor cells have a
higher sensitivity to it. Its molecular structure can be embedded
into the DNA double strand to form a stable complex, leading to the
inhibition of DNA replication and RNA synthesis, thereby causing
cell death or arrest in the S or G2 phase (14). The results of the current study
determined that the growth of K562 and K562/R cells was inhibited
after ADR treatment. K562 cells were arrested in the S phase and
K562/R cells were arrested in G1 phase. The effect of
cell cycle arrest was weaker in K562/R cells than in K562 cells. In
the process of cell division, the S phase is primarily responsible
for DNA replication; cells were arrested in the S phase, showing
that DNA replication was inhibited and required repairing, and the
more cells that were blocked in the S phase, the more DNA damage
that required repairing. In the comet assay, drug-sensitive cells
had a greater number of DNA ICLs than drug-resistant cells.
The results of the RT-qPCR showed that K562/R
drug-resistant cells expressed a higher level of FANCD2 and FANCF
mRNA than K562 cells. The experimental results are similar to the
results of Yarde et al (15), which revealed that the FA/BRCA
pathway is involved in the drug-resistance of multiple myeloma
cells. In addition, the current study determined that the protein
expression levels of FANCD2-L/S and FANCF in K562 and K562/R cell
lines were increased in a time- and dose dependent manner; however,
a higher expression level of FANCD2-L/S and FANCF was observed in
drug-resistant cells compared with drug-sensitive cells. Lundholm
et al (16) determined that
FANCD2 ubiquitination was enhanced in cisplatin-resistant non-small
cell lung cancer stem cells. Additionally, the level of FANCD2
ubiquitination has been shown to be reduced in drug-resistant
multiple myeloma cells after the reversal of cell drug-resistance
(8). Therefore, FANCF and
ubiquitinated FANCD2 proteins may be involved in the development of
drug-resistance in leukemia cells.
The primary function of the FA pathway is to repair
DNA ICLs, therefore, in the current study the DNA damage of the two
cell lines was detected using a comet assay after ADR treatment.
Drug-resistant cells developed fewer DNA ICLs than drug-sensitive
cells. Therefore it was hypothesized that drug-resistant cells may
have stronger DNA repair ability.
In conclusion, the results of the current study
showed that drug-resistant leukemia cells have enhanced FANCF
expression and FANCD2 ubiquitination. Therefore, the FA pathway,
which FANCF and FANCD2 are involved in, may be a potential
mechanism for ADR resistance in leukemia cells.
Acknowledgments
This study was supported by grants from the Research
Foundation of Hunan Provincial Science and Technology Department
(no. 2012FJ3138) and the Postgraduates Innovation Foundation of
Hunan Province (no. CX2011B065).
References
|
1
|
Wu DP: Leukemia. Internal Medicine.
9:600–616. 2009.In Chinese.
|
|
2
|
Chu JX, Yang CL and Yang TY: Leukemia
epidemiology report of China. Tianjing Med J. 6:323–326. 1982.In
Chinese.
|
|
3
|
Wang Y, Xu LH, Duan ZH, Cao KY, Luo JG, Xu
Y and Shi PJ: Effects of ADM on cell apoptosis and expression of
FAK mRNA in K562. J Trop Med. 12:1195–1198. 2012.In Chinese.
|
|
4
|
Moldovan GL and D’Andrea AD: How the
fanconi anemia pathway guards the genome. Annu Rev Genet.
43:223–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim H and D’Andrea AD: Regulation of DNA
cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev.
26:1393–1408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Romick-Rosendale LE, Lui VW, Grandis JR
and Wells SI: The Fanconi anemia pathway: repairing the link
between DNA damage and squamous cell carcinoma. Mutat Res.
743–744:78–88. 2013. View Article : Google Scholar
|
|
7
|
Chen Q, Van der Sluis PC, Boulware D,
Hazlehurst LA and Dalton WS: The FA/BRCA pathway is involved in
melphalan-induced DNA interstrand cross-link repair and accounts
for melphalan resistance in multiple myeloma cells. Blood.
106:698–705. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiao H, Xiao Q, Zhang K, Zuo X and
Shrestha UK: Reversal of multidrug resistance by curcumin through
FA/BRCA pathway in multiple myeloma cell line MOLP-2/R. Ann
Hematol. 89:399–404. 2010. View Article : Google Scholar
|
|
9
|
Chen CC, Taniguchi T and D’Andrea A: The
Fanconi anemia (FA) pathway confers glioma resistance to DNA
alkylating agents. J Mol Med (Berl). 85:497–509. 2007. View Article : Google Scholar
|
|
10
|
Taniguchi T, Tischkowitz M, Ameziane N, et
al: Disruption of the Fanconi anemia-BRCA pathway in
cisplatin-sensitive ovarian tumors. Nat Med. 9:568–574. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
D’Andrea AD: Targeting DNA repair pathways
in AML. Best Pract Res Clin Haematol. 23:469–473. 2010. View Article : Google Scholar
|
|
12
|
Leveille F, Blom E, Medhurst AL, et al:
The Fanconi anemia gene product FANCF is a flexible adaptor
protein. J Biol Chem. 279:39421–39430. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Winter JP, van der Weel L, de Groot J,
et al: The Fanconi anemia protein FANCF forms a nuclear complex
with FANCA, FANCC and FANCG. Hum Mol Genet. 9:2665–2674. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X and Li DH: Adriamycin-induced
apoptosis of tumor cells research progress. China Practical Med.
07:247–249. 2010.
|
|
15
|
Yarde DN, Oliveira V, Mathews L, et al:
Targeting the Fanconi anemia/BRCA pathway circumvents drug
resistance in multiple myeloma. Cancer Res. 69:9367–9375. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lundholm L, Haag P, Zong D, et al:
Resistance to DNA-damaging treatment in non-small cell lung cancer
tumor-initiating cells involves reduced DNA-PK/ATM activation and
diminished cell cycle arrest. Cell Death Dis. 4:e4782013.
View Article : Google Scholar : PubMed/NCBI
|