Introduction
Congenital cataracts are defined as the presence of
complete or partial lens opacification within the first year of
life (1). Cataract of the eye lens
is the leading cause of blindness worldwide. A congenital cataract
is particularly severe as it may impair visual development. The
prevalence of congenital cataracts is ~6.31/100,000 individuals,
and ~30% of cases are inherited (2,3).
Congenital cataracts represent a clinically and
genetically heterogeneous lens disorder (4). Depending on the morphology,
congenital cataracts may be classified into several subtypes,
including whole lens, nuclear, lamellar, cortical, polar, sutural,
pulverulent, cerulean and coralliform (5). To date, >40 loci in the human
genome associated with various forms of congenital cataracts have
been identified, including at least 26 genes associated with
autosomal dominant congenital cataract or autosomal recessive
congenital cataract. Among these genes, the crystallin and connexin
genes appear to be the most commonly associated with congenital
cataracts, whereas approximately half of the mutations belong to
the crystalline genes (CRYAA, CRYAB, CRYBA1/A3,
CRYBB1, CRYBB2, CRYBA4, CRYGC,
CRYGD and CRYGS) and a quarter of the mutations
belong to connexin genes (GJA3 and GJA8) (6). The remainder include heat shock
transcription factor-4 (HSF4), aquaporin-0, v-maf
musculoaponeurotic fibrosarcoma oncogene homolog, paired-like
homeodomain 3, beaded filament structural protein-2, chromatin
modifying protein and lens intrinsic membrane protein 2 (4).
The present study investigated two Chinese families
with congenital cataracts. The aim of the present study was to
identify the genetic mutations of the two families by direct
sequencing. The crystallin and connexin genes, which are the most
commonly associated with cataracts, were selected as the main
candidate genes. The present study may extend the mutation spectrum
of congenital cataracts.
Materials and methods
Clinical examination and isolation of
genomic DNA
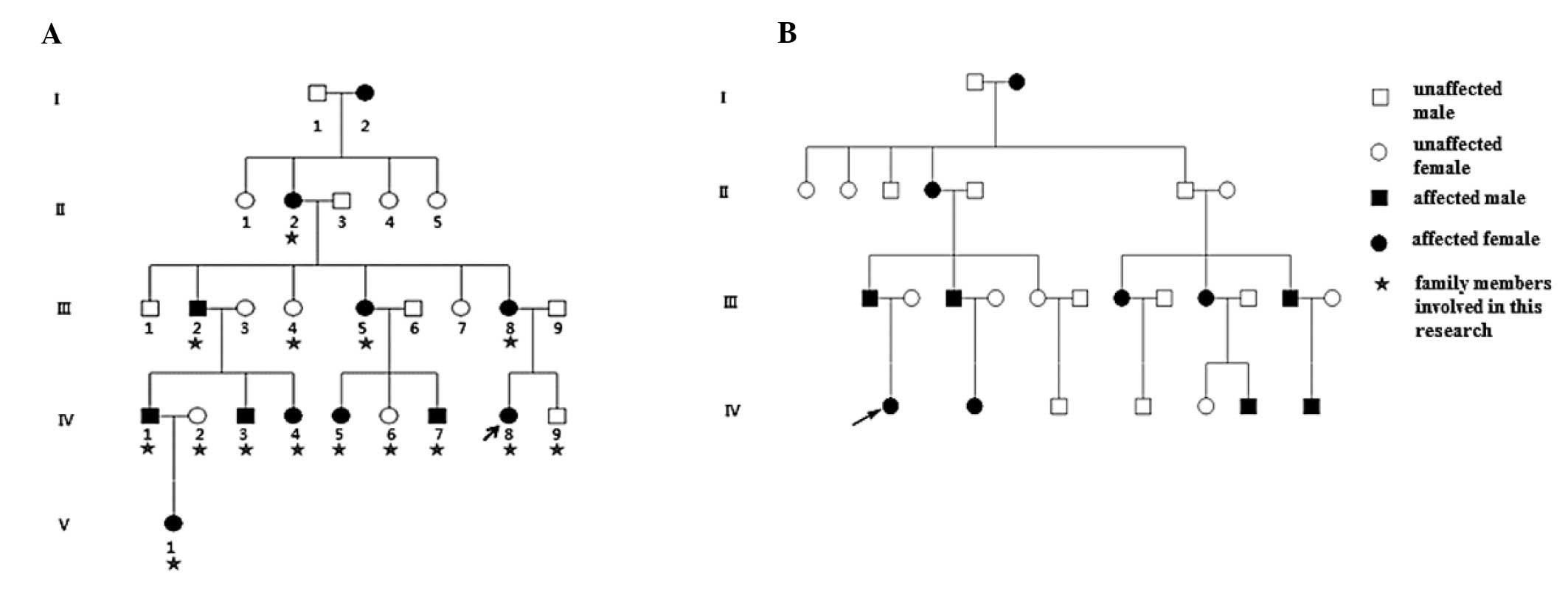
Family 1, a five-generation Chinese Han family, and
Family 2, a four-generation Chinese Han family, with autosomal
dominant congenital cataracts were recruited for the present study
from Peking University Third Hospital (Beijing, China; Fig. 1). A total of 100 healthy control
individuals were also recruited from Peking University Third
Hospital. The present study was approved by the ethics committee of
Peking University Health Science Center. Informed consent was
obtained from all participants. The present study followed the
principles of the Declaration of Helsinki (7). The congenital cataract-affected
status was determined by a history of cataract extraction or
ophthalmologic examination, and the participants underwent
ophthalmic examination, including visual acuity assessment,
slit-lamp examination and intraocular pressure measurement. The
phenotypes were documented using slit lamp photography (Topcon
SL-1E; Topcon Medical Systems, Inc., Oakland, NJ, USA).
Subsequently, 5 ml venous blood was obtained from each family
member and control, and was collected in a BD Vacutainer (BD
Biosciences, San Jose, CA, USA), containing EDTA. The genomic DNA
was extracted using a QIAamp DNA Blood Mini kit (Qiagen,
Germantown, MD, USA).
Mutation detection
All coding exons and flanking splicing junctions of
the candidate genes associated with congenital cataracts, including
CRYAA, CRYAB, CRYBA1, CRYBB1,
CRYBB2, CRYGC, CRYGD, CRYGS,
GJA3, GJA8 and CRYBA4 were amplified using
polymerase chain reaction (PCR), using the primers listed in
Table I. Each reaction mixture (25
µl) contained 20 ng genomic DNA, 1X PCR buffer, 1.5 mM
MgCl2, 0.2 mM dNTPs, 0.5 µM forward primer, 0.5
µM reverse primer and 2.5 Units Taq DNA polymerase (Qiagen,
Mississauga, ON, Canada). The following PCR program was used for
DNA amplification: 95°C for 5 min; followed by 35 cycles at 95°C
for 30 sec, 57–63°C for 30 sec (annealing temperature difference
according to primer), 72°C for 30 sec, and a final extension at
72°C for 10 min. The PCR products of the probands from each family
and one unaffected member were sequenced using an ABI3730 Automated
Sequencer (PE Biosystems, Foster City, CA, USA). The sequencing
results were analyzed using Chromas 2.33 (Technelysium Pty Ltd.,
South Brisbane, Australia) and were compared with the reference
sequence in the NCBI database (http://www.ncbi.nlm.nih.gov/). Finally, mutations was
screened for in the CRYAA and GJA8 genes from the
family members and 100 ethnically matched controls to confirm the
mutation.
 | Table IPrimers used for polymerase chain
reaction. |
Table I
Primers used for polymerase chain
reaction.
| Name | Forward (5′-3′) | Reverse (5′-3′) |
|---|
| CRYAA-1 |
AGCAGCCTTCTTCATGAGC |
CAAGACCAGAGTCCATCG |
| CRYAA-2 |
GGCAGGTGACCGAAGCATC |
GAAGGCATGGTGCAGGTG |
| CRYAA-3 |
GCAGCTTCTCTGGCATGG |
GGGAAGCAAAGGAAGACAGA |
| CRYAB-1 |
AACCCCTGACATCACCATTC |
AAGGACTCTCCCGTCCTAGC |
| CRYAB-2 |
CCATCCCATTCCCTTACCTT |
GCCTCCAAAGCTGATAGCAC |
| CRYAB-3 |
TCTCTCTGCCTCTTTCCTCA |
CCTTGGAGCCCTCTAAATCA |
| CRYBA1–1 |
GGCAGAGGGAGAGCAGAGTG |
CACTAGGCAGGAGAACTGGG |
| CRYBA1–2 |
AGTGAGCAGCAGAGCCAGAA |
GGTCAGTCACTGCCTTATGG |
| CRYBA1–3 |
AAGCACAGAGTCAGACTGAAGT |
CCCCTGTCTGAAGGGACCTG |
| CRYBA1–4 |
GTACAGCTCTACTGGGATTG |
ACTGATGATAAATAGCATGAACG |
| CRYBA1–5 |
GAATGATAGCCATAGCACTAG |
TACCGATACGTATGAAATCTGA |
| CRYBA1–6 |
CATCTCATACCATTGTGTTGAG |
GCAAGGTCTCATGCTTGAGG |
| CRYBB1–1 |
CCCTGGCTGGGGTTGTTGA |
TGCCTATCTGCCTGTCTGTTTCTC |
| CRYBB1–2 |
TAGCGGGGTAATGGAGGGTG |
AGGATAAGAGTCTGGGGAGGTGG |
| CRYBB1–3 |
CCTGCACTGCTGGCTTTTATTTA |
TCTCCAGAGCCCAGAACCATG |
| CRYBB1–4 |
CCAACTCCAAGGAAACAGGCATA |
CCTCCCTACCCACCATCATCTC |
| CRYBB1–5 |
TAGACAGCAGTGGTCCCTGGAGA |
AGCACTGGGAGACTGTGGAAGG |
| CRYBB1–6 |
CCTAGAAAAGGAAACCGAGGCC |
AGCGAGGAAGTCACATCCCAGTA |
| CRYBB2–1 |
GTTTGGGGCCAGAGGGGAGTGGT |
TGGGCTGGGGAGGGACTTTCAGTA |
| CRYBB2–2 |
CCTTCAGCATCCTTTGGGTTCTCT |
GCAGTTCTAAAAGCTTCATCAGTC |
| CRYBB2–3 |
GTAGCCAGGATTCTGCCATAGGAA |
GTGCCCTCTGGAGCATTTCATAGT |
| CRYBB2–4 |
GGCCCCCTCACCCATACTCA |
CTTCCCTCCTGCCTCAACCTAATC |
| CRYBB2–5 |
CTTACCCTTGGGAAGTGGCAATGG |
TCAAAGACCCACAGCAGACAAGTT |
| CRYGC-1 |
TGCATAAAATCCCCTTACCG |
CCTCCCTGTAACCCACATTG |
| CRYGC-2 |
TGGTTGGACAAATTCTGGAAG |
CCCACCCCATTCACTTCTTA |
| CRYGD-1 |
CAGCAGCCCTCCTGCTAT |
GGGTCCTGACTTGAGGATGT |
| CRYGD-2 |
GCTTTTCTTCTCTTTTTATTTCTGG |
AAGAAAGACACAAGCAAATCAGT |
| CRYGS-2 |
GAAACCATCAATAGCGTCTAAATG |
TGAAAAGCGGGTAGGCTAAA |
| CRYGS-3 |
AATTAAGCCACCCAGCTCCT |
GGGAGTACACAGTCCCCAGA |
| CRYGS-4 |
GACCTGCTGGTGATTTCCAT |
CACTGTGGCGAGCACTGTAT |
| GJA3-1 |
CGGTGTTCATGAGCATTTTC |
CTCTTCAGCTGCTCCTCCTC |
| GJA3-2 |
GAGGAGGAGCAGCTGAAGAG |
AGCGGTGTGCGCATAGTAG |
| GJA3-3 |
TCGGGTTCCCACCCTACTAT |
TATCTGCTGGTGGGAAGTGC |
| GJA8-1 |
CCGCGTTAGCAAAAACAGAT |
CCTCCATGCGGACGTAGT |
| GJA8-2 |
GCAGATCATCTTCGTCTCCA |
GGCCACAGACAACATGAACA |
| GJA8-3 |
CCACGGAGAAAACCATCTTC |
GAGCGTAGGAAGGCAGTGTC |
| GJA8-4 |
TCGAGGAGAAGATCAGCACA |
GGCTGCTGGCTTTGCTTAG |
| CRYBA4-1 |
GTCCTTTCCCTCCCTGCTAA |
AGGATGAGGATGGCATTCAG |
| CRYBA4-2 |
TAGCCCAGTCACTCCTGGAC |
CCTAGGATTCATGGGGACCT |
| CRYBA4-3 |
TTTGCAATCCCTGCTTTACC |
CTTCAGGAGGGCACAACAGT |
| CRYBA4-4 |
ACCCCTGAATGGTTGTGACT |
CTTGAAGTGGCGACATGAGA |
| CRYBA4-5 |
CAAATGGCAAGGTTTCTGGT |
GTCCCTCAAATTCTGCCTGA |
| CRYBA4-6 |
AGGGAATGGCATGATCAAAG |
GGCCTGAAGTAAATAGAAGAAAGG |
Bioinformatic analysis
The amino acid sequences of CRYAA and GJA8 from
several different species were obtained from the NCBI GenBank
(http://www.ncbi.nlm.nih.gov/genbank),
and conservation analysis was performed using CLC Main Workbench
4.5.1 Software (Aarhus, Denmark). The function impact of the
mutation was predicted using Polymorphism phenotyping (PolyPhen;
http://genetics.bwh.harvard.edu/pph2/).
Site-directed mutagenesis and plasmid
construction
The human CRYAA and GJA8 open reading
frame (ORF) cDNA was obtained from GeneCopoeia (Rockville, MD,
USA). Site-directed mutagenesis was performed to generate
CRYAA bearing the p.L139P mutation and GJA8 bearing
the p.D47N mutation, using a QuickChange Lightning Site-Directed
Mutagenesis kit (Stratagene, La Jolla, CA, USA). DNA sequencing was
used to confirm the introduced mutation (ABI 3730 Automated
Sequencer; Applied Biosystems, Foster City, CA, USA). The ORFs of
the wild-type (WT) and mutant (MT) sequences were amplified using
PCR from the cDNAs, and were inserted into the HindIII- and
XhoI-digested pEGFP-N1 vector (Invitrogen Life Technologies,
Carlsbad, CA, USA) to produce the pEGFP-CRYAA-WT, pEGFP-CRYAA-MT,
pEGFP-GJA8-WT and pEGFP-GJA8-MT expression plasmids. Each reaction
mixture (25 µl) contained 200 ng plasmids, 2X GC buffer, 0.2
mM dNTPs, 0.5 µM forward primer, 0.5 µM reverse
primer and 2.5 units of La-Taq DNA polymerase (Takara Bio., Inc.,
Beijing, China). The following PCR program was used for DNA
amplification: 95°C for 3 min; followed by 35 cycles at 95°C for 30
sec, 60°C for 30 sec, 72°C for 30 sec and a final extension at 72°C
for 10 min.
Cell culture and transfection
Hela cells were provided by Professor Fan Yong at
the Third Affiliated Hospital of Guangzhou Medical University
(Guangzhou, China). In each well of a six-well plate,
~10−6 cells were added once the cells grew to 100%
confluence. The Hela cells were maintained in Iscove's modified
Dulbecco's medium, supplemented with 10% fetal bovine serum, 100
mg/ml penicillin and 100 mg/ml streptomycin, in a humidified
atmosphere containing 5% CO2 at 37°C. Transfection was
performed using Lipofectamine 2000 (Invitrogen Life Technologies).
The Hela cells were seeded into six-well tissue culture plates 24 h
prior to transfection at ~60% confluence. The cells were
transfected with eithr the pEGFP-CRYAA-WT, pEGFP-CRYAA-MT,
pEGFP-GJA8-WT, pEGFP-GJA8-MT or GFP-control plasmid using
Lipofectamine 2000, according to the manufacturer's instructions.
At 48 h post-transfection, the cells were analyzed using
fluorescence microscopy (Nikon Eclipse TS-10; Nikon Instruments,
Amsterdam, Netherlands).
Results
Clinical evaluation
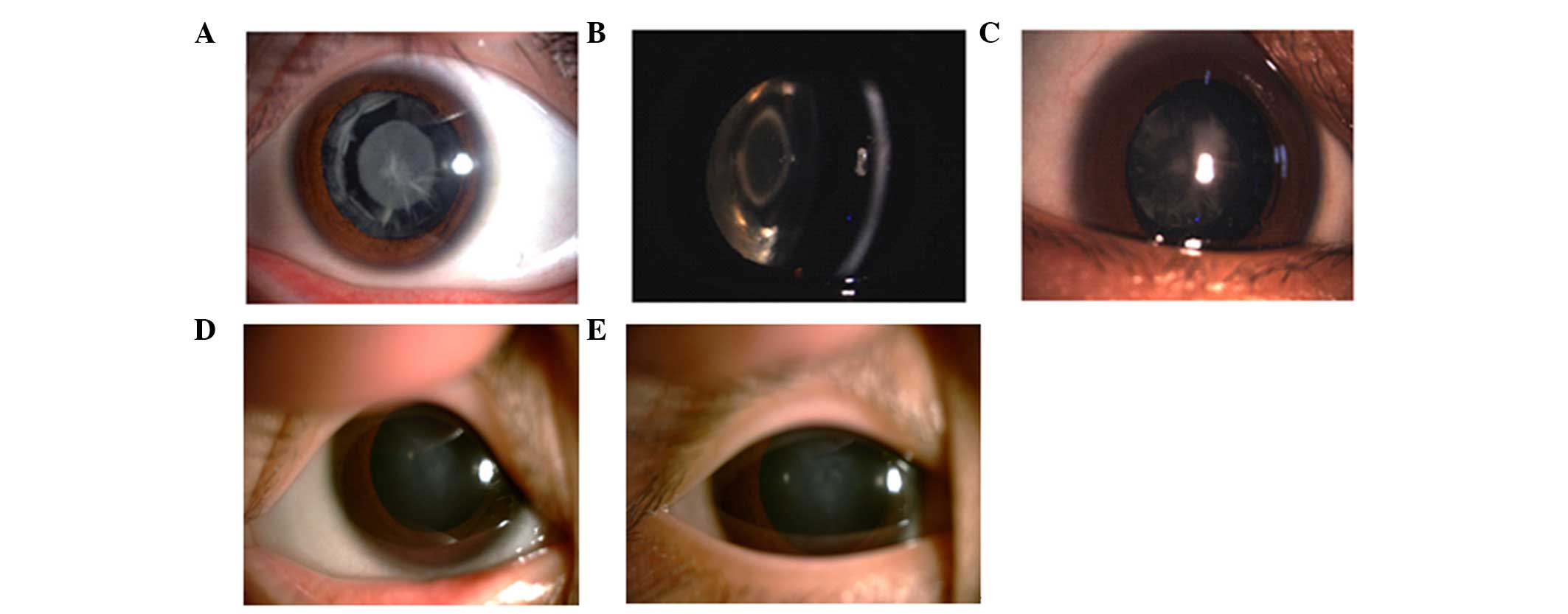
The slit-lamp examination revealed polymorphic
cataracts in Family 1 (Fig. 2A–C).
The proband in this family exhibited opacities involving the
nucleus and peripheral cortex, and the slit lamp image of
individual III:2 revealed a punctuate cataract in the central lens
and opacities involving the peripheral cortex. The images of
individual IV:4 revealed a nuclear cataract. The phenotypes of the
three individuals all differed. The slit lamp image of the proband
in Family 2 revealed nuclear cataracts (Fig. 2D and E). All affected individuals
in this family exhibited bilateral cataracts, and the slit-lamp
examination of the proband in this family revealed nuclear
cataracts in the left and right eyes.
Mutation analysis
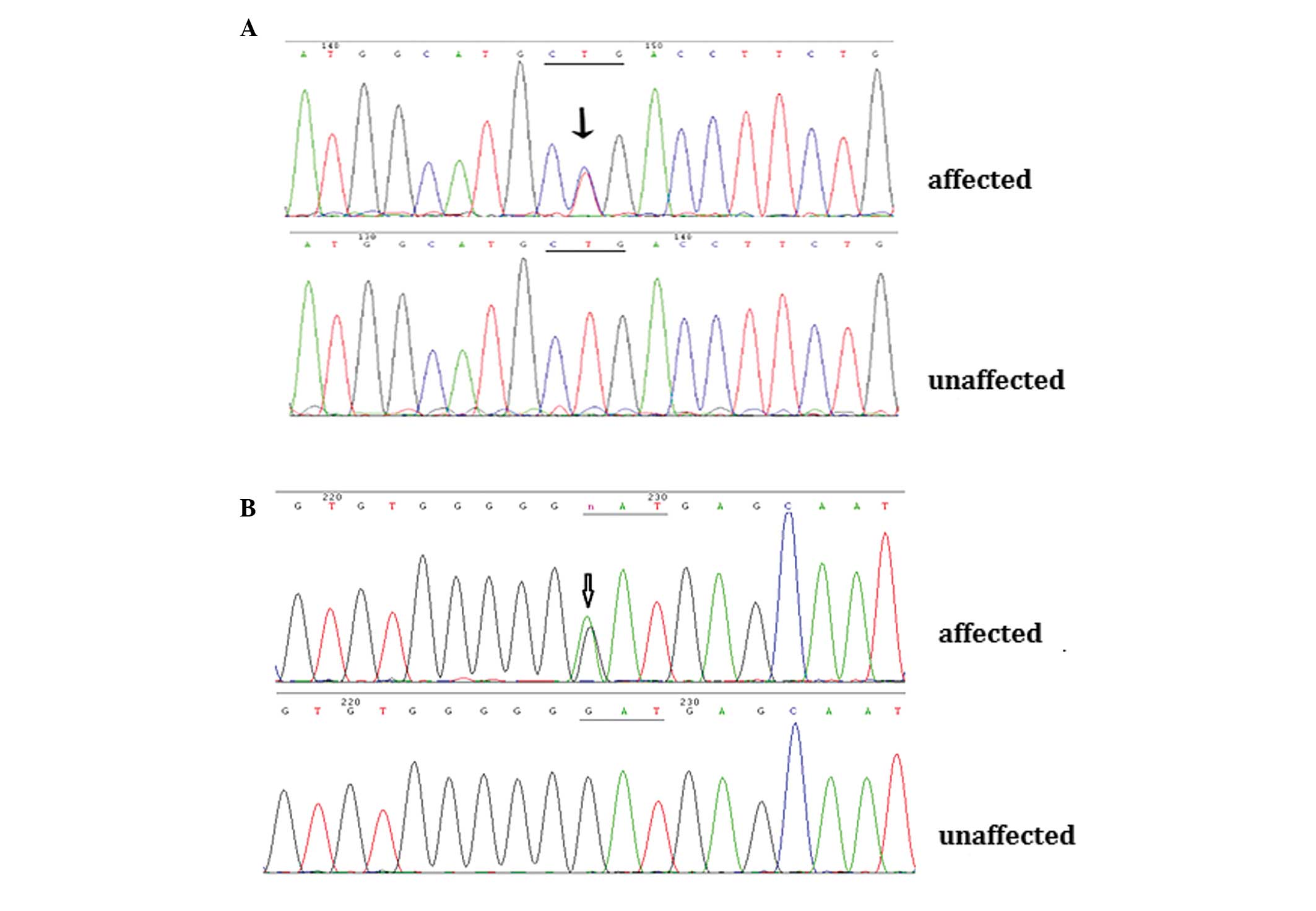
Through direct gene sequencing of the coding regions
of the candidate genes, a novel missense mutation, c.416 T>C
(p.L139P), was identified in the CRYAA gene in the affected
individuals from Family 1. In the affected members from Family 2,
the known mutation, c.139G>A (p.D47N), was detected in the
GJA8 gene (Fig. 3A and B).
These two mutations were not observed in any of the unaffected
family members or in the 100 unrelated control individuals.
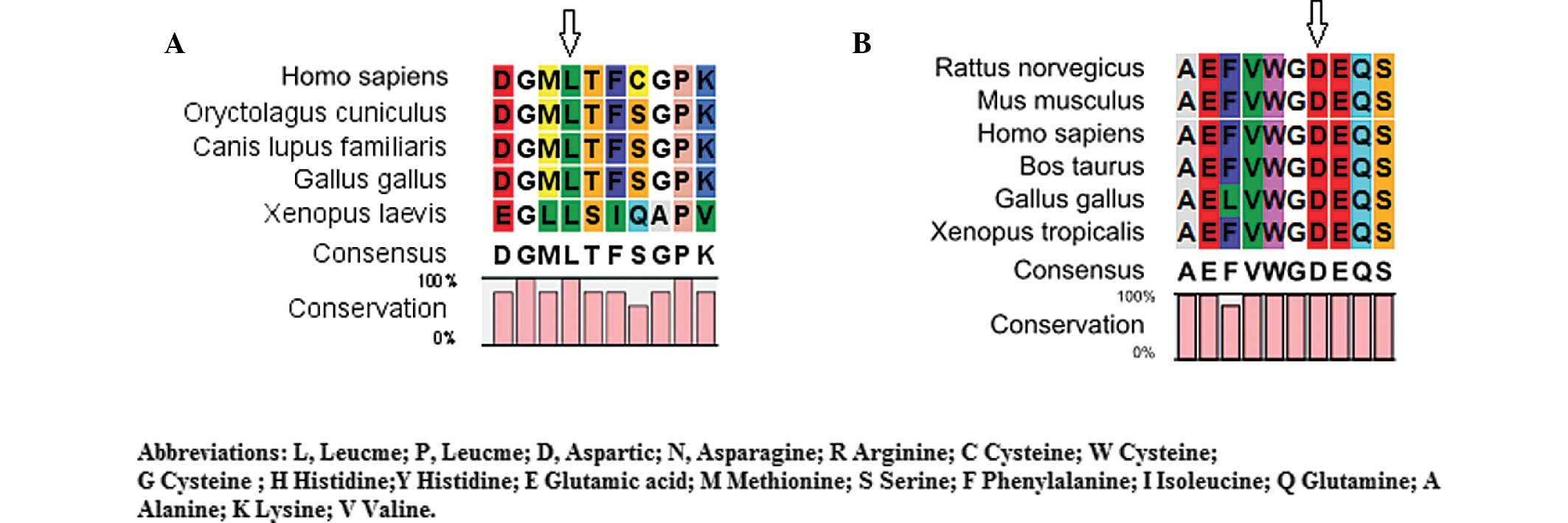
Bioinformatics analysis
The CLC Main Workbench software revealed that
leucine at amino acid position 139 of CRYAA and aspartic
acid at amino acid position 47 of GJA8 were highly conserved
among several species (Fig. 4A and
B). The PolyPhen analysis demonstrated that either L139P of
CRYAA or D47N of GJA8 produced a score of 1.000,
which was predicted to be 'probably damaging'.
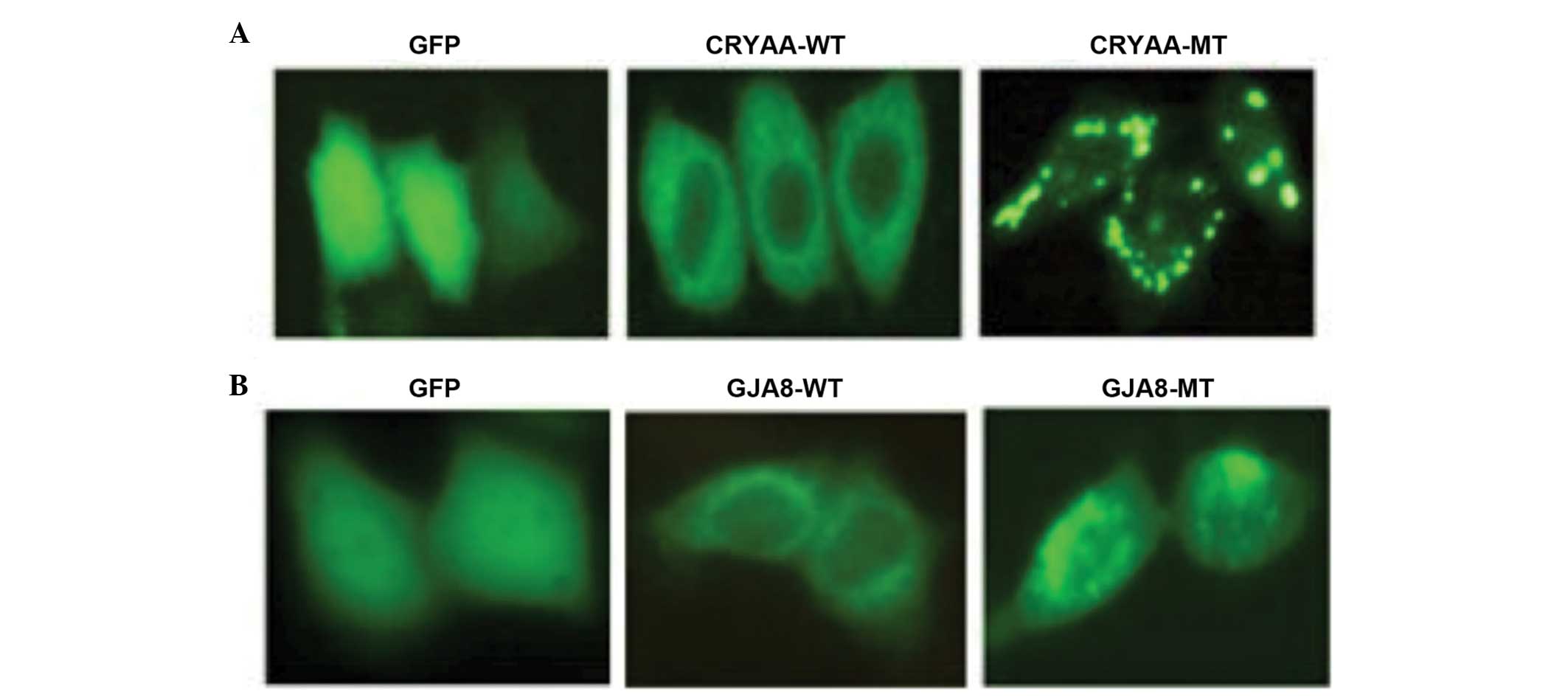
Functional analysis
The subcellular localization of the wild-type and
mutant proteins were assessed. The subcellular localization was
determined using C-terminal green fluorescent protein (GFP) fusion
constructs of CRYAA-WT, CRYAA-MT, GJA8-WT and GJA8-MT, followed by
fluorescence microscopy. GFP, as a control, was located in the
nucleus and cytoplasm. The cells transfected with CRYAA-WT
demonstrated a homogenous distribution of expression in the
cytoplasm alone, compared with CRYAA-MT. The expression of CRYAA-MT
in these cells revealed significant protein aggregation (Fig. 5A). It was likely that the protein
aggregation in the cytoplasm was due to protein conformational
changes, which resulted from the L139P mutation. In addition,
GJA8-WT was predominantly detected in the plasma membrane, whereas
GJA8-MT was aberrantly expressed in the cytoplasm (Fig. 5B), indicating that the D47N
mutation in GJA8 prevented its localization to the plasma
membrane.
Discussion
The lens is an avascular organ, which relies on
maintaining transparency to allow normal transmission of light to
focus images on the retina. The lens is comprised of two cell
types: Epithelial cells, which form a single layer along the
anterior surface, and fiber cells, which form the bulk of the
organ. The lens fiber cells, which differentiate from epithelial
cells throughout the lifespan of the organism, contain high
concentrations of small soluble proteins, termed crystallins.
Mature fiber cells have limited metabolic activities, and the
majority of the metabolic, synthetic and active transport machinery
in the lens is localized to the surface cells. Lens crystallin and
an extensive cell-cell communication system are important in
establishing and maintaining lens transparency. Damage to the lens
cells and/or proteins can cause opacities, which may result in a
decrease in vision and can eventually lead to blindness (8).
Previous studies and transgenic animal models have
indicated that mutations in crystallin genes may cause cataracts
(9,10). α-crystallin is the major protein of
the vertebrate eye lens and has a structural role in maintaining
lens transparency and an appropriate refractive index. It is also a
member of the small heat-shock-protein (sHSP) family, which are
stress-induced proteins and exhibit chaperone activity.
α-crystallin is composed of two particularly homologous subunits,
α-A (CRYAA) and α-B (CRYAB) (11).
The first exon of each gene encodes 60 amino acids, consisting of a
repeat of the 30 amino acid motif, and the second and the third
exons code for regions homologous to the sHsps (12). Several α-A crystallin mutations
have been previously reported, including R12C, R21W, R21L, R49C,
G98R, R54C, R116C and R116H (13–20).
With regards to secondary and tertiary structural changes, all the
mutants identified exhibit varying degrees of secondary and
tertiary structural changes, which can lead to protein
unfolding/misfolding and subsequently to the formation of protein
aggregates (18). In the present
study, the c.416T>C (p.L139P) mutation in CRYAA also formed
α-A-crystallin aggregates, therefore, this mutation may have
contributed to the development of cataracts in Family 1.
Since the lens is an avascular organ, intercellular
gap junction-mediated transportation of ion gradients and metabolic
materials, and intercellular communication are essential for organ
function and homeostasis (21,22).
Gap junction channels consist of connexin protein subunits and
three isoforms of the connexin gene family are expressed abundantly
in the vertebrate lens: GJA1 (Cx43), GJA3 (Cx46) and GJA8 (Cx50).
GJA1 is restrictively expressed in the lens epithelial cells. GJA3
and GJA8 are two connexin isoforms in the plasma membrane of fiber
cells (23,24). To date, several mutations in Cx46
have been reported to be associated with congenital cataracts with
different phenotypes. The amino acid at position 47 in connnexin 50
is a mutational hot-spot, and D47Y, D47H and D47N have been
reported previously (25–27). D47N mutants are loss-of-function
mutants, and the A mutant protein of Cx50 is unable to form
functional channels (28). The
present study identified a recurrent missense mutation D47N in Cx50
was associated with autosomal dominant nuclear cataracts in a
Chinese family. This mutation of Cx50 prevented its localization to
the plasma membrane. The aberrant localization may lead to a
capacity deficiency of Connnexin 50, forming functional
hemichannels and triggering a complex sequence of events, including
loss of membrane potential, disruption of transmembrane ion
gradients, subsequent decreased metabolic activity and decreased
cell growth (29,30).
In conclusion, the present study identified a novel
disease-causing mutation, c.416T>C (p.L139P), in CRYAA,
and a recurrent mutation, c.139G>A (p.D47N), in GJA8.
Functional analysis indicated that the two mutants led to marked
alteration compared with the wild-types. These findings extend the
mutation spectrum of CRYAA and provide further evidence that
the amino acid at position 47 is a mutational hot-spot and that
p.D47N is a common connexin 50 mutation.
Acknowledgments
This study was supported by the National Basic
Research Program of China (973 Program; no. 2014CB943203) and the
Special program of advanced technology of Beijing City Science
Committee (no. Z131100005213006). The authors would like to thank
the patients and their families for their involvement.
References
|
1
|
Bermejo E and Martínez-Frías ML:
Congenital eye malformations: clinical-epidemiological analysis of
1,124,654 consecutive births in Spain. Am J Med Genet. 75:497–504.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haargaard B, Wohlfahrt J, Fledelius HC,
Rosenberg T and Melbye M: A nationwide Danish study of 1027 cases
of congenital/infantile cataracts: etiological and clinical
classifications. Ophthalmology. 111:2292–2298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shiels A, Bennett TM and Hejtmancik JF:
Cat-Map: putting cataract on the map. Mol Vis. 16:2007–2015.
2010.PubMed/NCBI
|
|
4
|
Huang B and He W: Molecular
characteristics of inherited congenital cataracts. Eur J Med Genet.
53:347–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reddy MA, Francis PJ, Berry V,
Bhattacharya SS and Moore AT: Molecular genetic basis of inherited
cataract and associated phenotypes. Surv Ophthalmol. 49:300–315.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hejtmancik JF: Congenital cataracts and
their molecular genetics. Semin Cell Dev Biol. 19:134–149. 2008.
View Article : Google Scholar :
|
|
7
|
World Medical Association: World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beyer EC, Ebihara L and Berthoud VM:
Connexin mutants and cataracts. Front Pharmacol. 4:432013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Graw J: Genetics of crystallins: cataract
and beyond. Exp Eye Res. 88:173–189. 2009. View Article : Google Scholar
|
|
10
|
Hsu CD, Kymes S and Petrash JM: A
transgenic mouse model for human autosomal dominant cataract.
Invest Ophthalmol Vis Sci. 47:2036–2044. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Menko AS and Andley UP: αA-Crystallin
associates with α6 integrin receptor complexes and regulates
cellular signaling. Exp Eye Res. 91:640–651. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sharma KK, Kumar RS, Kumar GS and Quinn
PT: Synthesis and characterization of a peptide identified as a
functional element in alphaA-crystallin. J Biol Chem.
275:3767–3771. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Devi RR, Yao W, Vijayalakshmi P, Sergeev
YV, Sundaresan P and Hejtmancik JF: Crystallin gene mutations in
Indian families with inherited pediatric cataract. Mol Vis.
14:1157–1170. 2008.PubMed/NCBI
|
|
14
|
Gong B, Zhang LY, Pang CP, Lam DS and Yam
GH: Trimethylamine N-oxide alleviates the severe aggregation and ER
stress caused by G98R alphaA-crystallin. Mol Vis. 15:2829–2840.
2009.PubMed/NCBI
|
|
15
|
Hansen L, Yao W, Eiberg H, et al: Genetic
heterogeneity in microcornea-cataract: five novel mutations in
CRYAA, CRYGD and GJA8. Invest Ophthalmol Vis Sci. 48:3937–3944.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Litt M, Kramer P, LaMorticella DM, Murphey
W, Lovrien EW and Weleber RG: Autosomal dominant congenital
cataract associated with a missense mutation in the human alpha
crystallin gene CRYAA. Hum Mol Genet. 7:471–474. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mackay DS, Andley UP and Shiels A: Cell
death triggered by a novel mutation in the alphaA-crystallin gene
underlies autosomal dominant cataract linked to chromosome 21q. Eur
J Hum Genet. 11:784–793. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raju I and Abraham EC: Congenital cataract
causing mutants of alphaA-crystallin/sHSP form aggregates and
aggresomes degraded through ubiquitin-proteasome pathway. PLoS One.
6:e280852011. View Article : Google Scholar
|
|
19
|
Santhiya ST, Soker T, Klopp N, et al:
Identification of a novel, putative cataract-causing allele in
CRYAA (G98R) in an Indian family. Mol Vis. 12:768–773.
2006.PubMed/NCBI
|
|
20
|
Zhang LY, Yam GH, Tam PO, et al: An
alphaA-crystallin gene mutation, Arg12Cys, causing inherited
cataract-microcornea exhibits an altered heat-shock response. Mol
Vis. 15:1127–1138. 2009.PubMed/NCBI
|
|
21
|
Goodenough DA: Lens gap junctions: a
structural hypothesis for nonregulated low-resistance intercellular
pathways. Invest Ophthalmol Vis Sci. 18:1104–1122. 1979.PubMed/NCBI
|
|
22
|
Nielsen MS, Nygaard Axelsen L, Sorgen PL,
Verma V, Delmar M and Holstein-Rathlou NH: Gap junctions. Compr
Physiol. 2:1981–2035. 2012.
|
|
23
|
Gong X, Li E, Klier G, Huang Q, Wu Y, Lei
H, Kumar NM, Horwitz J and Gilula NB: Disruption of alpha3 connexin
gene leads to proteolysis and cataractogenesis in mice. Cell.
91:833–843. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rong P, Wang X, Niesman I, Wu Y, Benedetti
LE, Dunia I, Levy E and Gong X: Disruption of Gja8 (alpha8
connexin) in mice leads to microphthalmia associated with
retardation of lens growth and lens fiber maturation. Development.
129:167–174. 2002.PubMed/NCBI
|
|
25
|
Li J, Wang Q, Fu Q, et al: A novel
connexin 50 gene (gap junction protein, alpha 8) mutation
associated with congenital nuclear and zonular pulverulent
cataract. Mol Vis. 19:767–774. 2013.PubMed/NCBI
|
|
26
|
Lin Y, Liu NN, Lei CT, et al: A novel GJA8
mutation in a Chinese family with autosomal dominant congenital
cataract. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 25:59–62.
2008.PubMed/NCBI
|
|
27
|
Wang L, Luo Y, Wen W, Zhang S and Lu Y:
Another evidence for a D47N mutation in GJA8 associated with
autosomal dominant congenital cataract. Mol Vis. 17:2380–2385.
2011.PubMed/NCBI
|
|
28
|
Arora A, Minogue PJ, Liu X, et al: A novel
connexin 50 mutation associated with congenital nuclear pulverulent
cataracts. J Med Genet. 45:155–160. 2008. View Article : Google Scholar
|
|
29
|
Minogue PJ, Tong JJ, Arora A, et al: A
mutant connexin 50 with enhanced hemichannel function leads to cell
death. Invest Ophthalmol Vis Sci. 50:5837–5845. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sellitto C, Li L and White TW: Connexin 50
is essential for normal postnatal lens cell proliferation. Invest
Ophthalmol Vis Sci. 45:3196–3202. 2004. View Article : Google Scholar : PubMed/NCBI
|