Introduction
Ehlers-Danlos syndrome type IV (EDS IV; OMIM#
130050), also known as Ehlers-Danlos syndrome, vascular type, is an
autosomal dominant disorder characterized by abnormal type III
collagen, thin translucent skin, arterial/intestinal/uterine
fragility, extensive bruising and a characteristic facial
appearance (1). The prevalence of
EDS is estimated to range from 1/10,000 to 1/25,000 and EDS IV
represents 5–10% of cases (2). As
a result of spontaneous vascular/intestinal rupture, the overall
life expectancy of patients with EDS IV is 48 years (3). Mutations in COL3A1, the gene that
encodes type III collagen, have been associated with EDS IV. By
either sequencing COL3A1 from blood samples or using biochemical
tests, detection of abnormal type III collagen can identify >95%
of individuals with EDS IV (4).
The two methods are used to aid in the clinical diagnosis of EDS IV
(4).
Vascular complications are common in patients with
EDS IV, while aortic aneurysm is a rare condition. Only five
mutations of COL3A1 [c.505C>T (5), c.907G>A (6), c.2356G>A (7), c.2633G>A (8) and c.1815+5G>A (9)] have been reported to result in aortic
aneurysm, and no variants associated with ascending aortic aneurysm
have previously been reported.
The aim of the current study was to identify the
first COL3A1 mutation associated with ascending aortic aneurysm in
an EDS IV patient. In addition, this study summarizes all the
previously reported variants of COL3A1.
Patients and methods
Ethical approval and informed
consent
The current study has been approved by, and
conducted according to the instructions of the Ethics Committee of
Central South University (Changsha, China). The patient and the
other members of his family provided written informed consent for
publication.
Patient presentation
A 38-year-old male was admitted to the Department of
Cardiothoracic Surgery, The Second Xiangya Hosptial (Changsha,
China) due to detection of an aortic aneurysm combined with aortic
incompetence by echocardiography in medical examination. Physical
examination revealed that the patient had no notable symptoms
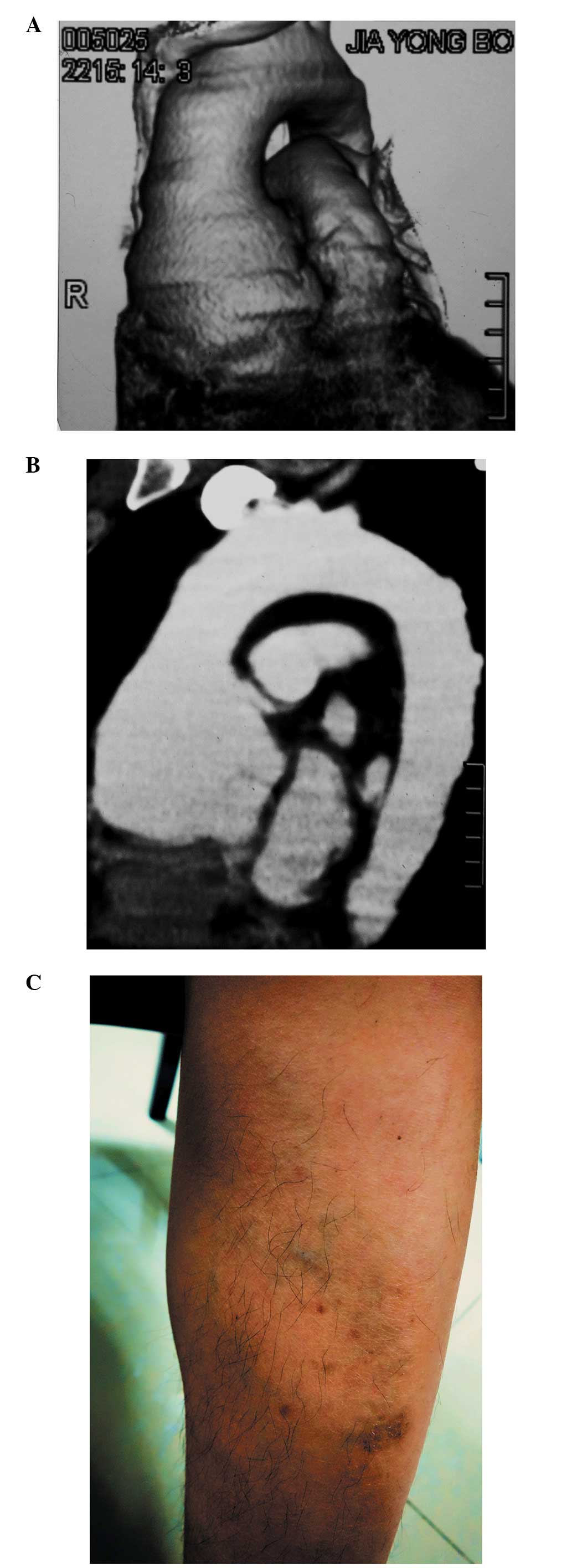
except for thin translucent skin (Fig.
1C) and a soft diastolic murmur of the aortic valve. There were
no associated events in the patient’s past medical history. The
results of an ECG and a chest x-ray were normal. His father,
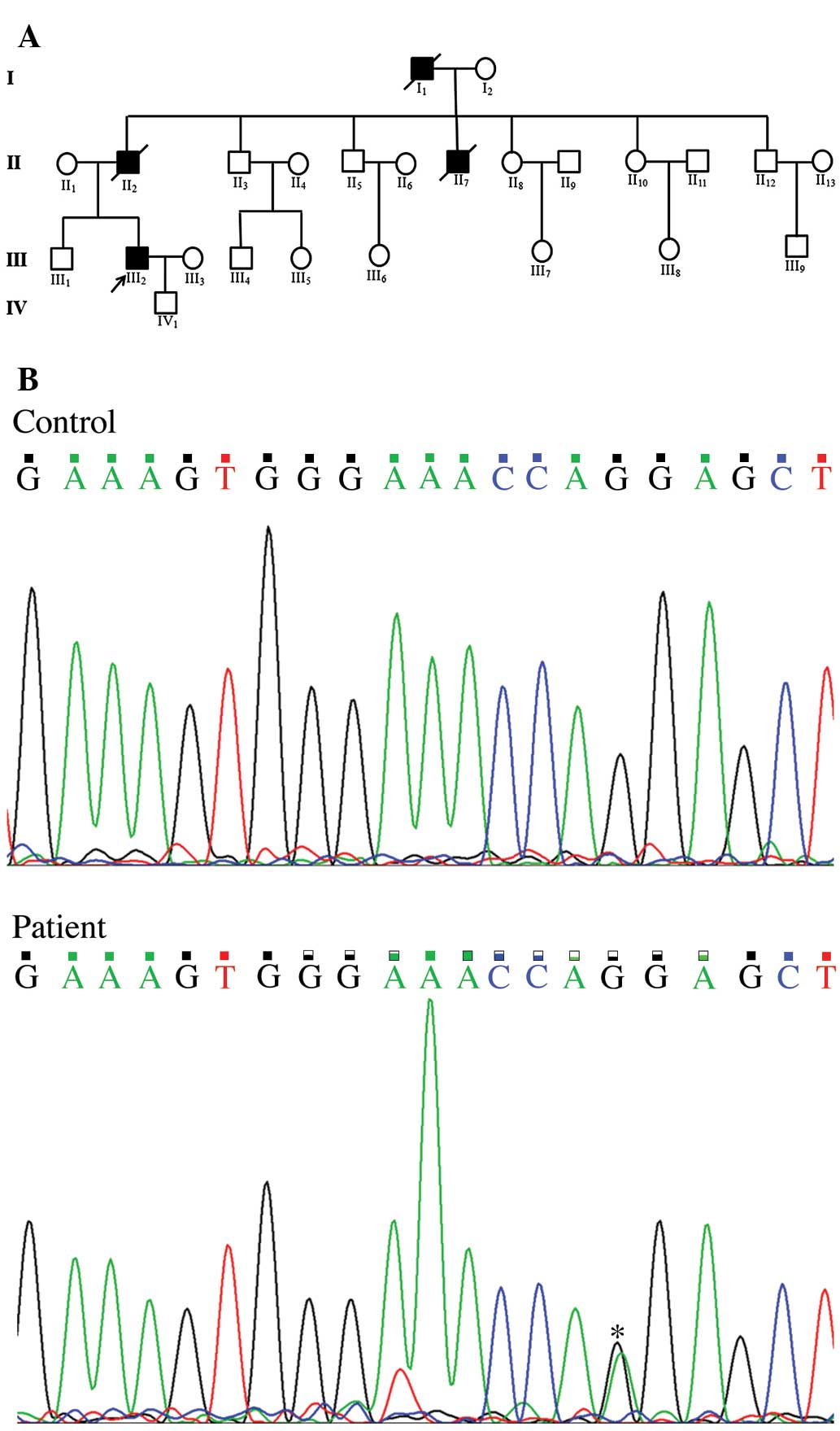
grandfather and one of his four uncles suffered from sudden
mortality (Fig. 2A) due to aortic
rupture. No other family members, including his son, had any
similar conditions or experiences.
Imaging examination
Computerized tomographic angiography (CTA) of the
heart and thoracic aorta was performed in the Second Xiangya
Hospital of Central South University (Changsha, China).
Molecular genetic analysis
Genomic DNA was prepared from the peripheral blood
of the patient and his family members using a DNeasy Blood &
Tissue kit (Qiagen, Valencia, CA, USA) using the QIAcube automated
DNA extraction system (Qiagen, Hilden, Germany). A total of 20
μl QIAGEN Proteinase and 200 μl sample (whole blood
in phosphate-buffered saline; Wako Pure Chemical Industries, Ltd.,
Osaka, Japan) was added into a 1.5 ml microcentrifuge tube. Buffer
AL (200 μl; in DNeasy Blood & Tissue Kit) was then added
and the sample was mixed by pulse vortexing (HYQ-2121A; Crystal
Technology & Industries, Inc, Dallas, TX, USA) for 15 sec. The
sample was then incubated at 56°C for 10 min and centrifuged at
8,000 x g for 1 min to remove drops from the inside of the lid.
Subsequently, 200 μl ethanol (96–100%; Sigma-Aldrich, St.
Louis, MO, USA) was added to the sample and it was pulse vortexed
again for 15 sec then centrifuged at 6,000 x g for 1 min. The
sample was then added to the the QIAamp Mini spin column without
wetting the rim, it was centrifuged at 6000 x g for 1 min, 500
μl Buffer AW1 was added and it was centrifuged again at 6000
x g for 1 min. A total of 500 μl Buffer AW2 was then added
without wetting the rim, and the sample was centrifuged twice at
20,000 x g for 3 min. A total of 200 μl Buffer AE was then
added and the sample was incubated at room temperature for 1 min,
and subsequently centrifuged at 6000 x g for 1 min. All buffers
were from the DNeasy Blood & Tissue kit.
All coding exons of FBN1 (NM_000138.4), TGFBR1
(NM_004612.2), TGFBR 2 (NM_003242.5), MYH11 (NM_022844.2), ACTA2
(NM_001141945.1), SLC2A10 (NM_030777.3), NOTCH1 (NM_017617.3) and
COL3A1 (NM_000090.3) were amplified by the polymerase chain
reaction (PCR) System 9700 (Applied Biosystems Life Technologies,
Foster City, CA, USA). The amplification used 25 μl reaction
mixture, which consisted of 0.3 mM deoxyribonucleotide
triphosphates (BioTeke Corporation, Beijing, China), 1X PCR buffer
(10 mM Tris-hydrochloric acid pH 9.0, 50 mM potassium chloride,
0.1% Triton X-100 and 0.01% w/v gelatin; BioTeke Corporation), 2.0
mM magnesium chloride (BioTeke Corporation), 0.5 μM each
primer (forward and reverse; created by Beijing Genomics
Institution, Beijing, China), 1.5 U Taq polymerase and 50 ng
genomic DNA. The thermal cycling conditions were as follows:
Initial denaturation at 95°C for 4 min, 35 cycles of amplification
consisting of denaturation at 95°C for 1 min, primer annealing at
X°C for 30 sec and primer extension at 72°C for 1 min. A final
extension step was performed at 72°C for 7 min. The PCR products
were sequenced by the ABI 3100 Genetic Analyzer (Applied Biosystems
Life Technologies). The results of sequencing were presented using
Chromas software, version 2.1.1 (http://technelysium.com.au/?page_id=13). The overlap
peaks frequently indicated variants and according to the base
sequence around the variant, the location of the variant in the
gene was identified. Continuous overlap peaks indicated frameshift
variants and single overlap peak may have indicated nonsense,
synonymous or missense variants. The identified mutation was
searched in Pubmed (http://www.ncbi.nlm.nih.gov/pubmed/), Google scholar
(http://scholar.google.com/), dbSNP
(http://www.ncbi.nlm.nih.gov/SNP/) and
the Exome Sequencing Project (ESP; http://evs.gs.washington.edu/EVS/) (10), and mutations were only considerd as
candidate mutations if they had never been previously reported in
humans, and were absent from dbSNP and ESP. Three online protein
prediction software packages, Polyphen-2 (polymorphism phenotyping)
(11), SIFT (Sorting Intolerant
From Tolerant) (12) and Mutation
Taster (13) were used to evaluate
the effect of the novel mutations. In the current study, frameshift
and nonsense variants were not observed and G984R in COL3A1 was the
only candidate mutation. All of the three protein prediction
software packages considered this variant to be damaging and it was
located in a conserved region in COL3A1. This mutation also led to
the replacement of one glycine in the (Gly-Xaa-Yaa) n repeat of the
collagen triple helix, which was considered pathogenic in various
previous studies (3,14–21).
Thus, G984R in COL3A1 was identified as a pathogenic mutation in
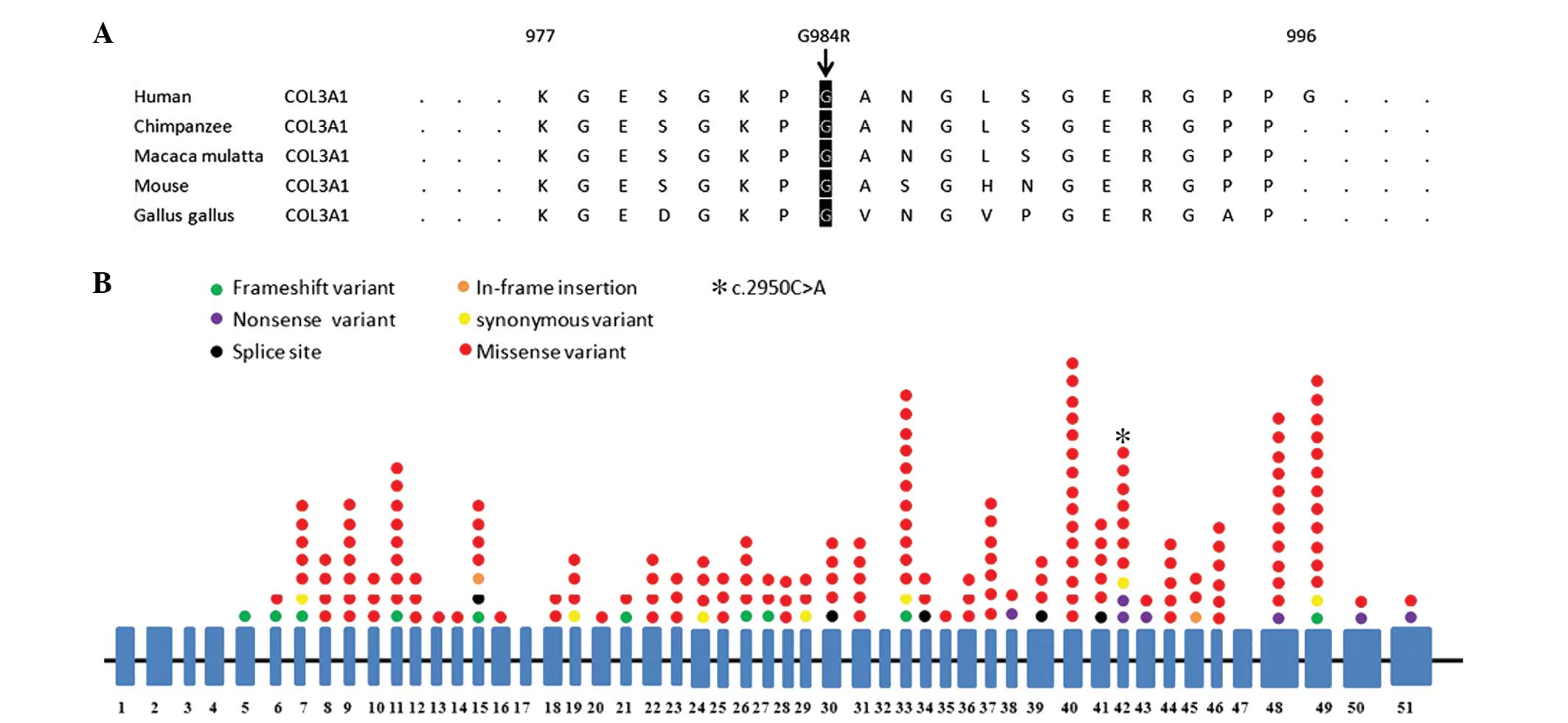
the current study. The amino acid sequence of human COL3A1 from
positions 977 to 996 was compared with similar regions in the
chimpanzee, Macaca mulatta, mouse and Gallus gallus
using Ensembl (http://asia.ensembl.org/index.html).
Variants summary
The variants previously reported in COL3A1 were
searched in Pubmed in order to identify mutations associated with
aortic aneurysm. All of the variants were matched in dbSNP and ESP
to identify variants previously associated with disease in healthy
populations. Mutations leading to aortic aneurysm were evaluated by
Polyphen-2, SIFT and Mutation Taster.
Results
Result of computerized tomographic
angiography (CTA)
The images from the CTA revealed that the root of
the ascending aorta was dilated and had formed a sacculated aortic
aneurysm (Fig. 1A and B) with a
diameter of >7 cm.
Novel mutation and functional
prediction
Sequencing the PCR products of the eight aortic
aneurysm-related genes, mentioned in Patients and methods,
identified the nucleotide A instead of G at position 2950 of the
coding sequence (c.2950G>A, Fig.
2B) in COL3A1, leading to an amino acid change from glycine to
arginine at position 984 of the protein (p.G984R). This
c.2950G>A mutation was only present in the patient and not in
his family members or our control cohorts as well as dbSNP and ESP.
A search of Pubmed and Google Scholar revealed that this mutation
has not been previously reported. The prediction results of
Polyphen-2, SIFT and Mutation Taster were probably damaging,
damaging and disease causing, respectively (Table I). Alignment of multiple COL3A1
protein sequences across species showed the G984 affected amino
acid is located in the conserved amino acid region in different
mammals (Fig. 3A).
 | Table IOverview of six mutations associated
with aortic aneurysm. |
Table I
Overview of six mutations associated
with aortic aneurysm.
| Mutation | Exon | Mutation effect | Protein change | dbSNP | ESP | Mutation Taster | SIFT | Polyphen-2 | Reference | Disease |
|---|
| c.505>T | 6 | Missense | L169F | rs111391222 | − | Polymorphism (score:
0.60) | Tolerated (score
0.731) | − | 22 | AAA |
| c.907G>A | 14 | Missense | G303R | rs121912919 | + | Disease causing
(score: 3.41) | Damaging (score
0.000) | − | 23 | AAA |
| c.2356G>A | 35 | Missense | G786R | rs113485686 | − | Disease causing
(score: 3.41) | Damaging (score
0.000) | − | 10 | TAA/AAA |
| c.2633G>A | 39 | Missense | R878H | − | − | Disease causing
(score: 0.79) | Damaging (score
0.600) | − | 11 | TAA |
| c.1815+5G>A | − | Splice site | − | rs146652498 | − | − | − | − | 12 | TAA |
| c.2950G>A | 42 | Missense | G984R | − | − | Disease causing
(score: 3.41) | Damaging (score
0.000) | Probably damaging
(score 1.000) | Present study | TAA |
Variants review
A literature review revealed that 268 variants,
including 66 variants in the introns and 204 variants in the exons
(Fig. 3B), were identified in
COL3A1. Of the 268 mutations, 257 variants were previously reported
to be pathogenic. In total, 1.9% (5/257) of the mutations were
found in the ESP. Of the exon variants, 159 of 204 variants lead to
the replacement of one glycine in the (Gly-Xaa-Yaa)n repeat of the
collagen triple helix. All of the 159 missense variants have been
previously reported as pathogenic and were not found in the ESP
database. In total, five mutations [c.505C>T (5), c.907G>A (6), c.2356G>A (7), c.2633G>A (8) and c.1815+5G>A (9)] were reported as causes of aortic
aneurysm. The results of functional prediction by Polyphen-2, SIFT
and Mutation Taster are listed in Table I.
Discussion
Due to vascular/intestinal rupture, EDS IV is the
most severe type among the different types of EDS. According to the
Villefranche nosology (1)
(Table II), the combination of
two major criteria is highly specific to this disease. The patient
described in this study had an aortic aneurysm combined with thin
translucent skin. Hence, his clinical diagnosis was EDS IV. The
molecular genetic analysis revealed a mutation in COL3A1, which
confirmed the diagnosis. In order to avoid aneurysmal rupture, the
ascending aorta of the patient was replaced with an artificial
aorta and three weeks later, following recovery, the patient was
discharged from hospital.
| Table IIDiagnostic criteria for EDS IV caused
by abnormal type III collagen. |
Table II
Diagnostic criteria for EDS IV caused
by abnormal type III collagen.
| Major criteria | Minor criteria |
|---|
| Thin translucent
skin | Acrogeria |
|
Arterial/intestinal/uterine fragility or
rupture | Hypermobility of
small joints
Tendon and muscle rupture |
| Extensive
bruising | Clubfoot |
| Characteristic facial
appearance | Early-onset varicose
veins
Pneumothorax
Gingival recession
Positive family history/sudden death
Arteriovenous, carotid-cavernous sinus fistula |
Since Richards et al (24) initially described a variant of
COL3A1 in EDS IV patients, 268 variants, including 66 intron
variants and 204 exon variants (Fig.
3B), were identified in COL3A1. Of the exon variants, 159/204
variants led to the replacement of one glycine in the
(Gly-Xaa-Yaa)n repeat of the collagen triple helix. All of the 159
missense variants have been previously reported as pathogenic and
were not present in the ESP database. In total, five mutations
[c.505C>T (5), c.907G>A
(6), c.2356G>A (7), c.2633G>A (8) and c.1815+5G>A (9)] were reported as causes of aortic
aneurysm. The results of functional prediction by Polyphen-2, SIFT
and Mutation Taster are listed in Table I. However, they were all found in
the ESP database. Whole exome data from the National Heart, Lung,
and Blood Institute Grand Opportunity (NHLBI GO) ESP provided
sequencing results of all protein-coding regions in 6,503
individuals without heart disease (12). Any reported pathogenic mutation
identified in the ESP database may be false positive. This idicates
that these five EDS IV-associated mutations may be SNPs or
non-pathogenic.
To the best of our knowledge, the c.2950G>A
mutation identified in this study has not been previously reported,
and is not present in the ESP database and control cohorts. This
mutation led to an amino acid change from glycine to arginine at
position 984 of the protein (p.G984R), which may damage the
formation of the triple helix. The prediction results of the
Polyphen-2, SIFT and Mutation Taster programs were that the
mutation may be damage or disease causing. A review of the
literature revealed that all the missense variants leading to the
replacement of one glycine in the (Gly-Xaa-Yaa)n repeat of the
collagen triple helix were previously reported to be pathogenic.
Hence, c.2950G>A is considered to be the cause of EDS IV in this
patient, which expands the spectrum of COL3A1 mutations and may
make a contribution to the genetic counseling of EDS IV in the
future.
COL3A1 encodes a procollagen molecule, proa1 (III);
basic collagen synthesis requires three polypeptide procollagen
chains, referred to as α chains, to be folded tightly into a triple
helix (3). Every third amino acid
in the protein triple helix is a glycine, and substitution of a
glycine slows down formation of the triple helix (25). This may lead to collagen III
breakdown or over modification. Collagen III is a major component
of the extracellular matrix in skin, blood vessels and a variety of
internal organs. Abnormal collagen III may result in thin
translucent skin and thin artery walls, which leads to a number
vascular complications, including aneurysm, artery dissection and
so on. Vascular complications are commonly observed in EDS IV,
while ascending aortic aneurysm is extremely rare. Hetzer et
al (26) reported an isolated
giant ascending aortic aneurysm in an infant with EDS IV. The
infant underwent replacement of the ascending aorta and proximal
aortic arch. Genetic analysis was not performed. To the best of our
knowledge, this is the only other report of an EDS IV patient with
ascending aortic aneurysm, which means the novel mutation
identified in our study is the first COL3A1 mutation reported in
association with ascending aortic aneurysm.
To date, including the novel mutation identified in
this study, only six mutations in COL3A1 have been found to be
associated with aortic aneurysms (5–9).
With the exception of c.907G>A identified in ESP, the mutations
are distributed in different regions of COL3A1, and the correlation
between aortic aneurysm and these specific mutations remains
unknown. Thus, these associations require further
investigation.
In conclusion, the results of this study revealed a
novel COL3A1 mutation (p.G984R) in an EDS IV patient with ascending
aortic aneurysm, which expands the spectrum of COl3A1 mutations and
may aid with the genetic counseling and diagnosis of EDS IV in the
future.
Acknowledgments
The authors would like to thank Miss Wang Jian for
her assistance in collecting blood samples, and additionally would
like to thank Mr Fan Liangliang and Mr Huang Hao for their
assistance in the statistical analysis.
References
|
1
|
Beighton P, De Paepe A, Steinmann B,
Tsipouras P and Wenstrup RJ: Ehlers-Danlos syndromes: revised
nosology, Villefranche, 1997. Ehlers-Danlos National Foundation
(USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet.
77:31–37. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Germain DP: Ehlers-Danlos syndrome type
IV. Orphanet J Rare Dis. 2:322007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pepin M, Schwarze U, Superti-Furga A and
Byers PH: Clinical and genetic features of Ehlers-Danlos syndrome
type IV, the vascular type. N Engl J Med. 342:673–680. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pepin M and Byers P: Ehlers-Danlos
syndrome type IV. Gene Reviews. 2011, http://www.ncbi.nlm.nih.gov/books/NBK1494/.
[Accessed November 20, 2013].
|
|
5
|
Anderson DW, Tromp G, Kuivaniemi H, et al:
A type III procollagen gene mutation in a patient with late onset
aneurysms. Matrix Biology. 14:3921994. View Article : Google Scholar
|
|
6
|
Tromp G, Wu Y, Prockop DJ, et al:
Sequencing of cDNA from 50 unrelated patients reveals that
mutations in the triple-helical domain of type III procollagen are
an infrequent cause of aortic aneurysms. J Clin Invest.
91:2539–2545. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kontusaari S, Tromp G, Kuivaniemi H,
Romanic AM and Prockop DJ: A mutation in the gene for type III
procollagen (COL3A1) in a family with aortic aneurysms. J Clin
Invest. 86:1465–1473. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kathiravel U, Keyser B, Hoffjan S, et al:
High-density oligonucleotide-based resequencing assay for mutations
causing syndromic and non-syndromic forms of thoracic aortic
aneurysms and dissections. Mol Cell Probes. 27:103–108. 2013.
View Article : Google Scholar
|
|
9
|
Sakai H, Suzuki S, Mizuguchi T, et al:
Rapid detection of gene mutations responsible for non-syndromic
aortic aneurysm and dissection using two different methods:
resequencing microarray technology and next-generation sequencing.
Hum Genet. 131:591–599. 2012. View Article : Google Scholar
|
|
10
|
Exome Variant Server, NHLBI GO Exome
Sequencing Project (ESP). Seattle, WA: (URL: http://evs.gs.washington.edu/EVS/)
[Accessed November 20, 2013].
|
|
11
|
Sunyaev S, Ramensky V and Bork P: Towards
a structural basis of human non-synonymous single nucleotide
polymorphisms. Trends Genet. 16:198–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: Mutation Taster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pepin M, Schwarze U, Superti-Furga A and
Byers PH: Clinical and genetic features of Ehlers-Danlos syndrome
type IV, the vascular type. N Engl J Med. 342:673–680. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Drera B, Zoppi N, Ritelli M, et al:
Diagnosis of vascular Ehlers- Danlos syndrome in Italy: clinical
findings and novel COL3A1 mutations. J Dermatol Sci. 64:237–240.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eder J, Laccone F, Rohrbach M, Giunta C,
Aumayr K, Reichel C and Trautinger F: A new COL3A1 mutation in
Ehlers–Danlos syndrome type IV. Exp Dermatol. 22:231–234. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kroes HY, Pals G and van Essen AJ:
Ehlers-Danlos syndrome type IV: unusual congenital anomalies in a
mother and son with a COL3A1 mutation and a normal collagen III
protein profile. Clin Genet. 63:224–227. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Palmeri S, Mari F, Meloni I, et al:
Neurological presentation of Ehlers-Danlos syndrome type IV in a
family with parental mosaicism. Clin Genet. 63:510–515. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sa YJ, Kim YD, Moon SW, Kim CK and Ki CS:
Occlusive vascular Ehlers-Danlos syndrome accompanying a congenital
cystic adenomatoid malformation of the lung: report of a case. Surg
Today. 43:1467–1469. 2013. View Article : Google Scholar
|
|
20
|
Rebelo M, Ramos L, Lima J, et al:
Ehlers-Danlos syndrome type IV in association with a (c.970G>A)
mutation in the COL3A1 gene. Acta Med Port. 24:1079–1086. 2011.
|
|
21
|
Kashizaki F, Hatamochi A, Kamiya K,
Yoshizu A and Okamoto H: Vascular-type Ehlers-Danlos syndrome
caused by a hitherto unknown genetic mutation: a case report. J Med
Case Rep. 7:352013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tan ZP, Huang C, Xu ZB, Yang JF and Yang
YF: Novel ZFPM2/FOG2 variants in patients with double outlet right
ventricle. Clin Genet. 82:466–471. 2012. View Article : Google Scholar
|
|
23
|
Chen J, Li B, Yang Y, et al: Mutations of
the TGFBR2 gene in Chinese patients with Marfan-related syndrome.
Clin Invest Med. 33:E14–E21. 2010.PubMed/NCBI
|
|
24
|
Richards AJ, Ward PN, Narcisi P, et al: A
single base mutation in the gene for type III collagen (COL3A1)
converts glycine 847 to glutamic acid in a family with
Ehlers-Danlos syndrome type IV: an unaffected family member is
mosaic for the mutation. Hum Genet. 89:414–418. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizuno K, Boudko S, Engel J and Bächinger
HP: Vascular Ehlers-Danlos Syndrome mutations in type III collagen
differently stall the triple helical folding. J Biol Chem.
288:19166–19176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hetzer R, Delmo Walter EM, Meyer R and
Alexi-Meskishvili V: Isolated giant aortic aneurysm in an infant:
Ehlers-Danlos Syndrome Type IV. Ann Thorac Surg. 86:632–634. 2008.
View Article : Google Scholar : PubMed/NCBI
|