Introduction
Cardiac hypertrophy is an important compensatory
mechanism for pathophysiological states, involving an increase in
size and/or thickness of the ventricles of the heart. Cardiac
hypertrophy is an adaptive response to increased workload or to
defects in the efficiency of the contractile machinery of the
heart. Sustained hypertrophic stimulation, however, is a
maladaptive response causing adverse effects, including
cardiomyocyte fetal gene re-expression and cardiac interstitial
fibrosis (1,2). Cardiac hypertrophy is a
well-established risk factor of cardiovascular mortality, and can
significantly increase the incidence of cardiovascular events,
including sudden death, ventricular arrhythmia, myocardial ischemia
and heart failure, and can thus contribute to an increase in the
rate of mortality (3). The
mechanistic process of cardiac hypertrophy remains to be fully
elucidated. Current therapies for cardiac hypertrophy have
predominantly focused on the regulation of hemodynamics (4); however, an effective method to
prevent and treat cardiac hypertrophy remains to be developed.
Pharmacological interventions targeting the molecular changes
involved in cardiac hypertrophy may be a promising approach for the
prevention of cardiac hypertrophy and progression to heart
failure.
3,3′-Diindolylmethane (DIM) is a Traditional Chinese
Medicine that is extracted from Brassica plants. It is the major
in vivo product derived from the acid-catalyzed condensation
of indole-3-carbinol (I3C). Numerous studies have suggested that
DIM has various properties, including eliminating free radicals,
antioxidant and anti-angiogenic effects and promoting apoptosis in
tumor cells (5–7). DIM can affect mitogen-activated
protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/Akt and the
nuclear factor-KB (NF-κB) signaling pathways (6,8,9), and
functions of anti-cancer agents (8), anti-angiogenic (6) and anti-inflammatory (9) processes. In a previous study by our
group (10), it was shown that DIM
prevented the development of cardiac hypertrophy and could revert
established cardiac hypertrophy through the promotion of AMPKα and
inhibition of mechanistic target of rapamycin (mTOR) signaling
in vivo. Furthermore, the cardioprotective effects of DIM
were ameliorated in mice lacking functional 5′ AMP-activated
protein kinase α (AMPKα)2 (10).
Angiotensin II (Ang II) has a central function in
the regulation of the cardiovascular system (11). Activation of the renin-angiotensin
system (RAS), and the subsequent generation of Ang II, are
important mediators of myocardial fibrosis, pathological
hypertrophy and heart failure (12,13).
The purpose of the present study was to use an in vitro
model of hypertrophy, induced by 1 μM Ang II in cultured rat
cardiac H9c2 cells, to evaluate the effects of DIM on the cellular
response.
Materials and methods
Materials
The primary antibodies included phosphorylated
(p)-AMPKα (sc- 01631; Santa Cruz, Santa Cruz, CA, USA), p-c-Jun,
total (T-)c-Jun, T-AMPKα (BS4046, BS1061, BS6271, respectively;
Bioworld Technology Inc., St. Louis Park, MN, USA) and p-mTOR
(2971), T-mTOR (2983; Cell Signalling Technology, Inc.), p-p70S6K
(9234), T-p70S6K (2708), p-S6 (2215), T-S6 (2217), p-eIF4E (9741),
T-eIF4E (2067P), p-4E-BP1 (2855), T-4E-BP1 (9644),
p-mitogen-activated protein kinase kinase (MEK)1/2 (9154), T-MEK1/2
(9122), p-extracellular signal-regulated kinase (ERK)1/2 (4370),
T-ERK1/2 (4695), p-p38 (511), T-p38 (9212), p-c-Jun N-terminal
kinase (JNK)1/2 (4668P), T-JNK1/2 (9258) and GAPDH (2118) (all
purchased from Cell Signalling Technology, Inc., Danvers, MA, USA).
DIM was purchased from Sigma-Aldrich (St. Louis, MO, USA; D9568-5G)
and dissolved in dimethyl sulfoxide (RNBC0311; Sigma-Aldrich) for
the in vitro bioassay.
Cell culture
Rat cardiac H9c2 cells (Cell Bank of the Chinese
Academy of Sciences, Shanghai, China) were cultured in Dulbecco’s
modified Eagle’s medium (DMEM, C1 1995; Gibco-BRL, Carlsbad, CA,
USA) supplemented with 10% fetal bovine serum (FBS; Gibco-BRL,
10099-133), 100 U/ml penicillin/100 mg/ml streptomycin (Gibco-BRL,
15140) and 5% CO2 at 37°C. The media was exchanged every
1–2 days and subcultured to 70–80% confluency. Cells were plated at
an appropriate density according to each experimental design. H9c2
cells were seeded in six-well plates at a density of
0.25×106 cells/well. Following 24 h adherence, the
culture medium was changed to serum-free DMEM 12 h prior to the
experiment. Cells were incubated with DIM (1, 5, and 10 μM)
in serum-free DMEM at 37°C with or without 1 μM AngII
(A9525; Sigma-Aldrich) for 24 or 48 h.
Immunofluorescence
Cells were analyzed for cardiac α-actinin expression
by immunofluorescence, in order to identify rat cardiac H9c2 cells
and assess cardiomyocyte hypertrophy. The cells were washed with
phosphate-buffered saline (PBS), fixed with RCL2® (RCL2-CS24L;
ALPHELYS, Plaisir, France) and permeabilized in 0.1% Triton™ X-100
in PBS. The cells were then incubated with anti-α-actinin (05-384;
Millipore, Billerica, MA, USA) at a dilution of 1:100 in 1% goat
serum. The cells were then incubated with Alexa FluorH 488 goat
anti-mouse immunoglobulin (Ig)G (A11001; Invitrogen Life
Technologies, Carlsbad, CA, USA) secondary antibody. The cells on
coverslips were mounted onto glass slides with Slow Fade Gold
antifade reagent with DAPI (S36939; Invitrogen Life Technologies).
The outline of 40 cells from each group were visualized by light
microscopy (BX51TRF; Olympus Corporation, Tokyo, Japan). A single
cell was measured using a quantitative digital image analysis
system (Image-Pro Plus version 6.0; Media Cybernetics, Rockville,
MD, USA).
Quantitative polymerase chain reaction
(qPCR)
qPCR was used to detect RNA expression levels of
hypertrophic and fibrotic markers. Total RNA was extracted from
frozen, pulverized mouse cardiac tissue using TRIzol™ (15596-026;
Roche Diagnostics, Mannheim, Germany). RNA yield and purity were
evaluated using a SmartSpec Plus Spectrophotometer (Bio-Rad,
Hercules, CA, USA), comparing the A260/A280 and A230/260 ratios.
The RNA (2 μg of each sample) was reverse-transcribed into
cDNA using oligo(DT) primers and the Transcriptor First Strand cDNA
Synthesis kit (04896866001; Roche Diagnostics). The PCR products
were quantified using a LightCycler 480 SYBR® Green 1
Master Mix (04707516001; Roche Diagnostics). Following an initial 5
min denaturation step at 95°C, a total of 42 primer-extension
cycles were carried out. Each cycle consisted of a 10 sec
denaturation step at 95°C, a 20 secannealing step at 60°C, and a 20
sec incubation at 72°C for extension. Then a final extension step
was performed at 72 °C for 10 min. The double standard curve was
used to quantify the PCR results. Calibrator Normalized Ratio =
(concentration of sample target/concentrations of sample
reference)/(concentration of calibrator target/concentration of
calibrator reference). The results were normalized against GAPDH
gene expression. The sequences of the oligonucleotide primers
(Sangon Biotech, Shanaghai, China) were as follows: : Atrial
natriuretic peptide (ANP) forward, 5′-AAAGCAAACTGAGGGCTCTGCTCG-3′,
reverse, 5′-TTCGGTACCGGAAGCTGTTGCA-3′; brain natriuretic peptide
(BNP) forward, 5′-CAGCAGCTTCTGCATC GTGGAT-3′, reverse,
5′-TTCCTTAATCTGTCGCCGCTGG-3′; myosin heavy chain β (β-MHC) forward,
5′-TCTGGACAGCTC CCCATTCT-3′, reverse, 5′-CAAGGCTAACCTGGAGAA
GATG-3′; and GAPDH forward, 5′-GACATGCCGCCTGGAGA AAC-3′, and
reverse, 5′-AGCCCAGGATGCCCTTTAGT-3′.
Western blot analysis
Cultured cardiac H9c2 cells were lysed in
radioimmunoprecipitation (RIPA) lysis buffer (720 μl RIPA,
20 μl PMSF (1 mM), 100 μl complete, 100 μl
cOmplete (Roche, Indianapolis, IN, USA, 04693124001), 100 μl
phostop (Roche, 04906837001), 50 μl NaF (1 mM), 10 μL
Na3VO4, per ml) and 30 μg cell lysate
was used for protein separation by 10% SDS-PAGE. The proteins were
then transferred to polyvinylidene difluoride (PVDF) membranes
(Millipore). Specific protein expression levels were normalized to
the GAPDH protein levels of the total cell lysate and cytosolic
proteins on the same PVDF membranes. The following primary
antibodies were used: p-AMPKα, p-c-Jun, T-c-Jun, T-AMPKα, p-mTOR,
T-mTOR, p-p70S6K, T-p70S6K, p-S6, T-S6, p-eIF4E, T-eIF4E, p-4E-BP1,
T-4E-BP1, p-mitogen-activated protein kinase kinase (MEK)1/2, T-
MEK 1/2, p-extracellular signal-regulated kinase (ERK)1/2,
T-ERK1/2, p-p38, T-p38, p-c-Jun N-terminal kinase (JNK)1/2,
T-JNK1/2 and GAPDH. The primary antibodies were diluted at 1:1,000,
with the exception of p-AMPKα, which was diluted 1:200. Antibody
incubation was performed overnight with gentle shaking at 4°C.
Quantification of the western blots was performed using an Odyssey
infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA). The
secondary antibodies, goat anti-rabbit IRdye® 800 CW
(LI-COR, 926-32211) IgG and goat anti-mouse IRdye® 800
CW (LI-COR, 926-32210), were used at a 1:10,000 dilution at 37°C in
Odyssey blocking for 1 h. The blots were scanned using an infrared
Li-Cor scanner, allowing for simultaneous detection of two targets
(phosphorylated and total protein) within the same experiment.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Differences among groups were determined by two-way
analysis of variance followed by Tukey’s post-hoc test. Comparisons
between two groups were performed using an unpaired Student’s
t-test. Statistical analyses were conducted using SPSS version 13.0
(SPSS Inc., Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
DIM attenuates cardiac H9c2 cell
hypertrophy
An in vitro model using Ang II (1 μM)
in cultured rat cardiac H9c2 cells was established in order to
confirm the effects of DIM on cardiac hypertrophy. Following
stimulation with DIM (1, 5, and 10 μM) with or without Ang
II (1 μM) for 24 h, cells were characterized for cardiac
α-actinin expression by immunofluorescence, to assess cardiomyocyte
hypertrophy. H9c2 cells showed an enlarged cell surface area as
compared with those induced by DIM (1, 5, and 10 μM) with
Ang II for 24 h (Fig. 1A and B).
qPCR demonstrated that DIM (1, 5, and 10 μM) markedly
decreased the induction of ANP, BNP and β-MHC mRNA expression
induced by Ang II (1 μM), most significantly in cells
treated with 5 μM DIM (Fig.
1C). There were no significant differences between the DIM 5
μM and DIM 10 μM groups. Additionally, qPCR
demonstrated that cells treated with 5 μM DIM for 6, 12, and
24 h exhibited a markedly decreased induction of ANP, BNP and β-MHC
mRNA expression by Ang II (Fig.
1D). These findings indicated that DIM attenuated cell
hypertrophy in vitro.
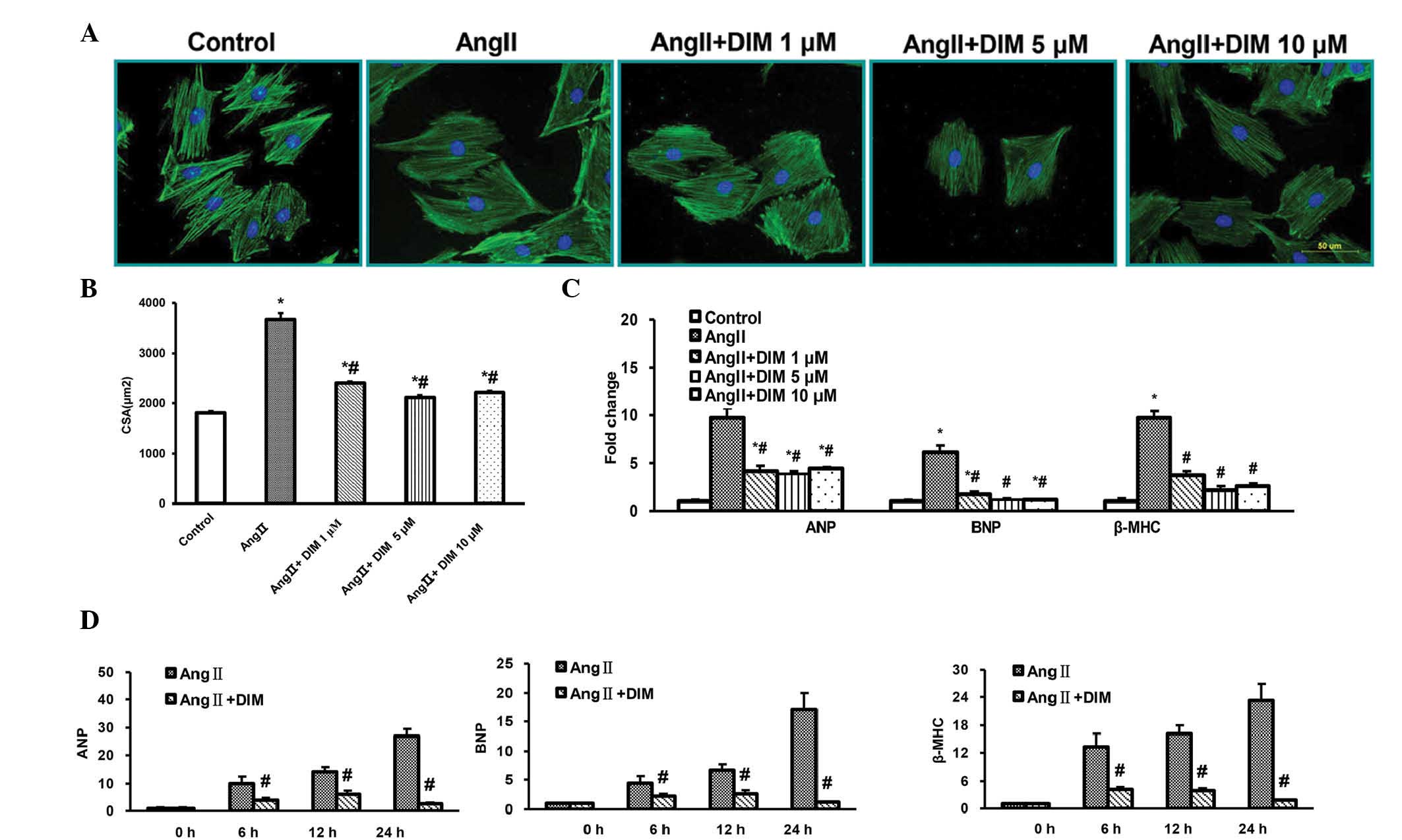 | Figure 1DIM attenuates cardiac H9c2 cell
hypertrophy in vitro. (A and B) Effects of DIM on the
reduction in size of H9c2 cells induced by Ang II (1 μM) for
24 h. (A) Representative immunofluorescent staining. Scale bar, 10
μm. (B) Quantitative analysis of immunofluorescence.
*P<0.05 vs control group. #P<0.05 vs Ang II group.
(C) qPCR analysis of the mRNA levels of ANP, BNP and β-MHC induced
by DIM (1, 5, 10 μM) with Ang II (1 μM) for 24 h.
*P<0.05 vs control group. #P<0.05 vs Ang II group.
(D) qPCR analysis of the mRNA levels of ANP, BNP and β-MHC induced
by DIM (5 μM) with Ang II (1 μM) at the time-points
indicated. #P<0.05 vs Ang II group at the same
time-point. ANP, atrial natriuretic peptide; BNP, brain natriuretic
peptide; Ang II, angiotensin II; DIM, 3,3′-diindolylmehane; β-MHC,
myosin heavy chain β; CSA, cell surface area; qPCR, quantitative
polymerase chain reaction analysis. |
DIM promotes p-AMPKa activity and
inhibits mTOR signaling in response to Ang II in vitro
In a previous study by our group, it was identified
by western blotting that DIM could enhance the phosphorylation of
AMPKα and block mTOR signaling induced by pressure overload in
vivo (7). In the present
study, the activation of AMPKα and mTOR signaling by Ang II (1
μM) in H9c2 cells treated with DIM (5 μM) or PBS for
0, 15, 30 and 60 min was evaluated for the first time, to the best
of our knowledge. The activation of AMPKα was increased in H9c2
cells treated with 5 μM DIM, as compared with that in
response to 1 μM AngII alone. The levels of phosphorylated
mTOR, p70S6K, S6, eIF4E and 4E-BP1 were significantly increased in
H9c2 cells in response to 1 μM Ang II. However, the
increased levels of mTOR, p70S6K, S6, eIF4E and 4E-BP1 were reduced
in cells treated with 5 μM DIM (Fig. 2A and B). These findings indicated
that DIM attenuated cell hypertrophy, predominantly through
stimulating p-AMPKα activity and inhibiting mTOR signaling in
vitro.
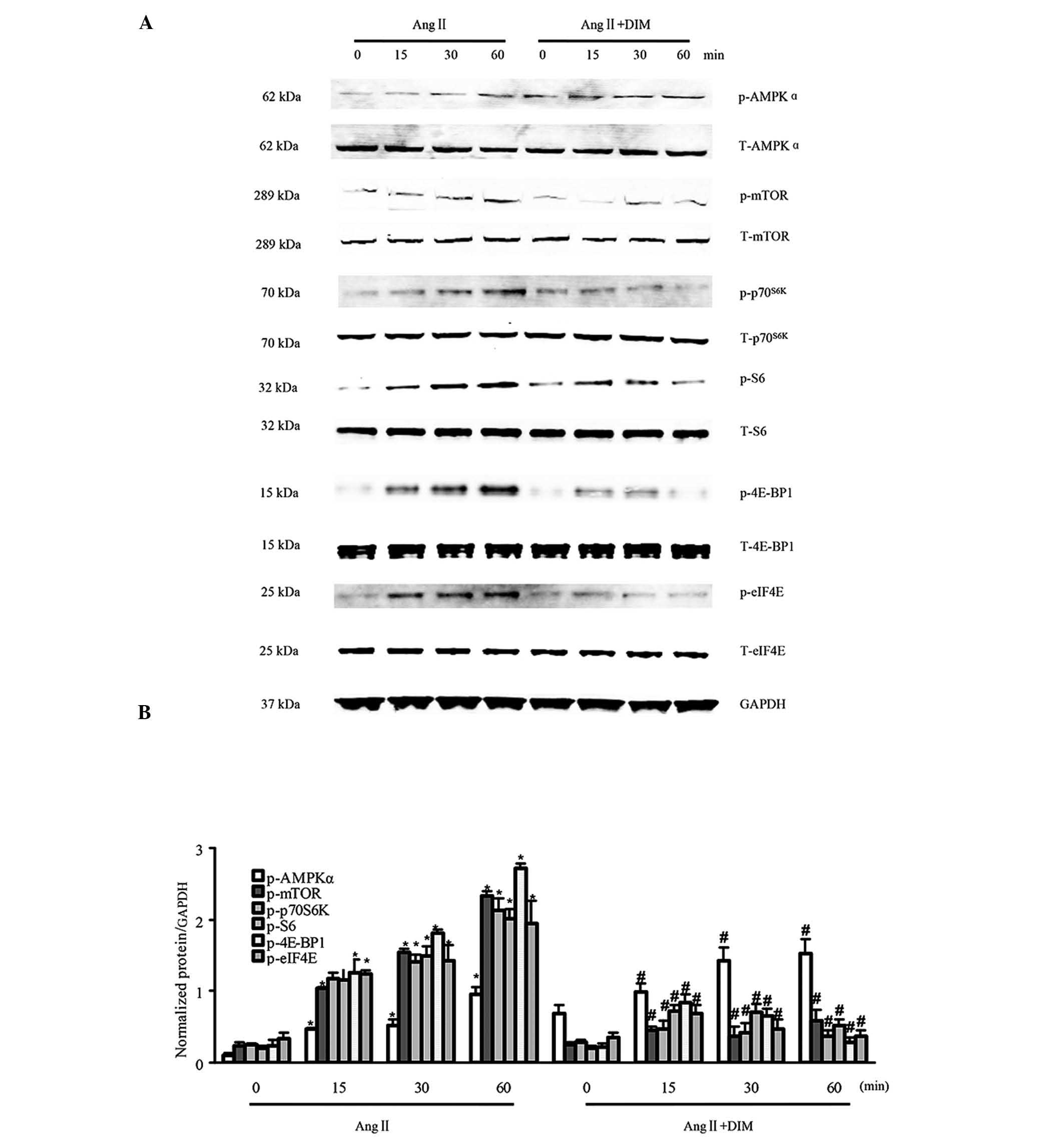 | Figure 2DIM enhances p-AMPKα activity and
inhibits mTOR signaling in response to hypertrophic stimuli in
vitro. (A and B) The levels of total (T-) and phosphorylated
(p-)AMPKα, mTOR, p70S6K, S6, eIF4E, and 4E-BP1 expression in H9c2
cells treated with DIM (5 μM) with Ang II (1 μM) at
the time-points indicated. (A) Representative blots. (B)
Quantitative results. *P<0.05 vs Ang II group at time-point 0.
#P<0.05 vs Ang II group at the same time-point. Ang
II, angiotensin II; DIM, 3,3′-diindolylmehane; p-AMPKα,
phosphorylated 5′ AMP-activated protein kinase α; mTOR, mammalian
target of rapamycin. |
DIM inhibits MAPK signaling in response
to hypertrophic stimuli in vitro
MAPK signaling has a central function in cardiac
hypertrophy (14). The state of
activation of MAPK signaling in H9c2 cells treated with 5 μM
DIM or PBS for 0, 15, 30, and 60 min by 1 μM Ang II, was
therefore evaluated. It was identified that the phosphorylated
levels of MEK1/2, ERK1/2, p38, JNK1/2 and c-Jun were significantly
increased in H9c2 cells in response to Ang II (1 μM).
However, the increased levels of MEK1/2, ERK1/2, JNK1/2 and c-Jun
were attenuated in cells treated with 5 μM DIM, whereas p38
was similarly activated in DIM- and PBS-treated groups (Fig. 3A and B). The results indicated that
DIM significantly ameliorated cardiac hypertrophy through direct
inhibition of the MAPK signaling pathway in vitro.
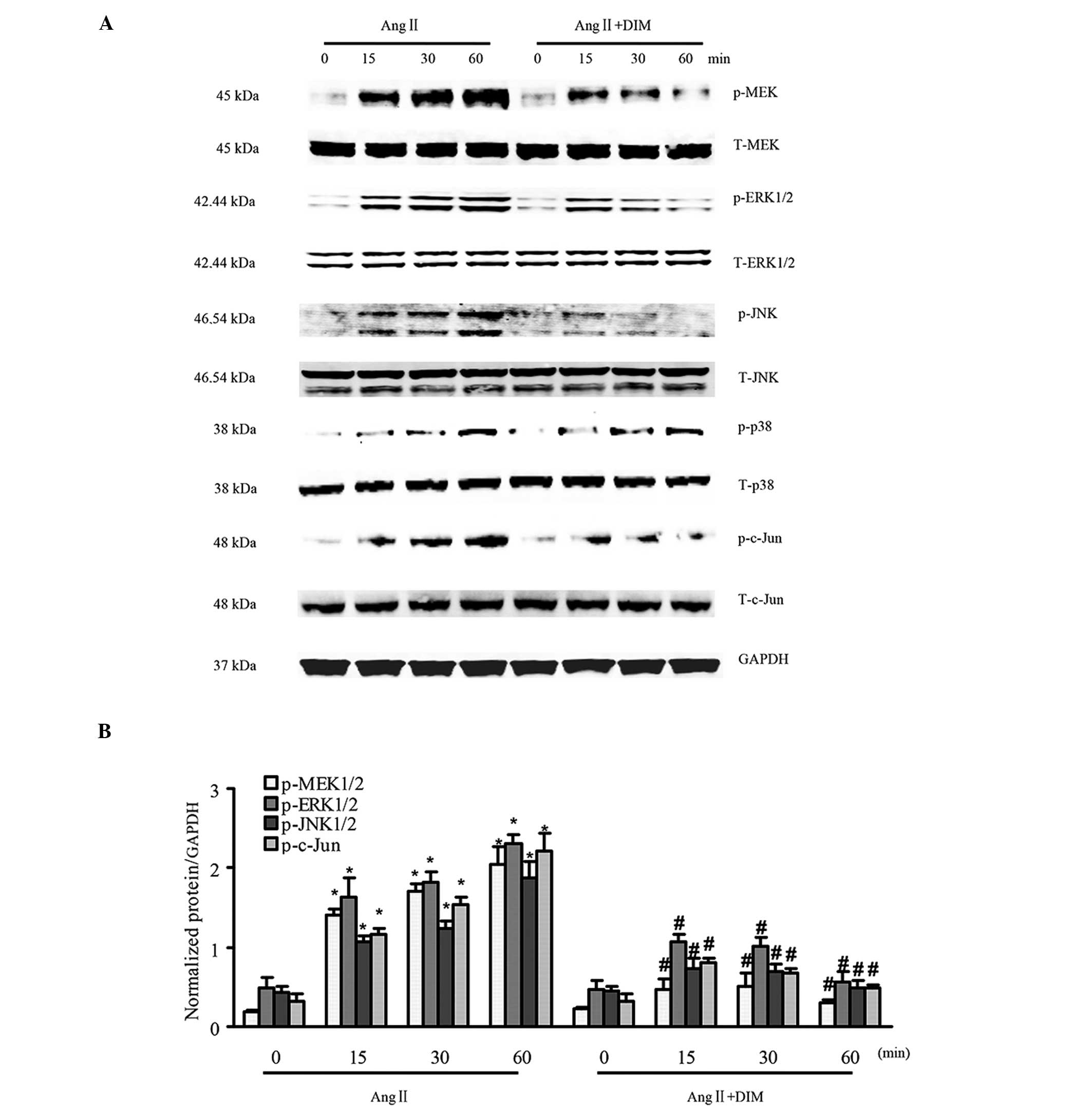 | Figure 3DIM inhibits MAPK signaling induced
by Ang II. (A and B) The levels of total (T-) and phosphorylated
(p-)MEK1/2, ERK1/2, p38, JNK1/2 and c-Jun expression in H9c2 cells
treated with DIM (5 μM) with Ang II (1 μM) at the
indicated time-points. (A) Representative blots. (B) Quantitative
results. *P<0.05 vs Ang II group at time-point 0.
#P<0.05 vs AngII group at the same time-point. Ang
II, angiotensin II; DIM, 3,3′-diindolylmehane; MAPK,
mitogen-activated protein kinase; p-MEK, phosphorylated
mitogen-activated protein kinase kinase; ERK, extracellular
signal-regulated kinase; JNK, c-Jun N-terminal kinase. |
Discussion
In the present study, the role of DIM in cultured
rat cardiac H9c2 cells with induced hypertrophy by Ang II in
vitro was examined. The results demonstrated that DIM
significantly ameliorated cardiac cell hypertrophy by stimulating
p-AMPKα activity and disrupting mTOR and MAPK signaling in response
to Ang II stimuli in vitro. These findings suggested that
DIM has a protective role in the regulation of rat cardiac H9c2
cell hypertrophy induced by Ang II in vitro.
H9c2 cells have been used as an alternative source
for non-proliferating adult cardiomyocytes, originating from the
ventricular muscle of the embryonic rat heart (15,16).
H9c2 cells display similarities to adult cardiomyocytes in their
morphology, protein expression, signaling and
hypertrophy-associated characteristics (15). Previous studies have cultured
primary rat neonatal cardiomyocytes (PNCM) and H9c2 cells and
treated the cells with Ang II and endothelin-1 (ET-1) to promote a
hypertrophic response (17). Ang
II and ET-1 are closely associated hypertrophic stimuli and have
been observed to cause a similar increase in cell size,
rearrangement of contractile proteins and upregulation of
hypertrophic genes in both PNCM and H9C2 cells (15). These findings validate the
importance of H9C2 cells as a model for in vitro studies of
cardiac hypertrophy (15). The
present study used an in vitro model of hypertrophy with 1
μM Ang II in cultured rat cardiac H9c2 cells, to demonstrate
the effects of DIM on the cellular phenotype. It was shown that DIM
(1, 5, and 10 μM) inhibited the enlargement of the cell
surface area and decreased the induction of ANP, BNP and β-MHC mRNA
expression induced by Ang II. The effect was most significant in
the 5 μM DIM-treated group. In addition, 5 μM DIM
markedly decreased the induction of ANP, BNP and β-MHC mRNA
expression induced by Ang II for 6, 12, and 24 h. There were no
significant differences between the 5 and 10 μM DIM-treated
groups. In a previous study by our group (10), it was demonstrated that DIM
attenuated cardiac hypertrophy with improvements to the
cardiomyocyte cross-sectional area in both the forward and reverse
experiments, and blunted the expression of hypertrophic markers
(10).
Diverse intracellular signaling pathways affecting
nuclear factors and the regulation of gene expression, are
implicated in the pathological processes involved in cardiac
hypertrophy, including AMPK/mTOR (18), MAPKs (19), PI3K/AKT (20) and NF-κB (21). AMPK is highly expressed in cardiac
myocytes and functions as a trimer of the α(1–2)
catalytic subunit with the β(1–2) and
γ(1–3) regulatory subunits (22). A previous study (23) has demonstrated that
5-aminoimidazole-4-carboxamide riboside, an AMPK activator,
attenuated cardiac hypertrophy induced by pressure overload in
vivo. Another study (18)
showed that mice deficient of the major α catalytic subunit in
cardiac muscle, AMPKα2, were protected from
pressure-overload-induced left ventricular remodeling, which was
associated with regulating mTOR/p70S6K signaling (18). In our previous study (10), it was found that DIM not only
prevented the development of cardiac hypertrophy, but also reversed
the established cardiac hypertrophy by regulating AMPKα and mTOR
signaling in vivo. The cardioprotective effects of DIM were
ameliorated in mice lacking functional AMPKα2 (10).
The MAPK signaling pathway consists of a sequence of
successively functioning kinases that ultimately result in the dual
phosphorylation and activation of p38, JNKs and ERKs (24). MEK-ERK1/2 signaling is sufficient
to promote cardiac hypertrophy in vivo (25). However, pharmacological inhibition
of MEK1-ERK1/2 may diminish the beneficial effects that are
associated with compensated hypertrophy, whereas the inhibition of
p38 and/or JNK may lead to a maladaptive hypertrophic response over
a longer period (20). The status
of MAPK signaling was evaluated in the presented hypertrophic
model. This study showed that MEK, ERK1/2, JNK1/2 and c-Jun
activation were inhibited in DIM-treated hearts and H9c2 cells (5
μM) as compared with the control groups stimulated with Ang
II (1 μM). The findings indicated that the inhibitory
effects of DIM on cardiac hypertrophy are partly mediated through
MAPK signaling.
In conclusion, the present study indicated that DIM
protected against rat cardiac H9c2 cell hypertrophy in response to
Ang II. These observations have revealed new insights into the
pathogenesis of cardiac remodeling and may have considerable
implications for the development of strategies for the treatment of
cardiac hypertrophy and heart failure through the application of
DIM. Additional studies are required to explore the potential
clinical uses of DIM.
Acknowledgments
This work was supported by the National Nature
Science Foundation of China (nos. 30901628, 30972954, 81000036 and
81000095) and the Fundamental Research Funds for the Central
Universities of China (nos. 5107002 and 2012302020212).
References
|
1
|
Blaxall BC, Tschannen-Moran BM, Milano CA
and Koch WJ: Differential gene expression and genomic patient
stratification following left ventricular assist device support. J
Am Coll Cardiol. 41:1096–1106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuwahara K and Nakao K: New molecular
mechanisms for cardiovascular disease: transcriptional pathways and
novel therapeutic targets in heart failure. J Pharmacol Sci.
116:337–342. 2011. View Article : Google Scholar
|
|
3
|
Proud CG: Ras, PI3-kinase and mTOR
signaling in cardiac hypertrophy. Cardiovasc Res. 63:403–413. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duan C, LM, Zhang J and Ma R: RASSF1A: A
potential novel therapeutic target against cardiac hypertrophy.
Prog Biophys Mol Biol. 113:284–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jayakumar P, PK and Sankaran M:
Attenuation of hyperglycemiamediated oxidative stress by
indole-3-carbinol and its metabolite 3,3′-diindolylmethane in
C57BL/6J mice. J Physiol Biochem. 70:525–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kunimasa K, Kobayashi T, Kaji K and Ohta
T: Antiangiogenic effects of indole-3-carbinol and
3,3′-diindolylmethane are associated with their differential
regulation of ERK1/2 and Akt in tube-forming HUVEC. J Nutr.
140:1–6. 2010. View Article : Google Scholar
|
|
7
|
Azmi AS, AA, Banerjee S, Rangnekar VM,
Mohammad RM and Sarkar FH: Chemoprevention of pancreatic cancer:
Characterization of Par-4 and its modulation by 3,3′
diindolylmethane (DIM). Pahrm Res. 25:2117–2124. 2008. View Article : Google Scholar
|
|
8
|
Xue L, Firestone GL and Bjeldanes LF: DIM
stimulates IFNgamma gene expression in human breast cancer cells
via the specific activation of JNK and p38 pathways. Oncogene.
24:2343–2353. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cho HJ, Seon MR, Lee YM, et al:
3,3′-Diindolylmethane suppresses the inflammatory response to
lipopolysaccharide in murine macrophages. J Nutr. 138:17–23.
2008.
|
|
10
|
Zong J, Deng W, Zhou H, et al:
3,3′-Diindolylmethane protects against cardiac hypertrophy via
5′-adenosine monophosphate-activated protein kinase-α2. PLoS One.
8:e534272013. View Article : Google Scholar
|
|
11
|
Matsubara H: Renin-angiotensin system in
human failing hearts: message from nonmyocyte cells to myocytes.
Circ Res. 88:861–863. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weber KT and Brilla CG: Pathological
hypertrophy and cardiac interstitium. Fibrosis and
renin-angiotensin-aldosterone system Circulation. 83:1849–1865.
1991.
|
|
13
|
Mehta PK and Griendling KK: Angiotensin II
cell signaling: physiological and pathological effects in the
cardiovascular system. Am J Physiol Cell Physiol. 292:C82–C97.
2007. View Article : Google Scholar
|
|
14
|
Zhou H, Shen DF, Bian ZY, et al:
Activating transcription factor 3 deficiency promotes cardiac
hypertrophy, dysfunction, and fbrosis induced by pressure overload.
PLoS One. 6:e267442011. View Article : Google Scholar
|
|
15
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9C2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In vitro Cell Dev Biol
Anim. 47:125–131. 2011. View Article : Google Scholar
|
|
16
|
Dick E, Rajamohan D, Ronksley J and
Denning C: Evaluating the utility of cardiomyocytes from human
pluripotent stem cells for drug screening. Biochem Soc Trans.
38:1037–1045. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kimes BW and Brandt BL: Properties of a
clonal muscle cell line from rat heart. Exp Cell Res. 98:367–381.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang P, Hu X, Xu X, et al: AMP activated
protein kinase-alpha2 deficiency exacerbates
pressure-overload-induced left ventricular hypertrophy and
dysfunction in mice. Hyper tension. 52:918–924. 2008. View Article : Google Scholar
|
|
19
|
Zhou H, Bian ZY, Zong J, et al: Stem cell
antigen 1 protects against cardiac hypertrophy and fibrosis after
pressure overload. Hypertension. 60:802–809. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan L, Huang H, Tang QZ, et al:
Breviscapine protects against cardiac hypertrophy through blocking
PKC-alpha-dependent signaling. J Cell Biochem. 109:1158–1171.
2010.PubMed/NCBI
|
|
22
|
Arad M, Seidman CE and Seidman JG:
AMP-activated protein kinase in the heart: role during health and
disease. Circ Res. 100:474–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li HL, Yin R, Chen D, et al: Long-term
activation of adenosine monophosphate-activated protein kinase
attenuates pressure-overload-induced cardiac hypertrophy. J Cell
Biochem. 100:1086–1099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garrington TP and Johnson GL: Organization
and regulation of mitogen-activated protein kinase signaling
pathways. Curr Opin Cell Biol. 11:211–218. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zong J, Zhang DP, Zhou H, et al: Baicalein
protects against cardiac hypertrophy through blocking MEK-ERK1/2
signaling. J Cell Biochem. 2013. View Article : Google Scholar
|














