Introduction
Clostridium difficile was first detected in
the fecal flora of healthy newborns in the mid-1930s (1) and considered to be non-pathogenic
until identified as the primary cause of pseudo-membranous colitis
in 1978 (2). C. difficile
is a gram-positive anaerobic, spore-forming bacterium, and accounts
for 15–30% of all episodes of antibiotic-associated diarrhea (AAD)
and 95–100% of cases of pseudomembranous colitis (PMC) (3,4). The
morbidity, mortality and relapse rates of the diseases caused by
C. difficile have markedly increased in hospital and
community settings due to the spread of hypervirulent strains,
including BI/NAP1/027, restriction endonuclease analysis type BI,
North American pulsed-field gel electrophoresis type 1 and
polymerase chain reaction ribotype 027 (5,6), and
due to the improper administration of antibiotics in the past
decade (7,8). Based on several reports from the US,
Canada and Europe, the incidence of C. difficile infection
(CDI) has increased between 2- and 4-fold in the past decade,
particularly in elderly patients exposed to health care settings,
including long-term care facilities and hospitals (9–11).
Children and peripartum females, previously described as low risk
populations for CDI, now show increased incidence (12). CDI-induced mortality rates have
markedly increased between 3,000/year in 1999–2000 and 14,000/year
in 2006–2007 (13). The risk of
recurrence following an initial episode of CDI is 20%, which
increases up to 60% following a third episode (14). CDI remains the most common cause of
hospital-acquired diarrhea, with the number of hospitalized
patients diagnosed with any CDI discharge increasing between
139,000 in 2000 and 336,600 in 2009, at a cost of $1,000,000,000
annually (15). In addition, CDI
has surpassed methicillin-resistance Staphylococcus aureus
as the most common hospital and healthcare facility-associated
infection (16). C.
difficile toxin A (TcdA), an enterotoxin, and C.
difficile toxin B (TcdB), a cytotoxin, are the major virulence
factors of C. difficile, and contribute to its pathogenicity
by inducing mucosal inflammation and diarrhea (17). In addition to these toxins, C.
difficile may produce a number of other putative virulence
factors, including C. difficile binary toxin (CDT),
fibronectin binding protein A, fimbriae, S-layer protein A, Cwp84
cysteine protease and Cwp66 and CwpV adhesions (17).
A previous nationwide epidemiological study
demonstrated that isolation of an A−B+
Clostridium (C.) difficile strain, which produces TcdB, but
not TcdA, has spread widely (18–20)
and is more frequent in east Asian countries (8,19,21–23).
Initially, TcdA was considered to be the most important factor
responsible for diarrheal disease (20), however, several observations in
variety of animal models have demonstrated this is not always the
case. In hamsters and mice, intragastric administration of TcdA,
but not TcdB, resulted in intestinal fluid accumulation, diarrhea,
hemorrhage and mortality (24).
However, previous reports have demonstrated that
A−B+ C. difficile strains can lead to
similar symptoms, ranging between mild diarrhea and severe
pseudomembranous colitis (25,26).
Diagnosis of CDI via the detection of TcdB is required in
microbiological laboratories in order to control these strains of
C. difficile, however, to date no such diagnostic reagents
have been developed in China. Therefore, rapid diagnosis of C.
difficile in patients with AAD and PMC is important for guiding
the treatment and control of nosocomial infection spread. The
primary objective of the present study was to establish hybridoma
cell lines stably secreting anti-CDB3 monoclonal antibodies (mAbs),
which may be used for the development of double-antibody sandwich
(ds)-ELISA. In addition, the present study aimed to detect TcdB in
diarrhea stools using ds-ELISA, in order to establish an effective
method for the future diagnosis of CDI.
Materials and methods
Ethical statement
Six female BALB/c mice (age, 6–8 weeks old; weight,
~20 g) were purchased from the Laboratory Animal Center of the
Central South University (Changsha, China) and housed under
pathogen-free conditions at 18–28°C, 40–70% humidity with free
access to standard chow and water. All experiments were performed
according to the guidelines of the Animal Ethics Committee of the
Central South University. The mice were sacrificed using rapid
cervical dislocation to minimize animal suffering.
Expression of glutathione S transferase
(GST)-CDB3 recombinant protein
The recombinant plasmid pGEX-4T-1-CDB3 of
Escherichia coli BL21 (DE3) was constructed in the Clinical
Laboratory in Xiangya Hosipital of Central South University. It was
transformed into an BL21(DE3) competent strain (American Type
Culture Collection, Manassas, VA, USA), and the E. coli
strains was induced. The optimal expression of recombinant proteins
was achieved by regulating the concentration of bacteria prior to
induction [optical density (OD), 600], along with the induction
temperature, induction time and the concentration of
isopropyl-β-D-thiogalactopyran oside (IPTG; Sigma-Aldrich, St.
Louis, MO, USA), details of which are shown in Table I. Following induction, the bacteria
were harvested and centrifuged at 4°C for 15 min at 4,449 × g, and
the pellet was resuspended in lysis buffer (Beijing
Donglinchangsheng Biotechnology Co., Ltd., Beijing, China)
containing 0.15 M NaCl, 0.05 M Tris-HCl, 0.1% SDS (Beijing
Donglinchangsheng Biotechnology Co., Ltd.), 1% Triton X-100, 1%
sodium deoxycholate (pH 7.2), followed by lysis by sonication in
ice and centrifugation at 4°C for 10 min at 10,012 × g. C.
difficile strains expressing the GST-CDB3 were also grown, as
described above. The expression of the GST-CDB3 recombinant protein
was analyzed using 10% SDS-PAGE. The SDS-PAGE gel was placed in
Coomassie Brilliant Blue dye (Sangon Biotech, Co., Ltd., Shanghai,
China) overnight in room temperature,then washed with distilled
water three times.
 | Table IL9(34) orthogonal test for the optimal
expression of recombinant proteins. |
Table I
L9(34) orthogonal test for the optimal
expression of recombinant proteins.
| Level | Concentration IPTG
(mmol/l) | Induction time
(h) | Concentration of
bacteria (OD 600) | Induction
temperature (°C) |
|---|
| 1 | 0.6 | 10 | 0.6 | 20 |
| 2 | 0.8 | 12 | 0.8 | 30 |
| 3 | 1.0 | 16 | 1.0 | 37 |
Purification and identification of
recombinant protein GST-CDB3
The GST-CDB3 recombinant protein was purified using
a GST Sefinose™ kit (Sangon Biotech, Co., Ltd.) and dialyzed in 1X
phosphate-buffered saline (PBS) at 4°C overnight, prior to being
condensed to a high concentration. The recombinant protein
concentration was determined using a Bicinchoninic Acid (BCA)
Protein Assay kit (Thermo Fischer Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's instructions. The expression
of the recombinant protein was analyzed using western blotting, as
previously described (27).
Briefly, the affinity purified recombinant GST-CDB3 protein was
incubated at 72°C for 10 min in 1X NuPage SDS loading buffer
(Beijing Donglinchangsheng Biotechnology Co., Ltd.), prior to being
separated by 10% SDS-PAGE under native and denaturing conditions,
and then transferred onto a nitrocellulose membrane (EMD Millipore,
Billerica, MA, USA). Following blocking with 5% fat-free milk at
37°C for 2 h, the membrane was incubated with mouse anti-GST
monoclonal primary antibody (1:2,000; P09211; EMD Millipore) at
37°C for 2 h, followed by incubation with polyclonal horseradish
peroxidase (HRP)-conjugated goat anti-mouse secondary antibody
(1:1,000; M8645; Sigma-Aldrich) at 37°C for 1 h. Between each
incubation step, the membranes were washed five times with PBS and
0.2% Tween 20 (PBST; Beijing Donglinchangsheng Biotechnology Co.,
Ltd.). The bands were visualized using an enhanced
chemiluminescence (ECL) system (Pierce Biotechnology, Inc.,
Rockford, IL, USA).
Immunization of mice
BALB/c mice were used for hybridoma production, as
previously described (28).
Briefly, 8-week-old female BALB/c mice were intraperitoneally
injected three times with 50 µg purified GST-CDB3
recombinant protein at 2 week intervals. Complete and incomplete
Freund's adjuvant (1:1; Sigma-Aldrich) was used to emulsify the
protein for the first and subsequent immunizations. At 10 days
following the third immunization, ELISA (Sangon Biotech, Co., Ltd.)
was performed, as previously described (29), to confirm that the serum contained
antibodies targeting recombinant protein GST-CDB3, and to detect
the antibody titer of mouse serum (using the tail vein procedure).
Recombinant protein GST-CDB3 was used as a coating antigen, 1%
BSA-PBS as a blocking solution, mouse serum as the primary
antibody, and HRP-conjugated goat anti-mouse immunoglobulin (Ig) G
polyclonal antibodies (1:3,000; Sigma-Aldrich) as the secondary
antibody. The mouse with the highest titer specific for GST-CDB3,
determined using indirect ELISA, was then administered with 100
µg recombinant protein GST-CDB3 without adjuvant 1 week
later. Subsequently, 3 days after the last immunization, the spleen
was removed and macerated to obtain B lymphocytes, and then washed
twice with RPMI-1640 medium (Gibco Life Technologies, Carlsbad, CA,
USA).
Hybridoma production
Myeloma cells and spleen lymphocytes were prepared
for fusion, as previously described (30). The SP2/0 mouse myeloma cell line
(Cancer Research Laboratory, Central South University) in the
logarithmic growth phase and with good morphological appearance was
used as a fusion partner, the SP2/0 cells were cultured in 10%
fetal bovine serum (FBS; GE Healthcare Life Sciences, Logan, UT
USA) and RPMI-1640 medium at 37°C in an atmosphere containing 5%
CO2, prior to treatment with 8-azaguanine
(Sigma-Aldrich). The feeder cells of the peritoneal macrophages
were collected from normal BALB/c mice by peritoneal washing with
RPMI-1640 medium supplemented with hypoxanthine (1×104
M), aminopterin (4×107 M) and thymidine
(1.6×105 M; Sigma-Aldrich) and seeded into 96-well
plates 1 day prior to fusion. The splenic lymphocytes and SP2/0
cells were fused at a ratio of 5–10:1. This protocol involved the
incubation of the cells in 1 ml 50% (w/w) polyethylene
glycol/dimethylsulfoxide 3350 (Sigma-Aldrich) at 37°C for 1 min,
followed by washing with RPMI-1640 medium for 5 min. The hybridomas
were seeded into 96-well plates containing the feeder cells and
screened using hypoxanthine-aminopterin-thymidine medium. Growth of
the hybridomas was regularly observed by an optical microscope
(BIO500 PH; Olympus, Tokyo, Japan).
Screening of positive clones and
cloning
At 2 weeks following fusion, the cell clones
producing anti-CDB3 antibody were screened and identified by
indirect ELISA, using the (0.89 mg/ml) GST-CDB3 recombinant protein
and 1.21 mg/ml GST protein as coating antigens, 1% BSA-PBS as a
blocking solution, the supernatant of each hybridoma cell clone as
the primary antibody, and HRP-conjugated goat anti-mouse IgG
polyclonal antibodies (1:3,000; Sigma-Aldrich) as the secondary
antibody. The optical density (OD) was read at 450 nm using a
microplate reader (Tianshi Technologies, Co., Ltd., Beijing, China
and considered positive when the ratio of OD450 experimental to
OD450 control was greater than three-fold. The wells, which were
positive for the GST-CDB3 recombinant protein, but negative for GST
protein were considered positive, and were sub-cloned using
limiting dilution, as previously described (31). Further cloning was performed
between three and five times in hypoxan-thine-thymidine medium
(Sigma-Aldrich) for 2 weeks until all selected hybridomas were
positive. Finally, the positive clones in the 96-well plates were
transferred into 24-well plates in RPMI-1640 medium supplemented
without FBS. The strain of hybridoma cells, which established
stable growth with the highest positive expression was selected,
expanded and preserved at −70°C.
Preparation and titer determination of
anti-CDB3 mAb
The production of hybridomas from ascitic fluid was
performed, as previously described by Harlow et al (31). The BALB/c mice were
intraperitoneally injected with 0.5 ml incomplete Freund's adju
vant. After 7 days, the mice were intraperitoneally injected with
5×106 hybridoma cells diluted in D-Hank's buffered
solution (Sigma-Aldrich), which exhibited optimal results following
indirect ELISA. The ascitic fluid was collected after 2 weeks and
centrifuged for 5 min at 2,780 × g to remove cellular deposition
and other sediments. The anti-CDB3 mAb supernatant was then diluted
into a series of gradients (1:100, 1:200, 1:400, 1:800, 1:1,600,
1:3,200, 1:6,400, 1:12,800, 1:25,600, 1:51,200, 1:102,400 and
1:204,800), and indirect ELISA was performed using recombinant
protein GST-CDB3 as a coating antigen, 1% BSA-PBS as a blocking
solution, the serial dilutions of supernatant as the primary
antibody, and HRP-conjugated goat anti-mouse IgG polyclonal
antibodies (1:3,000; Sigma-Aldrich) as the secondary antibody, in
order to detect the titer of anti-CDB3 mAb. The OD was read at 450
nm.
Isotype determination and purification of
anti-CDB3 mAb
The antibody isotyping of anti-CDB3 mAb was
performed using an SBA Clonotyping system/HRP ELISA kit
(SouthernBiotech, Birmingham, AL, USA) containing goat anti-mouse
IgG1, IgG2a, IgG2b, IgG3, IgA, IgM, Igκ and Igλ, according to the
manufacturer's instructions. The anti-CDB3 IgG antibodies (5 ml)
were added with an equivalen volume of precooled ammonium sulfate
overnight in 4°C, and then centrifuged at 4°C for 15 min at 1,738 ×
g. The supernatant was decanted, and the precipitation was
solubilized in PBS (0.02 M; 5 ml) and purified using Protein G
affinity chromatography (GE Healthcare Life Sciences), according to
the manufacturer's instructions. The positive components confirmed
by indirect ELISA were dialyzed and condensed to a high
concentration. Reduction was performed using mercaptoethanol
(Sigma-Aldrich) and separation was performed using 10% SDS-PAGE.
Concentration was determined using a BCA Protein Assay kit.
Determination of affinity using
ELISA
The present study determined affinity levels using
ELISA, as previously described (32). Briefly, ELISA plates pre-coated
with 5 µg/ml GST-CDB3 recombinant protein were blocked with
2% FBS at 37°C for 2 h. Serial dilutions of the purified mAb were
incubated at 37°C for 2 h. The plates were then rinsed and
incubated with HRP-conjugated goat anti-mouse IgG poly-clonal
antibodies (1:3,000) at 37°C for 1 h. OD was measured at 450 nm and
curves were constructed using the OD450 values obtained from the
serial dilutions of the purified mAb, in order to determine the
relative affinity.
Western blot analysis of anti-CDB3 mAb
specificity
The GST-CDB3 recombinant protein was cleaved using
thrombin (Sigma-Aldrich) and separated using 10% SDS-PAGE. The
proteins were transferred onto a nitrocellulose membrane by
electroblotting, prior to being blocked with 5% fat-free milk at
37°C for 2 h. The membrane was incubated with purified anti-CDB3
mAb (1:1,000) overnight at 4°C. Following three washes with PBST,
the membrane was incubated with HRP-conjugated goat anti-mouse IgG
polyclonal antibodies (1:1,000; Sigma-Aldrich) in 37°C for 1 h.
Following further washing the membrane was visualized using an ECL
system.
Conjugation of mAb to HRP
The conjugation of mAb to HRP was performed, as
previously described (33). A
total of 2.6 mg HRP dissolved in 1.0 ml water and 0.1 ml sodium
periodate solution (0.1 mol/l) were combined, stirred for 20 min,
and dialyzed in 1 mM sodium acetate buffer (Sangon Biotech, Co.,
Ltd.; pH 4.4) overnight at 4°C. A total of 0.5 ml (0.16 M) was
subsequently added to the solution and stirred at 4°C for 1 h. A
total of 6 mg monoclonal 4A4G2 in 1 ml sodium carbonate buffer
(0.01 M, pH 9.6) was added and stirred for 2 h. A total of 0.1 ml
sodium borohydride solution (4 mg/ml) was added and dialyzed in
0.02 M PBS (pH 7.2) overnight at 4°C, then precipitated by the
addition of equal volumes of 100% ammonium sulfate (4°C, 4 h),
prior to centrifugation at 3,000 × g for 5 min, dialyzed against
0.01 M PBS (pH 7.4) overnight at 4°C, and stored at 4°C following
dilution with 500 ml/l glycerol (1:1).
Establishment and optimization of
ds-ELISA
A ds-ELISA for TcdB was established using 4A4G2 as a
HRP-labeled mAb and 2F8A6 as a capture mAb. A chessboard titration
method was designed to examine the optimal working concentration of
capture antibody and HRP-labeled mAb. Briefly, a 96-well
immunoplate was coated with purified anti-CDB3 mAb (2F8A6;
concentrations, 20 µg/ml, 10 µg/ml, 5 µg/ml,
2.5 µg/ml, 1.25 µg/ml and 0.625 µg/ml) in 0.05
M/l carbonate buffer (pH 9.6) overnight at 4°C. Following blocking
with 200 µl 2% BSA-PBS overnight at 4°C, the plate was
washed five times in PBST, and 100 µl serial dilutions of
GST-CDB3 recombinant protein in PBS were added to each well and
incubated at 37°C for 2 h, followed by a further five washes. At
total of 100 µl diluted HRP-conjugate anti-CDB3 mAb (1:50,
1:100, 1:200, 1:400, 1:800, 1:1,600, 1:3,200, 1:6,400 and 1:12,800;
detecting antibody) was subsequently added, and the plates were
incubated at 37°C for 1 h. The plates were then washed and
incubated at 37°C for 30 min in the dark, following which then the
OD450 was measured.
Stool sample collection
This study was conducted at, and approved by, the
Xiangya Hospital of Central South University (Hunan, China) between
June 2011 and November 2012. Patients with diarrhea were enrolled
which was defined as more than three loose watery stool passages
per day, on consecutive days or six times within 36 h. CDI was
diagnosed when the Clostridium difficile was isolated from
stool culture and identified by API20A (BioMerieux, Marcy l'Etoile,
France), the Clostridium difficile isolates were also
positive for toxin genes (tcdA, tcdB) as shown by multiplex PCR.
The non-CDC group was defined as a group of patients who did not
have diarrhea or had diarrhea but were negative for toxin A/B.
Analysis of clinical stool samples using
ds-ELISA
A total of 45 diarrhea stool samples, including 15
samples containing A+B+ toxigenic C.
difficile, 15 samples containing A+B+
toxigenic C. difficile and 15 samples negative for C.
difficile, were determined using anaerobic toxigenic culture.
Briefly, selective enrichment culture for C. difficile was
performed, and the levels of TcdA, TcdB, CDTA, CDTB were determined
using polymerase chain reaction (PCR). All experimental procedures
and specimen collections were approved by the Ethics Committee of
the Xiangya Hospital of Central South University. The expression
levels of TcdB in the stool samples were analyzed using the
ds-ELISA technique, as described above.
Fecal culture
Each stool sample (2 ml) was added to an equal
volume of absolute ethanol, mixed and left at room temperature for
1 h, then centrifuged (4,449 × g/10 min) and the pellet was
inoculated on selective Clostridium difficile
Moxalactam-Norfloxacin-Taurocholate agar (CDMN-TA; Oxoid Ltd.,
Cambridge, UK) supplemented with 7% horse blood after alcohol shock
(10), and incubated at 35±2°C in
an anaerobic atmosphere (85% nitrogen, 10% hydrogen and 5% carbon
dioxide) for 48 h. Suspect Clostridium difficile colonies
were subcultured and identified as Clostridium difficile on
the basis of their morphology on agar plates and by Gram staining
as well as the characteristic odor. In addition, oxygen resistance
experiments were conducted on specific colonies and confirmed by
API 20A (BioMerieux). The demonstration of toxin production was
conducted by multiplex PCR for the toxin genes (tcdA and tcdB) or a
positive toxin assay using the VIDAS® Clostridium
difficile Toxin A & B (BioMerieux, France). Strains were
stored at -80°C in cryobank tubes until further testing.
Multiplex PCR
Toxin gene DNA was isolated from colonies of
Clostridium difficile using a DNeasy blood and tissue kit
(Qiagen, Hilden, Germany) according to the manufacturer's
instructions. All isolates were tested for the presence of tcdA,
tcdB, cdtA and cdtB genes. The oligonucleotide primers used are
shown in Table II. The
amplification reactions were performed in 25 µl final volume
containing 12.5 µl Taq PCR Master mix (2X; Beijing Tiangen Biotech
Co., Ltd., Beijing, China), 1 µl each primer (10
µmol/l), 8.5 µl water and 2 µl DNA. The
reaction conditions of PCR are shown in Table III. Gels were run under standard
conditions on 1.0% agarose for 25 min at 100 V, using a 100–2,000
bp ladder (Fermentas, Pittsburgh. PA, USA) as size standard and
stained with ethidium bromide for visualization.
| Table IIPrimer sequences of oligonucleotides
was used for conventional PCR. |
Table II
Primer sequences of oligonucleotides
was used for conventional PCR.
| PCR product | Primer | Sequence
(5′–3′) | Fragment length
(bp) |
|---|
| tcdA | NK1 |
5-GGACATGGTAAAGATGAATTC-3 | 546 |
| NK2 |
5-CCCAATAGAAGATTCAATATTAAGCTT-3 | |
| tcdB | NK104 |
5-GTGTAGCAATGAAAGTCCAAGTTTACGC-3 | 204 |
| NK105 |
5-CACTTAGCTCTTTGATTGCTGCACCT-3 | |
| cdtA | Cdta pos |
5-TGAACCTGGAAAAGGTGATG-3 | 375 |
| Cdta rev |
5-AGGATTATTTACTGGACCATTTG-3 | |
| cdtB | Cdtb pos |
5-CTTATTGCAAGTAAATACTGAG-3 | 510 |
| Cdtb rev |
5-ACCGGATCTCTTGCTTCAGTC-3 | |
| Table IIIPolymerase chain reaction
conditions. |
Table III
Polymerase chain reaction
conditions.
| PCR product | Reaction conditions
|
|---|
| Initial enzyme
activation | Cycles | Denaturation | Annealing | Elongation | Final
elongation |
|---|
| tcdA | 95°C, 1 min | 30 | 95°C, 15 sec | 62°C, 2 min | 72°C, 40 sec | 72°C, 10 min |
| tcdB | 95°C, 1 min | 30 | 95°C, 20 sec | 55°C, 30 sec | 60°C, 2 min | 72°C, 10 min |
| cdtA/cdtB | 94°C, 1 min | 30 | 94°C, 45 sec | 52°C, 1 min | 72°C, 80 sec | 72°C, 10 min |
Results
Expression of GST-CDB3 recombinant
protein
The optimal conditions for inducing the expression
of the GST-CDB3 recombinant protein, which was the optimal
concentration of bacteria prior to induction, was 0.8 nM, and that
of IPTG was 0.6 mM, determined using OD600. The optimal induction
duration was 12 h and the induction temperature was 30°C. The
GST-CDB3 recombinant protein was predominantly expressed as a
soluble protein.
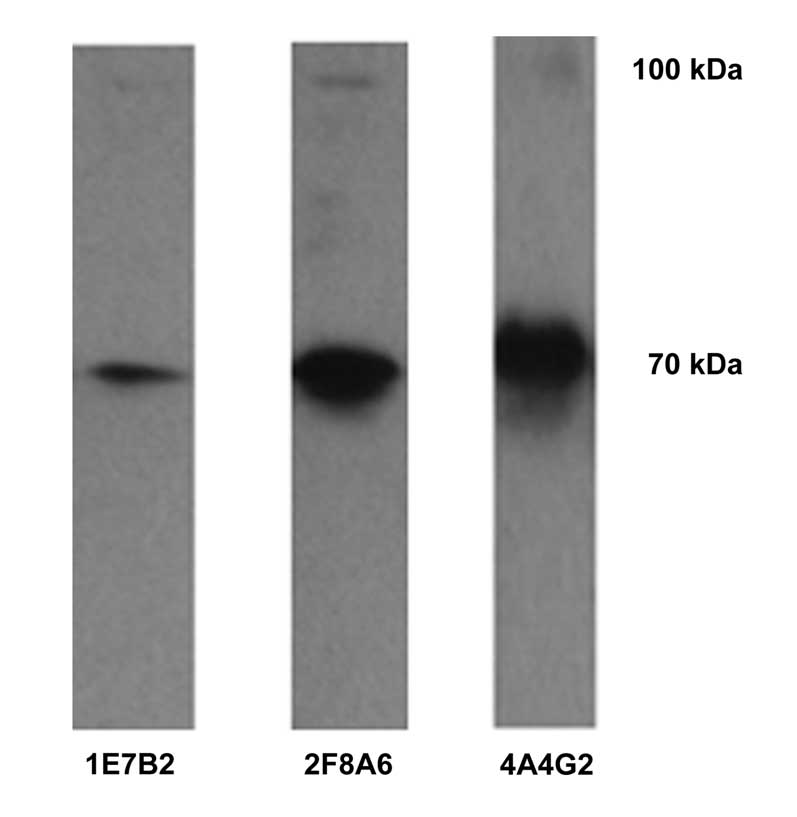
Purification and identification of
GST-CDB3 recombinant protein
The GST-CDB3 recombinant protein was purified using
a GST Sefinose™ kit under native conditions, identified using 10%
SDS-PAGE and subsequently stained using Coomassie Brilliant Blue
R250 (Fig. 1). The molecular
weight of the GST-CDB3 recombinant protein was 96 kDa. The protein
was then further identified using western blot analysis with
anti-GST tag antibody (Fig. 2).
The recombinant protein concentration was 0.89 mg/ml.
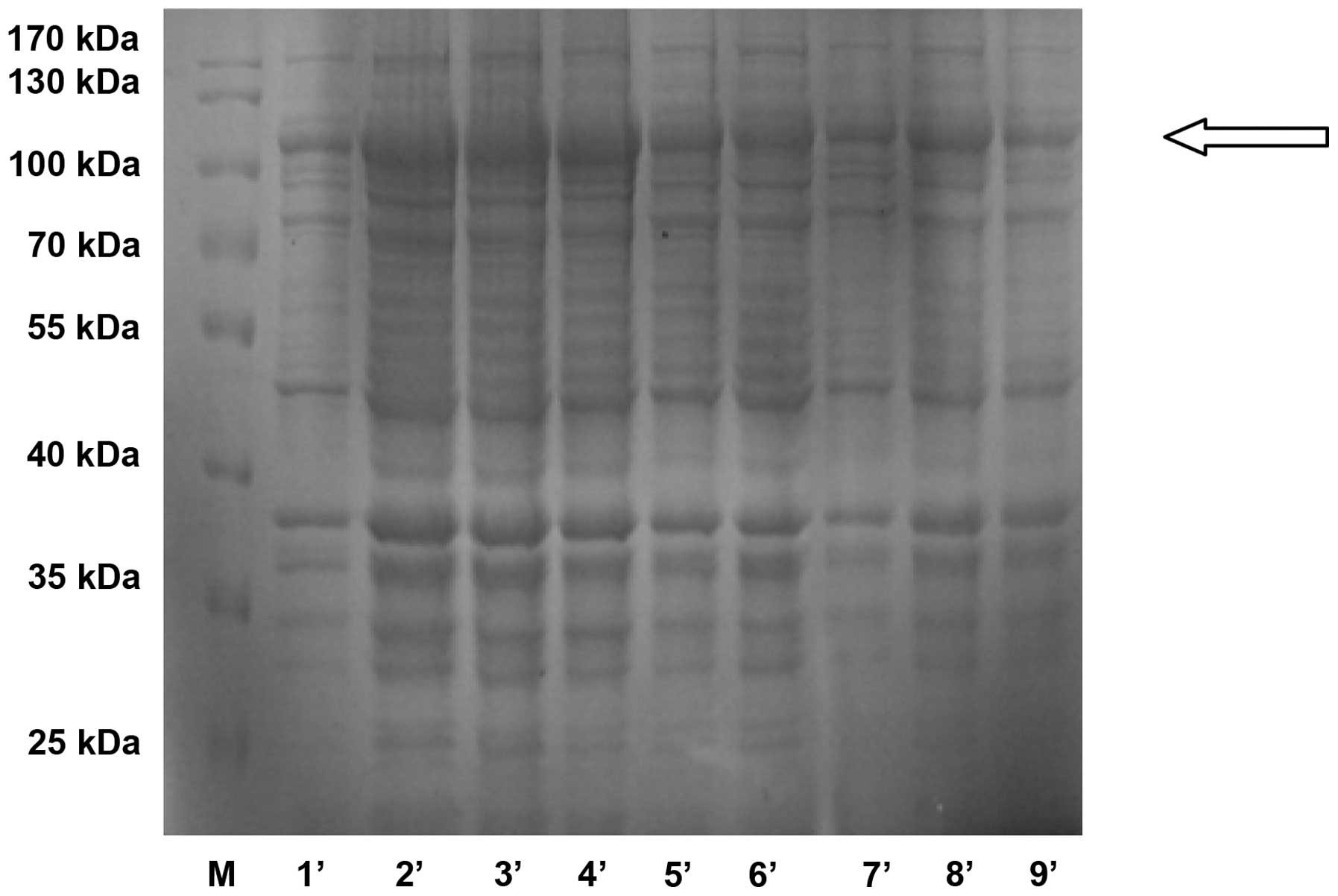 | Figure 1Expression of purified recombinant
GST-CDB3 protein in Escherichia coli was analyzed using 10%
SDS-PAGE. The proteins were visualized by Coomassie Blue R-250
staining. Lane 1, protein molecular weight marker; Lane 2, purified
GST-CDB3 recombinant protein; Lane 3, molecular size. M, protein
molecular weight marker; 1′, A1B1C1D1; 2′, A1B2C2D2; 3′, A1B3C3D3;
4′, A2B1C2D3; 5′, A2B2C3D1; 6′, A2B3C1D2; 7′, A3B1C3D2; 8′,
A3B2C1D3; and 9′, A3B3C2D1. A was the concentration of IPTG
(mmol/l); B was the induction time (h); C was the concentration of
bacteria (OD 600); D was induction temperature (°C). 1,2,3 was the
level of each, as shown in Table
1. GST, glutathione S transferase. |
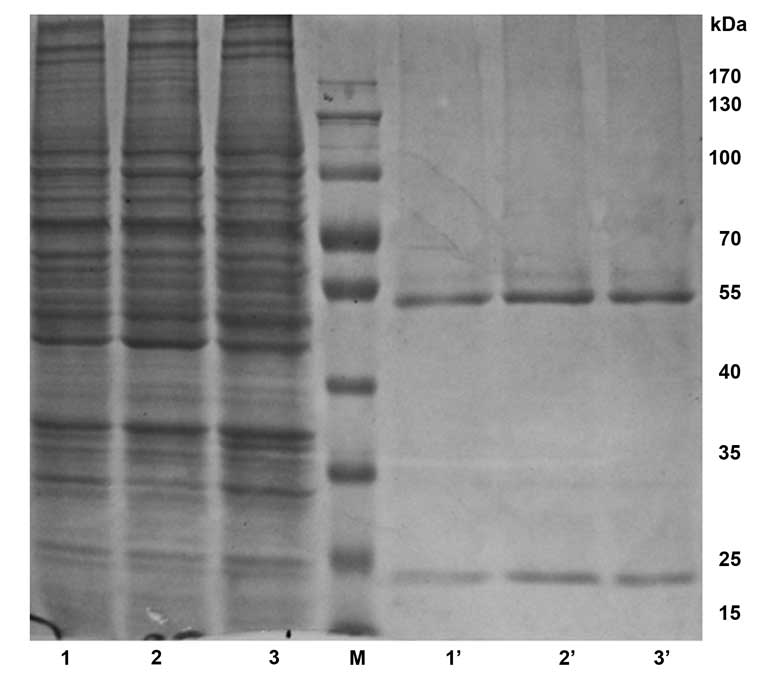
Preparation of anti-CDB3 mAb
In the present study, the rate of cell fusion was
98.4% (945/960), and a total of five (1E7B, 1F8D3, 2F8A6, 3B6F1 and
4A4G2) positive hybridoma cell lines stably secreting anti-CDB3 mAb
were identified: 1E7B2 IgG1 (λ), 1F8D3 IgM (κ), 2F8A6 IgG2a (κ),
3B6F1 IgM (λ) and 4A4G2 IgG1 (κ). The titer was assayed using
indirect ELISA, obtaining the following dilutions: 1E7B2
(1:25,600), 1F8D3 (1:12,800), 2F8A6 (1:51,200), 3B6F1 (1:25,600)
and 4A4G2 (1:51,200). The 1E7B2, 2F8A6 and 4A4G2 mAbs were
successfully purified by bitterness-ammonium sulfate and protein G
agarose gel electrophoresis. Following dialyzation and
condensation, the purity was identified using 10% SDS-PAGE, with a
purification of >90% (Fig. 3),
and the concentrations of the mAbs were 27.30 µg/ml, 64.37
µg/ml and 37.36 µg/ml, respectively. The molecular
weight was ~160 kDa.
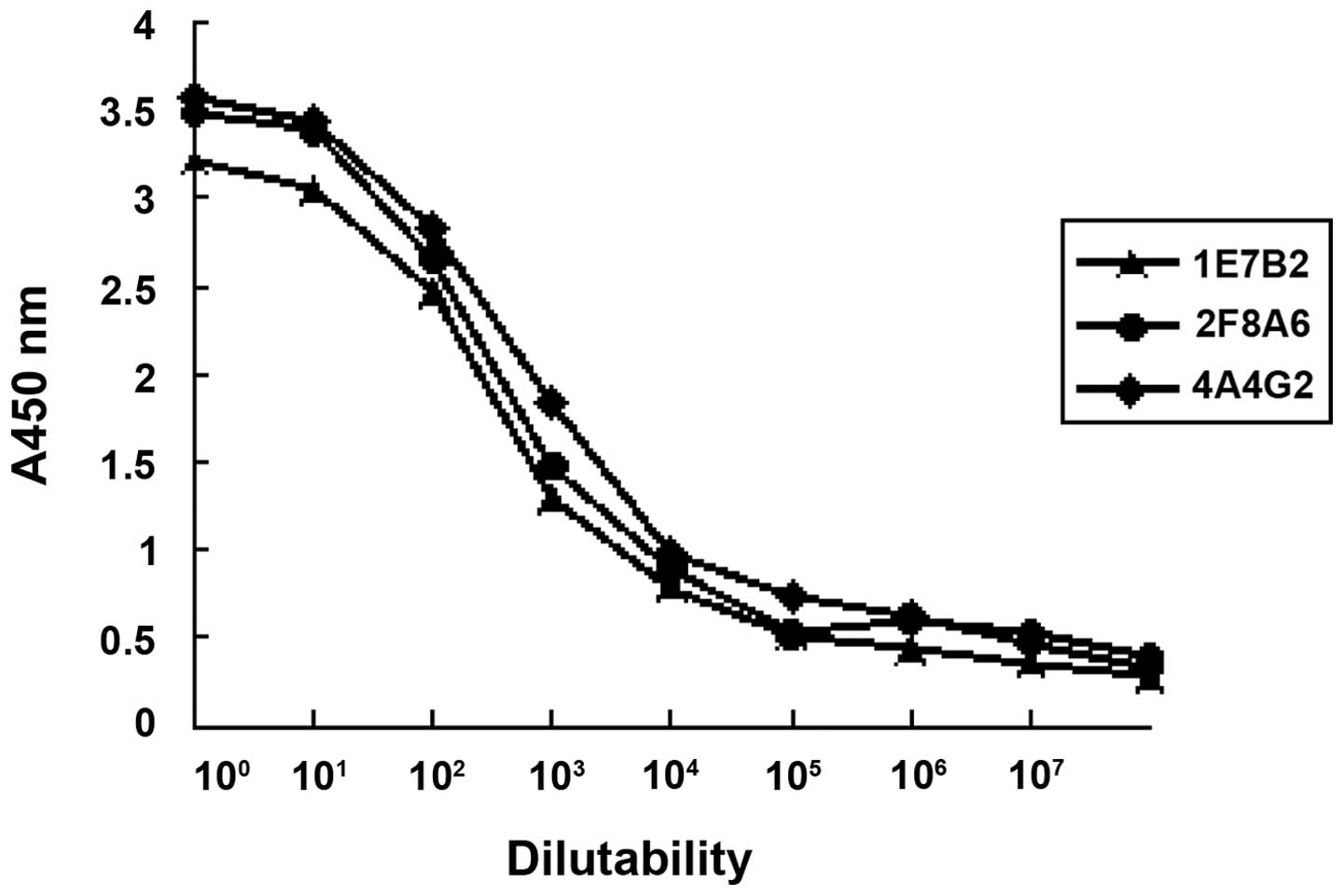
Affinity of anti-CDB3 mAb
The relative affinities of the mAbs were defined by
the antibody concentration at which the OD value reached half the
maximal signal at the plateau stage of antigen-antibody binding.
The order of relative affinity of the three selected mAbs were
4A4G2, 2F8A6, 1E7B2 (Fig. 4).
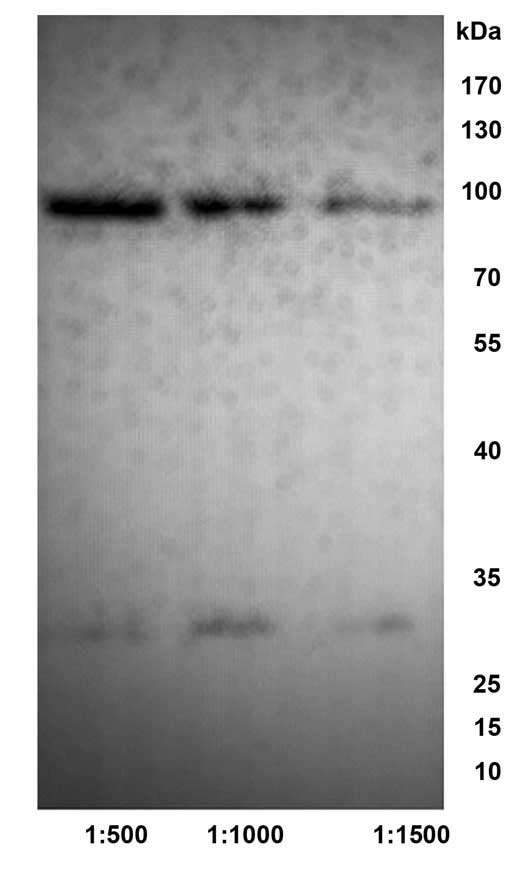
Specificity of anti-CDB3 mAb
Western blotting results demonstrated that the
1E7B2, 2F8A6 and 4A4G2 anti-CDB3 mAbs reacted specifically to the
large unit of the GST-CDB3 recombinant protein cleaved by thrombin
(molecular weight, ~70 kDa) and the uncleaved GST-CDB3 recombinant
protein (molecular weight, ~100 kDa), however, no reaction with the
tagged protein GST was observed (molecular weight, ~26 kDa)
(Fig. 5).
Establishment and optimization of
ds-ELISA
The anti-CDB3 mAb (4A4G2) was successfully labeled
with HRP by modified sodium periodate oxidation. The ds-ELISA was
and the optimal working concentration of capture antibody and
HRP-labeled mAb were demonstrated to be 20 µg/ml and 1:400,
respectively, as determined using chessboard titration.
Detection of TcdB in clinical stool
samples using ds-ELISA
Of the 15 specimens of
A+B+-type C. difficile, 11 were
positive for TcdB (73.33%), detected using ds-ELISA, 13 of the 15
A−B+-type C. difficile samples were
positive for TcdB (86.67%) and all of the 15 C. difficile
specimens were negative for TcdB.
Discussion
Rapid and accurate diagnosis of CDI is essential for
improving the outcomes of patients and for reducing horizontal
transmission in health care facilities. There are several types of
laboratory assessments aimed at detecting pathogens and their
toxins in diarrheal fecal specimens, each with their own qualities
and limitations. However, an optimal test for the diagnosis of
C. difficile-associated diseases remains to be established
(34,35). Curry (36) reported that the 'diagnosis of CDI
remains one of the most vexing difficulties for hospital
microbiology laboratories', due to the absence of a single accepted
gold standard. For several years, the cell culture cytotoxicity
neutralization assay (CCNA) was regarded as the standard method for
the diagnosis of CDI (34) due to
its correlation with PMC with a specificity >97%, and its
ability to detect toxins in the stool as levels as low as 10 pg.
However, the CCNA assay is expensive, lacks standardization among
laboratories, requires advanced skills and cell culture facilities
and has a 24–48 h turn around period. Several studies have reported
toxigenic culture (TC) as the method of choice for the diagnosis of
CDI, and is suggested to be the most sensitive method for CDI
detection (34,35,36).
Compared with TC, CCNA has only 67–79% sensitivity (36). The Society for Healthcare
Epidemiology of America and the Infectious Diseases Society of
America 2010 guidelines note that 'the sensitivity and specificity
of stool culture followed by identification of a toxigenic isolate,
as performed by an experienced laboratory provides the standard
against which other clinical tests should be compared' (3). However, this method is slow and
laborious, often requires 48–72 h to complete and, therefore, is
unlikely to be used in clinical laboratories as the standard method
for CDI diagnosis. Furthermore, successfully culturing C.
difficile-producing toxin without detection of toxin in the
diarrheal stool specimens has not been associated with poorer
clinical outcomes, compared with stools negative for toxigenic
C. difficile (35). Nucleic
acid amplification tests (NAATs) are the most recent methods used
for the detection of C. difficile. Available NAATs for the
identification of C. difficile genes are PCR, real-time PCR,
and loop-mediated isothermal amplification (37). These assessments detect the
chromosomal genes encoding TcdA and TcdB, or the toxin regulatory
gene directly from stool samples (38); however, despite detecting the toxin
gene, they do not detect the toxin in stool samples, raising
concerns over accurate diagnosis in asymptomatic carriers. In
addition, the use of PCRs may increase CDI incidence rates by
>50% (39). A glutamate
dehydrogenase antigen assay may be used as a rapid screening method
for the presence of C. difficile in stools. The assessment
is fast (15–45 min), convenient, inexpensive and sensitive,
however, it only documents the presence of C. difficile, not
the presence of toxigenic strains (20% of C. difficile
strains do not produce toxin) or the presence of toxin in stool
(40). Commercial rapid enzyme
immunoassays have been the most widely used diagnostic assessment
for the detection of TcdA and TcdB due to the fact that they are
technically simple to perform, provide rapid (same day) results and
are relatively inexpensive (34);
however, they may lead to more false-positive test results,
compared with the cyto-toxicity assay, as the polyclonal antibodies
against TcdA and TcdB used in immunoassays may target other
antigens at the same time (41).
These limitations may be solved with the use of mAbs, as they can
not only improve the specificity of the method, but also decrease
the number of false positive results.
A−B+ C. difficile
strains causing severe infections and outbreaks have been reported
to have increased significantly increased. The prevalence of
A−B+ strains among CDI isolates varies
depending on the country; in the majority of Europe and in North
America, the prevalence of A−B+ strains has
been reported to be only 0.2–8% (8,42),
however, a report from Shanghai revealed that
A−B+ strains were responsible for 33.3% of
all CDI cases in China (19),
which suggests that A−B+ strains are more
prevalent in Asia. TcdB is organized into four distinct structural
domains (43): The N-terminal
glucosyltransferase domain (residues 1–543), the cysteine protease
domain (residues 544–807), the membrane translo-cation (residues
808–1850) and the C-terminal oligopeptide repeat domain (residues
1851–2366). The short, homologous regions, termed combined
repetitive oligopeptides, found in TcdB are more divergent and less
frequent than those found in TcdA (43). TcdB is divided into three
fragments, which are separately expressed: CDB1 (amino acids 1–546)
contains the catalytic domain; CDB2 (amino acids 901–1750) harbors
the putative transmembrane domain; and CDB3 (amino acids 1751–2366)
is thought to be the receptor binding domain. Von Eichel-Streiber
et al (44) demonstrated
that anti-CDB3 exhibited a marked reaction with CDB3 and a weak
reaction with CDB1, but not with CDB2, concluding that the receptor
CDB3 binding domain, harboring repetitive peptide domains, was the
most antigenic region of the toxin.
The present study successfully established five
hybridoma cell lines (1E7B, 1F8D3, 2F8A6, 3B6F1 and 4A4G2), which
stably secreted anti-CDB3 mAb, by immunizing BALB/c mice with the
GST-CDB3 recombinant protein, which was charac-terized using
indirect ELISA and western blotting. The IgG isotype was
predominant [1E7B2 IgG1 (λ), 2F8A6 IgG2a (κ) and 4A4G2 IgG1 (κ)].
Indirect ELISA demonstrated that the 2F8A6 and 4A4G2 mAbs exhibited
high titers of specific binding affinities for CDB3. Western
blotting revealed that the 2F8A6 and 4A4G2 mAbs specifically
recognized the CDB3 protein. Therefore, the purified 4A4G2 mAb was
labeled with HRP and selected as the detection antibody, and the
purified 2F8A6 mAb was selected as the coated antibody to establish
a ds-ELISA for the detection of TcdB in clinical diarrhea stool
samples. The concentration of the coated antibody was determined as
20 µg/ml, and the optimal working concentration of
HRP-labeled mAb was 1:400, determined using the chessboard
titration method in ds-ELISA. To the best of our knowledge, the
present study is the first to describe a ds-ELISA using
monospecific antibody as a capture antibody, and mAb as a detection
antibody. This type of ds-ELISA offers distinct advantages: The
antibodies utilized, including secondary antibodies, are
homogeneous, with specificity for CDB3 and are also less likely to
interact with closely associated proteins. Cross-reactivity of
secondary antibodies is also eliminated. The results of the present
study indicated that the sensitivity of the ds-ELISA was 73.33% to
A−B+ toxigenic C. difficile strains
and 86.67% to A−B+ toxigenic C.
difficile strains, with a specificity of 100% overall.
The isolation of A−B+ C.
difficile strains producing TcdB, but not TcdA, are widespread,
and are more frequent in east Asian countries. A diagnostic method
capable of rapidly detecting the presence of TcdB is required for
use in microbiological laboratories in order to control these
strains of C. difficile, however, no diagnostic reagents
have been available in China. The rapid diagnosis of C.
difficile is important and guides the treatment and control of
nosocomial spread of infection. To the best of our knowledge, the
present study is the first to develop novel mAbs specific to CDB3,
and a ds-ELISA kit with high specificity and sensitivity for the
rapid detection of TcdB was established. These provide useful tools
for the diagnosis of TcdB in CDI.
Acknowledgments
The present study was supported by funding from the
Research Fund Project of the Health Department of the Hunan
Province, China (grant no. 1132).
References
|
1
|
Hall IC and O'Toole E: Intestinal flora in
newborn infants with a description of a new pathogenic anaerobe. Am
J Dis Child. 49:390–402. 1935. View Article : Google Scholar
|
|
2
|
George WL, Sutter VL, Goldstein EJ, Ludwig
SL and Finegold SM: Aetiology of antimicrobial-agent-associated
colitis. Lancet. 1:802–803. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cohen SH, Gerding DN, Johnson S, Kelly CP,
Loo VG, McDonald LC, Pepin J and Wilcox MH; Healthcare Epidemiology
of America; Infectious Diseases Society of America: Clinical
practice guidelines for Clostridium difficile infection in adults:
2010 update by the society for healthcare epidemiology of America
(SHEA) and the infectious diseases society of America (IDSA).
Infect Control Hosp Epidemiol. 131:431–455. 2010. View Article : Google Scholar
|
|
4
|
Kelly CP: Current strategies for
management of initial Clostridium difficile infection. J Hosp Med.
7(Supple 3): S5–S10. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miller M, Gravel D, Mulvey M, Taylor G,
Boyd D, Simor A, Gardam M, McGeer A, Hutchinson J, Moore D, et al:
Health care-associated Clostridium difficile infection in Canada:
Patient age and infecting strain type are highly predictive of
severe outcome and mortality. Clin Infect Dis. 50:194–201. 2010.
View Article : Google Scholar
|
|
6
|
Kelly CP and LaMont JT: Clostridium
difficile - more difficult than ever. N Engl J Med. 359:1932–1940.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khanna S and Pardi DS: The growing
incidence and severity of Clostridium difficile infection in
inpatient and outpatient settings. Expert Rev Gastroenterol
Hepatol. 4:409–416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bauer MP, Notermans DW, van Benthem BH,
Brazier JS, Wilcox MH, Rupnik M, Monnet DL, van Dissel JT and
Kuijper EJ; ECDIS Study Group: Clostridium difficile infection in
Europe: A hospital-based survey. Lancet. 377:63–73. 2011.
View Article : Google Scholar
|
|
9
|
Gilca R, Hubert B, Fortin E, Gaulin C and
Dionne M: Epidemiological patterns and hospital characteristics
associated with increased incidence of Clostridium difficile
infection in Quebec, Canada, 1998–2006. Infect Control Hosp
Epidemiol. 31:939–947. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barbut F, Jones G and Eckert C:
Epidemiology and control of Clostridium difficile infections in
healthcare settings: An update. Curr Opin Infect Dis. 24:370–376.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuijper EJ, Coignard B and Tüll P; ESCMID
Study Group for Clostridium difficile; EU Member States; European
Centre for Disease Prevention and Control: Emergence of Clostridium
difficile-associated disease in North America and Europe. Clin
Microbiol Infect. 12(Suppl 6): 2–18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kutty PK, Woods CW, Sena AC, Benoit SR,
Naggie S, Frederick J, Evans S, Engel J and McDonald LC: Risk
factors for and estimated incidence of community-associated
Clostridium difficile infection, North Carolina, USA. Emerg Infect
Dis. 16:197–204. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hall AC, Curns AT, McDonald LC, Parashar
UD and Lopman BA: The roles of Clostridium difficile and norovirus
among gastroenteritis-associated deaths in the United States,
1997–2007. Clin Infect Dis. 55:216–223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Khanna S and Pardi DS: Clostridium
difficile infection: New insights into management. Mayo Clin Proc.
87:1106–1117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McDonald LC, Lessa F and Sievert D;
Centers for Disease Control and Prevention (CDC): Vital signs:
Preventing Clostridium difficile infections. MMWR Morb Mortal Wkly
Rep. 61:157–162. 2012.
|
|
16
|
Miller BA, Chen LF, Sexton DJ and Anderson
DJ: Comparison of the burdens of hospital-onset, healthcare
facility-associated Clostridium difficile infection and of
healthcare-associated infection due to methicillin-resistant
Staphylococcus aureus in community hospitals. Infect Control Hosp
Epidemiol. 32:387–390. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuehne SA, Cartman ST, Heap JT, Kelly ML,
Cockayne A and Minton NP: The role of toxin A and toxin B in
Clostridium difficile infection. Nature. 467:711–713. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goorhuis A, Legaria MC, van den Berg RJ,
Harmanus C, Klaassen CH, Brazier JS, Lumelsky G and Kuijper EJ:
Application of multiple-locus variable-number tandem-repeat
analysis to determine clonal spread of toxin A-negative Clostridium
difficile in a general hospital in Buenos Aires, Argentina. Clin
Microbiol Infect. 15:1080–1086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang H, Wu S, Wang M, Zhang Y, Fang H,
Palmgren AC, Weintraub A and Nord CE: Clostridium difficile
infections in a Shanghai hospital: Antimicrobial resistance, toxin
profiles and ribotypes. Int J Antimicrob Agents. 33:339–342. 2009.
View Article : Google Scholar
|
|
20
|
Lyerly DM, Krivan HC and Wilkins TD:
Clostridium difficile: Its disease and toxins. Clin Microbiol Rev.
1:1–18. 1988.PubMed/NCBI
|
|
21
|
Kim H, Riley TV, Kim M, Kim CK, Yong D,
Lee K, Chong Y and Park JW: Increasing prevalence of toxin
A-negative, toxin B-positive isolates of Clostridium difficile in
Korea: Impact on laboratory diagnosis. J Clin Microbiol.
46:1116–1117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Iwashima Y, Nakamura A, Kato H, Kato H,
Wakimoto Y, Wakiyama N, Kaji C and Ueda R: A retrospective study of
the epidemiology of Clostridium difficile infection at a University
Hospital in Japan: Genotypic features of the isolates and clinical
characteristics of the patients. J Infect Chemother. 16:329–333.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Elliott B, Squire MM, Thean S, Chang BJ,
Brazier JS, Rupnik M and Riley TV: New types of toxin A-negative,
toxin B-positive strains among clinical isolates of Clostridium
difficile in Australia. J Med Microbiol. 60:1108–1111. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pruitt RN and Lacy DB: Toward a structural
understanding of Clostridium difficile toxins A and B. Front. Cell
Infect Microbiol. 2:282012.
|
|
25
|
Komatsu M, Kato H, Aihara M, Shimakawa K,
Iwasaki M, Nagasaka Y, Fukuda S, Matsuo S, Arakawa Y, Watanabe M,
et al: High frequency of antibiotic-associated diarrhea due to
toxin A-negative, toxin B-positive Clostridium difficile in a
hospital in Japan and risk factors for infection. Eur J Clin
Microbiol Infect Dis. 22:525–529. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shin BM, Kuak EY, Yoo SJ, Shin WC and Yoo
HM: Emerging toxin A–B+ variant strain of Clostridium difficile
responsible for pseudomembranous colitis at a tertiary care
hospital in Korea. Diagn Microbiol Infect Dis. 60:333–337. 2008.
View Article : Google Scholar
|
|
27
|
Hadji-Ghasemi F, Gharagozlou S, Ghods R,
Roohi A, Khoshnoodi J and Shokri F: Generation and characterization
of a mouse monoclonal antibody with specificity similar to
staphylococcal protein A (SPA). Hybrid Hybridomics. 22:33–39. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hajighasemi F, Khoshnoodi J and Shokri F:
Development of two murine monoclonal antibodies recognizing human
nG1m(a)-like isoallotypic markers. Hybridoma (Larchmt). 27:473–479.
2008. View Article : Google Scholar
|
|
29
|
Hajighasemi F, Gharagozlou S, Ghods R,
Khoshnoodi J and Shokri F: Private idiotypes located on light and
heavy chains of human myeloma proteins characterized by monoclonal
antibodies. Hybridoma (Larchmt). 25:329–335. 2006. View Article : Google Scholar
|
|
30
|
Singh RP, Bandyopadhyay SK, Sreenivasa BP
and Dhar P: Production and characterization of monoclonal
antibodies to peste des petits ruminants (PPR) virus. Vet Res
Commun. 28:623–639. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harlow ED and Lane D: Antibodies: A
Laboratory Manual. Cold Spring Harbor Laboratory; New York: pp.
148–242. 1988
|
|
32
|
Hajighasemi F, Saboor-Yaraghi AA and
Shokri F: Measurement of affinity constant of anti-human IgG
monoclonal antibodies by an Elisa-based method. Iran Immunol.
1:154–161. 2004.
|
|
33
|
Jie Y, Luo HB and Lu DY: Modern
microbiological techology and its application. 1st edition. people
health press; Beijing: pp. 173–183. 1997
|
|
34
|
Curry S: Clostridium difficile. Clin Lab
Med. 30:329–342. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Crobach MJ, Dekkers OM, Wilcox MH and
Kuijper EJ: European Society of Clinical Microbiology and
Infectious Diseases (ESCMID): Data review and recommendations for
diagnosing Clostridium difficile-infection (CDI). Clin Microbiol
Infect. 15:1053–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wilcox MH: Overcoming barriers to
effective recognition and diagnosis of Clostridium difficile
infection. Clin Microbiol Infect. 18(Suppl 6): 13–20. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boyanton BL, Sural P, Loomis CR, Pesta C,
Gonzalez-Krellwitz L, Robinson-Dunn B and Riska P: Loop-mediated
isothermal amplification compared to real-time PCR and enzyme
immunoassay for toxigenic Clostridium difficile detection. J Clin
Microbiol. 50:640–645. 2012. View Article : Google Scholar :
|
|
38
|
Deshpande A, Pasupuleti V, Rolston DD,
Jain A, Deshpande N, Pant C and Hernandez AV: Diagnostic accuracy
of realtime polymerase chain reaction in detection of Clostridium
difficile in the stool samples of patients with suspected
Clostridium difficile infection: A metaanalysis. Clin Infect Dis.
53:e81–e90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Longtin Y, Trottier S, Brochu G,
Paquet-Bolduc B, Garenc C, Loungnarath V, Beaulieu C, Goulet D and
Longtin J: Impact of the type of diagnostic assay on Clostridium
difficile infection and complication rates in a mandatory reporting
program. Clin Infect Dis. 56:67–73. 2013. View Article : Google Scholar
|
|
40
|
Wilkins TD and Lyerly DM: Clostridium
difficile testing: After 20 years, still challenging. J Clin
Microbiol. 41:531–534. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Planche T, Aghaizu A, Holliman R, Riley P,
Poloniecki J, Breathnach A and Krishna S: Diagnosis of Clostridium
difficile infection by toxin detection kits: A systematic review.
Lancet Infect Dis. 8:777–784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cheknis AK, Sambol SP, Davidson DM, Nagaro
KJ, Mancini MC, Hidalgo-Arroyo GA, Brazier JS, Johnson S and
Gerding DN: Distribution of Clostridium difficile strains from a
North American, European and Australian trial of treatment for
Clostridium difficile infections: 2005–2007. Anaerobe. 15:230–233.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Albesa-Jové D, Bertrand T, Carpenter EP,
Swain GV, Lim J, Zhang J, Haire LF, Vasisht N, Braun V, Lange A, et
al: Four distinct structural domains in Clostridium difficile toxin
B visualized using SAXS. J Mol Biol. 396:1260–1270. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
von Eichel-Streiber C, Laufenberg-Feldmann
R, Sartingen S, Schulze J and Sauerborn M: Comparative sequence
analysis of the Clostridium difficile toxins A and B. Mol Gen
Genet. 233:260–268. 1992. View Article : Google Scholar : PubMed/NCBI
|