Introduction
Tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL) is a member of the TNF
superfamily. Four types of death receptors specific for TRAIL have
been found: Death receptor 4 (DR4), DR5 and decoy receptors DCR1
and DCR2. Death receptors mediate TRAIL-induced cell death, whereas
the decoy receptors inhibit death signaling (1). The increased sensitivity of
transformed cells to TRAIL-induced cell death compared with that of
normal cells suggests the potential of TRAIL to treat various
cancer types. Furthermore, TRAIL induces cell death in normal human
epidermal keratinocytes (2). In
addition, human TRAIL can induce tissue injury (as cell death,
inflammation) in human endothelial cells (3).
Hypoxia is a common environmental stressor.
Hypoxia-inducible factor 1 (HIF-1) is a transcriptional factor
composed of α- and β-sub-units that mediates changes in gene
expression at low oxygen concentrations (4). Targets of HIF-1α include cytokines
and growth factors, as well as molecules involved in angiogenesis,
glucose uptake and metabolism, and cell survival (5,6). In
addition, HIF-1α is essential for adaptation of cells to
environmental stress and has an important role in skin development
and wound healing (7,8). HIF-1α is strongly expressed in skin
epithelium. Human and mouse skin is hypoxic, with normal oxygen
levels of 1.5–5.0% (9,10). However, unlike numerous internal
tissue types, human epidermis gains much of its oxygen supply from
the atmosphere and experiences higher oxygen levels than those of
internal tissues (11). Of note,
hypoxia activates autophagic flux and induces clearance of the p62
protein, suggesting a role for p62 in the regulation of hypoxic
HaCaT-cell survival responses (12).
Autophagy is a strictly controlled program in which
parts of the cytoplasm are sequestered in double membrane
autophagosome vesicles, which fuse with lysosomes to form
autolysosomes (13). Lysosomes
degrade protein aggregates, aged proteins and cytoplasmic
organelles (14,15). Oxidative stress induces the
accumulation of high-weight protein aggregates containing the
autophagy marker protein p62 in autophagy-deficient keratinocytes
(16). In addition, blocking
autophagic flux significantly increased inflammatory cytokine
levels and p62 protein expression in primary human keratinocytes
(17). These studies suggested
that blocking autophagic flux (p62 protein accumulation) is
involved in increased inflammation and induced cell death.
In the present study, human HaCaT keratinocytes were
stimulated with hypoxia, and the TRAIL-induced expression of
autophagy markers, including LC3 and p62, as well as apoptosis were
assessed. Furthermore, the effects of the autophagic flux
inhibitors 3-methyladenine (3-MA) and chloroquine (CQ) on autophagy
marker expression and HaCaT cell viability were assessed. The
present study indicated that autophagy inhibitors may increase the
anti-cancer efficiency of TRAIL.
Materials and methods
Cell culture
HaCaT cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) and antibiotics (100
μg/ml gentamycin and 100 μg/ml
penicillin-streptomycin; Gibco; Thermo Fisher Scientific, Inc.). A
hypoxia chamber was used to create the low-oxygen environment
composed of 1% O2, 5% CO2 and 94%
N2.
Protein isolation and western blot
analysis
Total protein was isolated from harvested cells
(using lysis buffer comprising phenylmethanesulfonyl fluoride,
Na3VO4 and complement C), suspended in
phosphate-buffered saline (PBS). The pellets were re-suspended and
sonicated in buffer (Sigma-Aldrich, St. Louis, MO, USA) containing
20 mM Tris, pH 7.5, 1% Triton X-100, 1 mM EDTA, 1 mM ethylene
glycol tetraacetic acid (EGTA) as well as protease and phosphatase
inhibitors (Sigma-Aldrich). The lysates were subjected to western
blot analysis. Total protein from HaCaT cells was isolated by
homogenization in cold radioimmunoprecipitation assay buffer
(Sigma-Aldrich) containing 50 mM Tris, pH 7.5, 150 mM sodium
chloride, 1% NP-40, 0.5% sodium deoxycho-late, 0.1% SDS, 0.1 mM
EDTA and 0.1 mM EGTA, as well as the appropriate protease and
phosphatase inhibitors. The lysates were centrifuged and the
supernatants were subjected to western blot analysis.
Protein concentrations were estimated using a
protein assay (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Nitrocellulose (NC, Merck Millipore,
Milford, MA, USA) membranes were incubated with 5% nonfat milk to
block non-specific binding. The membranes were subsequently exposed
to antibodies that recognized polyclonal LC3 (cat. no. 4108; Cell
Signaling Technology, Inc., Danvers, MA, USA); monoclonal P62 (cat.
no. MABC32; Merck Millipore) and monoclonal HIF-1α (cat. no.
sc-53546; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The
primary antibodies were incubated at a dilution of 1:1,000 in 5%
bovine serum albumin or 5% non-fat milk for 18 h at 4°C. The
membranes were exposed to polyclonal goat anti-rabbit
immunoglobulin (Ig)G (cat. no. ADI-SAB-300-j; Enzo Life Sciences,
Inc., Farmingdale, NY, USA), polyclonal goat anti-mouse IgG (cat.
no. ADI-SAB-100-j; EnzoLife Sciences, Inc.) or anti-goat secondary
antibodies conjugated with horseradish peroxidase (dilution,
1:10,000 for all) in Tris-buffered saline containing Tween-20
(TBS-T) and 5% nonfat milk for 1 h at room temperature.
Electrophoresis was performed using an electrophoresis chamber
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). A Westsave Gold
enhanced chemiluminescence kit (AbFrontier; Young In Frontier Co.,
Ltd., Seoul, South Korea) was used, and the signals were detected
using a chemiluminescence detection system (Fusion Fx7 version
15.18; Vilber Lourmat, Eberhardzell, Germany) and exposed to X-ray
film.
Crystal violet assay
Cell viability was determined by crystal violet
staining (Sigma-Aldrich), as described previously (18). Briefly, HaCaT cells were
pre-incubated under hypoxic conditions (1% O2 for 24 h)
and exposed to 100–400 ng/ml TRAIL (AbFrontier; Young In Frontier
Co., Ltd.) for 6 h. The cells were pre-treated with autophagy
inhibitors [200 μM 3-MA (Sigma-Aldrich) or 50 μM CQ
(Sigma-Aldrich)] for 3 h and exposed to 200 ng/ml TRAIL for 6 h
under hypoxic (1% O2 for 24 h) or normoxic conditions.
Cell viability was calculated based on the relative dye intensity
compared with that of the controls.
Lactate dehydrogenase (LDH) assay
Cytotoxicity was assessed by the LDH assay using the
supernatant and a LDH Cytotoxicity Detection kit (Takara Bio Inc.,
Tokyo, Japan) according to the manufacturer's instructions. LDH
activity was determined by measuring the absorbance at a wavelength
of 490 nm using a SpectraMax M Series spectrophotometer.
Statistical analysis
Values are expressed as the mean ± standard error.
Results of different treatments and time courses were analyzed
using one-way analysis of variance. Comparisons between two groups
were analyzed by two-tailed Student's t-test, analysis of
variance and Duncan's multiple range test using the SAS statistical
package 9.1 (SAS Institute, Cary, NC, USA)
Results
Hypoxia inhibits TRAIL-induced apoptosis
of HaCaT cells
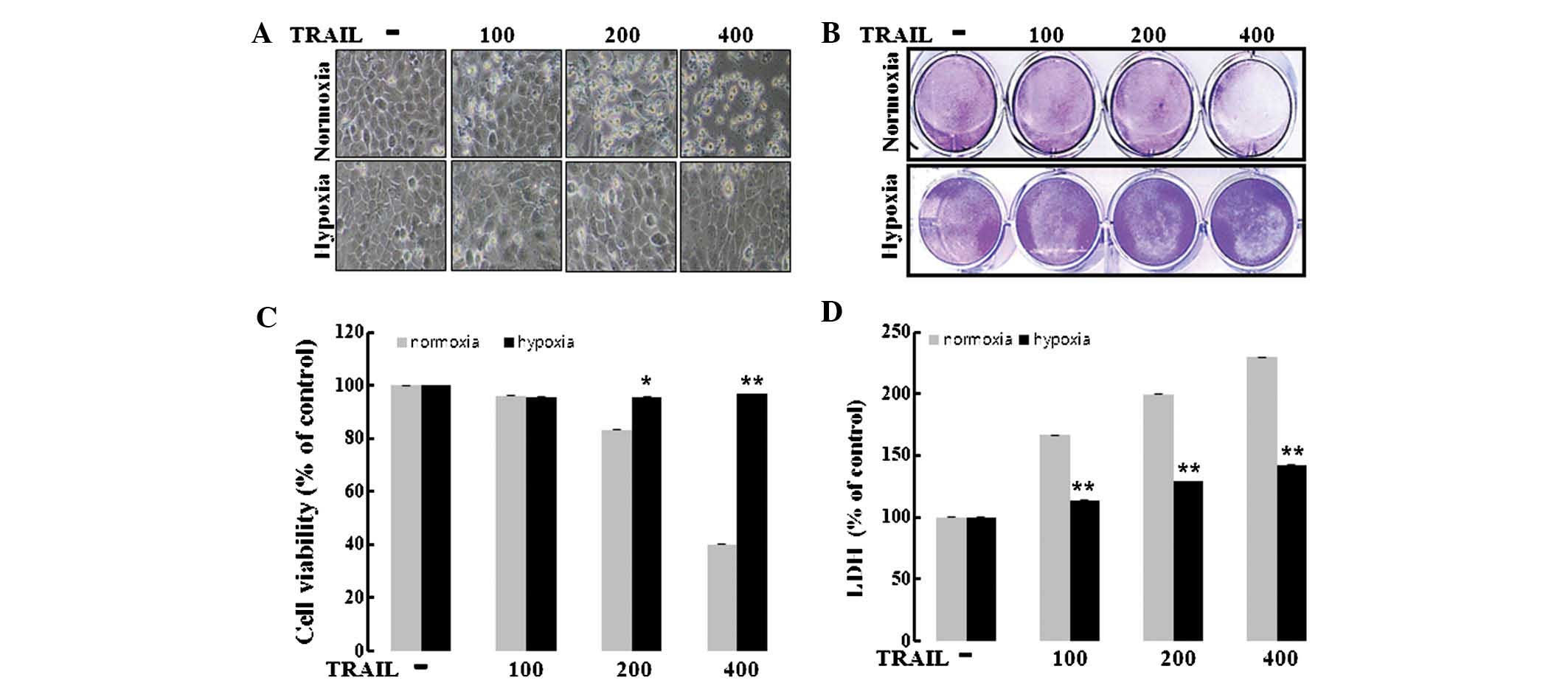
HaCaT cells were cultured under hypoxic or normoxic
conditions and treated with TRAIL (100–400 ng/ml). Morphological
examination of the cell population by microscopy (Eclipse TS100;
Nikon Corporation, Tokyo, Japan) indicated decreased TRAIL-induced
apoptosis under hypoxia as compared with normoxia (Fig. 1A). The influence of hypoxia on
TRAIL-induced apoptosis in HaCaT cells was then quantified by
crystal violet staining (Fig. 1B).
HaCaT cells were responsive to TRAIL treatment (10–60% reduction in
cell viability) under normoxic conditions, whereas under hypoxic
conditions, TRAIL only had a minor effect (5% reduction in cell
viability) on cell viability (Fig.
1C), indicating that hypoxia prevented TRAIL-induced
apop-tosis. Consistent with these results, the LDH assay also
showed that hypoxia prevented TRAIL-induced apoptosis (Fig. 1D). These results confirmed that
hypoxia prevented TRAIL-mediated apoptosis.
HIF-1α induces the expression of
autophagy markers in hypoxic HaCaT cells
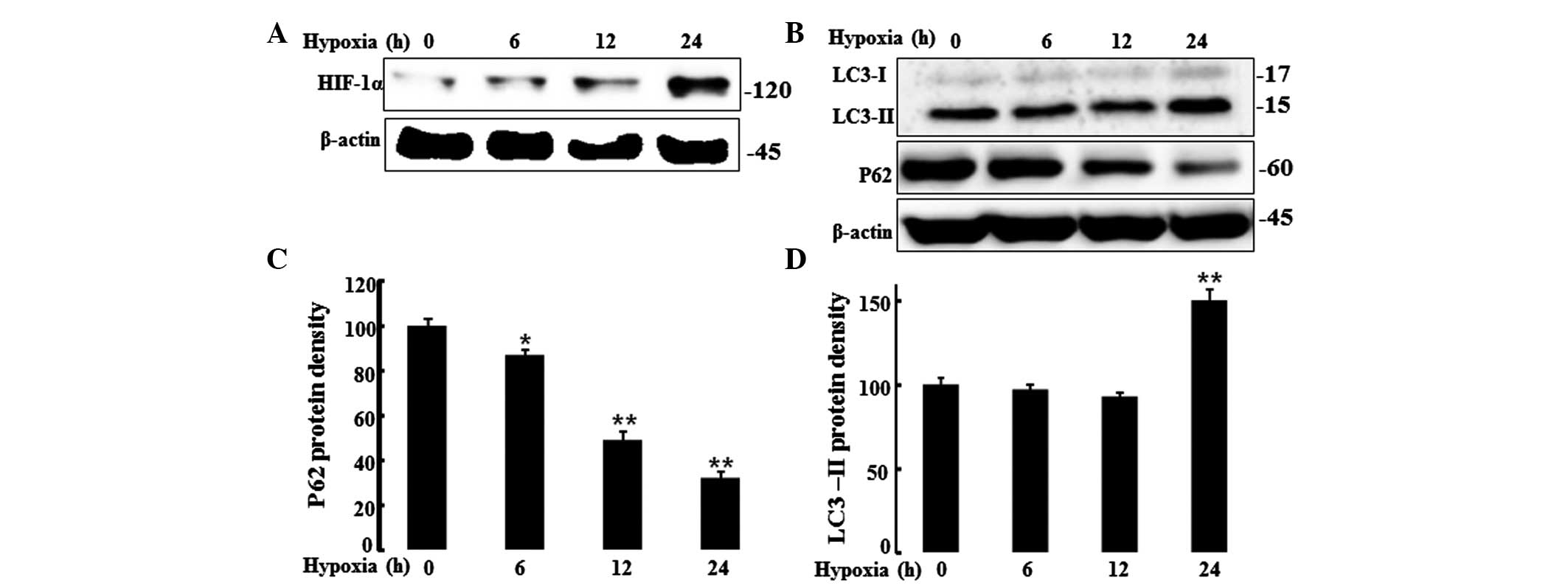
HIF-1α is a transcription factor and a major
regulator of cell adaptation to low oxygen levels. The present
study evaluated HIF-1α protein levels in HaCaT cells under hypoxic
and normoxic conditions by western blot analysis. Cells were
incubated under hypoxic conditions (1% O2; 0, 6, 12 or
24 h), and a western blot analysis was performed to determine
HIF-1α protein levels (Fig. 2A).
Cells displayed increased HIF-1α protein levels under the hypoxic
conditions but not under normoxia. It is known that mild hypoxia
activates autophagy in keratinocytes and reduces p62 protein levels
(12). Autophagy controls cell
survival, growth and cellular homeostasis as well as cellular
defense. Thus, the present study assessed the autophagy marker, LC3
and the protein, P62 that is cleared by autophagic flux, by western
blot analysis (Fig. 2B–D).
Decreased p62 protein levels were observed under hypoxic conditions
as compared to those under normoxic conditions. By contrast, the
autophagic flux marker, LC3-II was increased after 24 h of hypoxia.
These results indicated that hypoxia increased autophagic flux.
Autophagic flux inhibitors induce
apoptosis in hypoxic keratinocytes
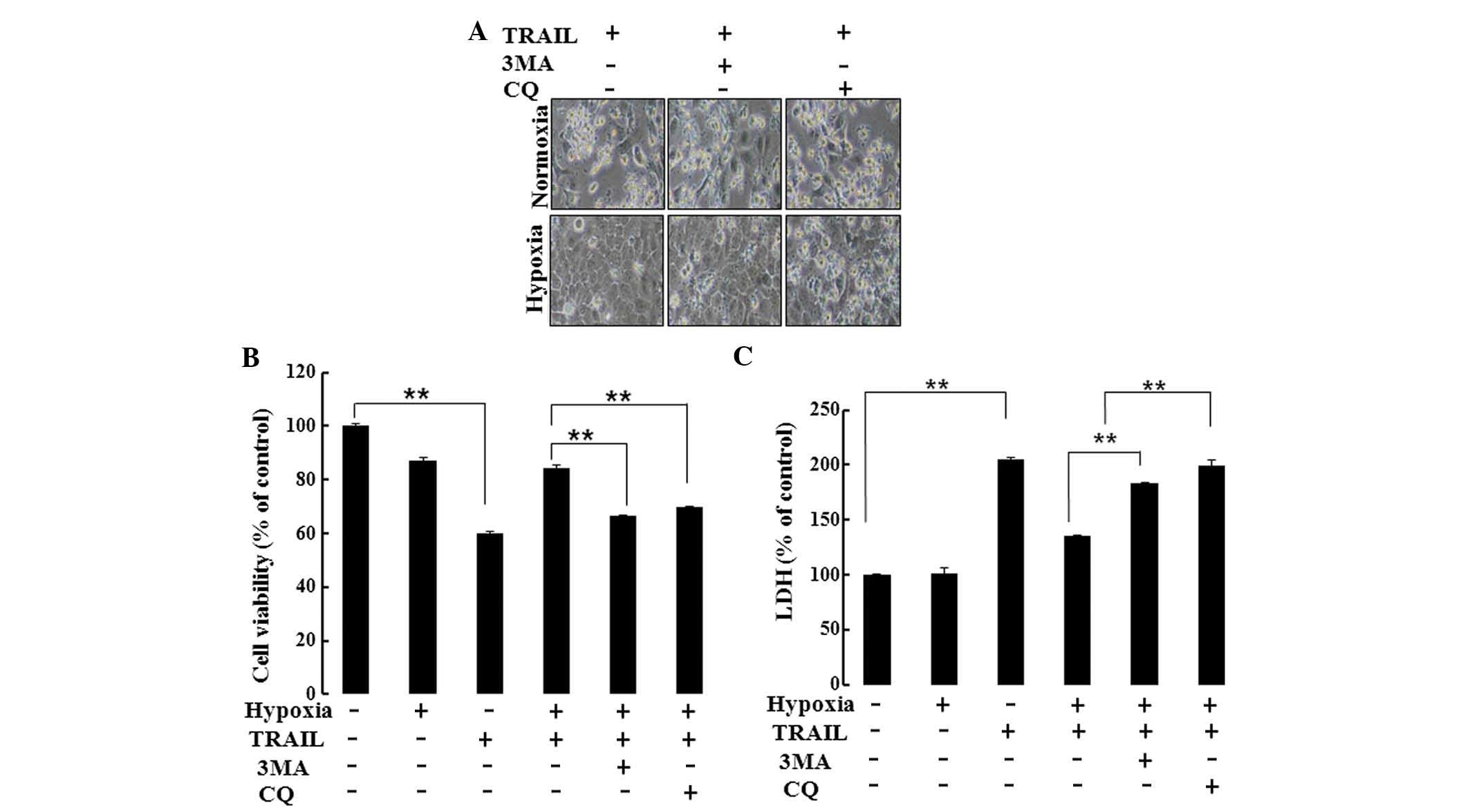
Autophagy inhibitors were utilized to inhibit
autophagic flux in order to examine the protective role of
autophagy in TRAIL-induced cell death. Hypoxia prevented
TRAIL-induced apoptotic death in cells not treated with autophagy
inhibitors. By contrast, treatment with the autophagy inhibitors
3-MA and CQ blocked hypoxic inhibition of TRAIL-induced apoptosis
(Fig. 3A). Cell viability and LDH
assays confirmed that hypoxia-mediated induction of autophagy
protected hypoxic HaCaT cells from TRAIL-induced apoptosis
(Fig. 3B and C). These results
demonstrate that TRAIL-induced apoptosis was blocked by autophagy
inhibitors.
Autophagic flux is blocked by autophagy
inhibitors
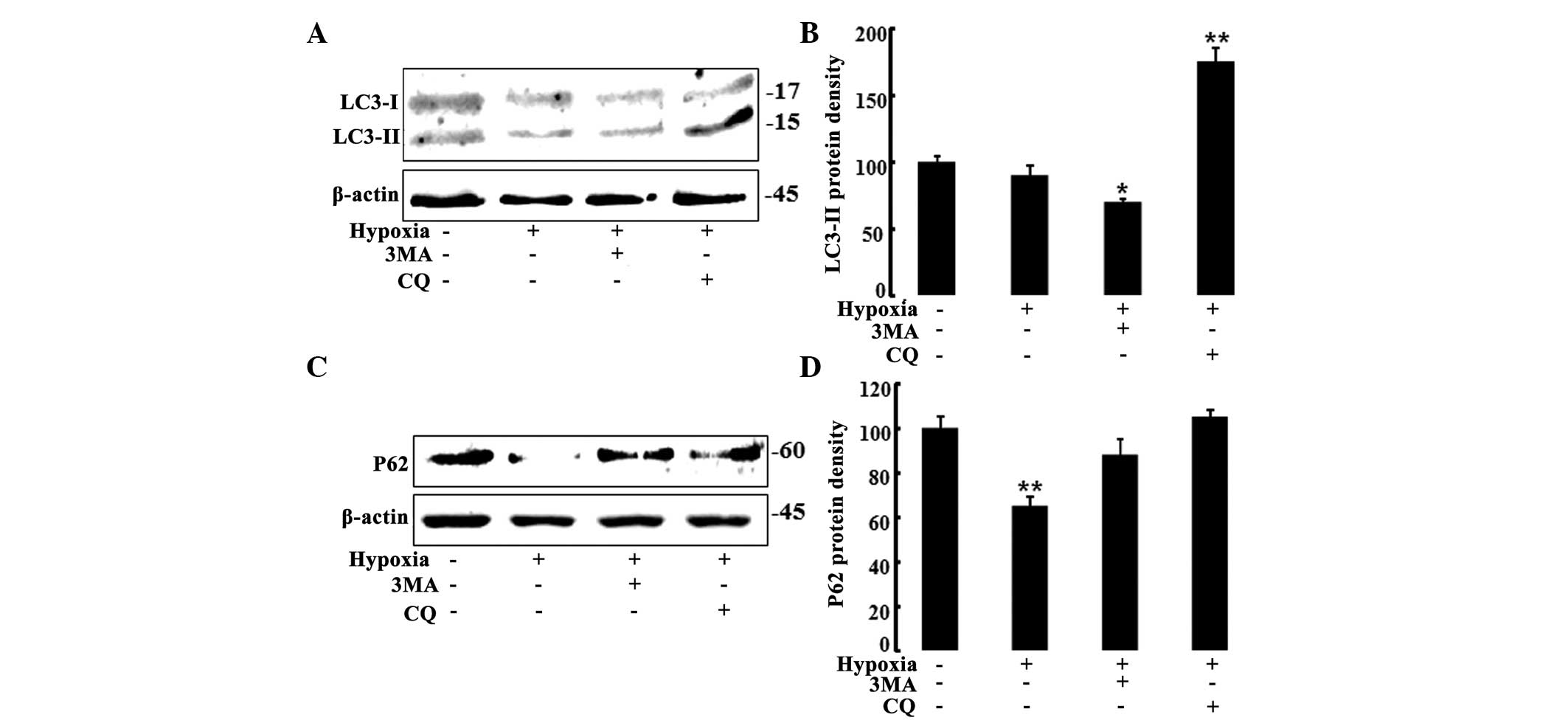
To confirm the inhibition of autophagic flux by the
autophagy inhibitors, expression of the autophagy marker, LC3 and
the levels of p62 protein were assessed by western blot analysis.
Treatment with 3-MA decreased the levels of LC3 in hypoxic HaCaT
cells, confirming the inhibition of autophagy; however, treatment
with CQ increased the levels of LC3-II (Fig. 4A and B). Autophagy
inhibitor-treated cells demonstrated increased p62 protein levels
(Fig. 4C and D). As 3-MA inhibits
conversion of LC3-I to LC3-II and CQ blocks fusion of
autophagolysosomes, treatment with 3-MA decreased the LC3-II levels
whereas CQ increased the LC3-II levels. These results confirmed
that autophagic flux was inhibited by the autophagy inhibitors,
3-MA and CQ.
Discussion
The main goal of the present study was to
demonstrate the role autophagy has in the inhibition of
TRAIL-induced apoptosis by hypoxia. The results suggested a
therapeutic potential for autophagic flux inhibitors in
chemotherapeutic intervention strategies that involve the use of
TRAIL for skin cancer.
TRAIL is a member of the TNF superfamily that
induces cancer cell death and normal human epidermal keratinocyte
death by death receptors, including DR4 and DR5 (2). Death receptors mediate TRAIL-induced
apoptosis, whereas the decoy receptors inhibit apoptosis induced by
TRAIL (1). TRAIL induces
cornification in normal human keratinocytes, which is a specific
form of programmed cell death (19). Early studies discovered that TRAIL
induces tissue injury (including apoptosis and inflammation) in
human endothelial cells and may influence tumor therapies using
TRAIL (2,3,20).
However, the roles of TRAIL treatment in human epidermal
keratinocytes are not well understood. The results of the present
study showed that TRAIL-treated HaCaT cells had a 30–60% reduced
viability under normoxia, whereas cell viability was not markedly
affected by TRAIL under hypoxic conditions (5% reduction).
Changes in oxygen levels are tightly linked to
metabolism, apoptosis, the cell cycle and cell signaling. HIF-1 is
a transcriptional factor with α- and β-sub-units that mediates
changes in gene expression under hypoxic conditions and is
essential for skin development and wound healing (7,8).
Furthermore, a recent study indicated that p62 protein expression,
an autophagic flux marker, decreases under hypoxic conditions
(12). The present study showed
that p62 expression decreased and LC3 expression increased under
hypoxic conditions. These results suggested that the expression of
HIF-1 induced autophagic flux in human epidermal keratinocytes and
inhibited TRAIL-induced apoptosis.
Furthermore, the autophagy inhibitors 3-MA and CQ
were used under hypoxic conditions to confirm the inhibition of
cell death by autophagic flux. HaCaT cells showed a reduced
resistance to the apoptotic effect of TRAIL during treatment with
the autophagic inhibitors under hypoxic conditions. These results
indicated that autophagic flux suppressed TRAIL-induced
apoptosis.
TRAIL treatment has been shown to induce
autophagy-dependent cell death in a variety of cancer cells
(21,22). The p62 protein, a marker of
autophagy, is important in the degradation of polyu-biquitinated
proteins via the autophagy pathway (23). Recently, high autophagic flux with
clearance of the p62 protein was detected in TRAIL-resistant cells;
however, TRAIL-sensitive cells exhibited low autophagic flux and
accumulation of the p62 protein (24). Data from the present study
demonstrated that hypoxia inhibits TRAIL-induced cell death (via
observation of the reduction of p62 protein), however treatment
with TRAIL plus an autophagy inhibitor increased TRAIL-induced
apoptosis, which was demonstrated by an accumulation of the p62
protein (Figs. 3A and 4D).
In conclusion, the results of the present study
suggested that the autophagic flux induced by hypoxia inhibits
TRAIL-induced apoptosis. To the best of our knowledge, the present
study was the first to identify that TRAIL-induced apoptosis was
inhibited by hypoxia in keratinocytes, but that autophagy
inhibitors were able to restore the susceptibility of keratinocytes
to TRAIL-induced apoptosis under hypoxic conditions. These findings
provided insight into the molecular mechanisms of keratinocyte
apoptosis and the beneficial effects of TRAIL in skin cancer
therapy. It is recommended that autophagy inhibitors are used to
enhance the efficiency of TRAIL in the treatment of skin
cancer.
Acknowledgments
The present study was supported by the National
Research Foundation of the Korea Grant funded by the Korean
Government (no. 2013R1A2A2A01009614).
References
|
1
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu NL, Lee TA, Tsai TL and Lin WW:
TRAIL-induced keratinocyte differentiation requires caspase
activation and p63 expression. J Invest Dermatol. 131:874–883.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li JH, Kirkiles-Smith NC, McNiff JM and
Pober JS: TRAIL induces apoptosis and inflammatory gene expression
in human endothelial cells. J Immunol. 171:1526–1533. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Semenza GL: Hypoxia-inducible factor 1:
control of oxygen homeostasis in health and disease. Pediatr Res.
49:614–617. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu Y, Kirito K, Yoshida K, Mitsumori T,
Nakajima K, Nozaki Y, Hamanaka S, Nagashima T, Kunitama M, Sakoe K
and Komatsu N: Inhibition of hypoxia-inducible factor-1 function
enhances the sensitivity of multiple myeloma cells to melphalan.
Mol Cancer Ther. 8:2329–2338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaelin WG Jr: Treatment of kidney cancer:
insights provided by the VHL tumor-suppressor protein. Cancer.
115(Suppl 10): S2262–S2272. 2009. View Article : Google Scholar
|
|
7
|
Tandara AA and Mustoe TA: Oxygen in wound
healing-more than a nutrient. World J Surg. 28:294–300. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
LaVan FB and Hunt TK: Oxygen and wound
healing. Clin Plast Surg. 17:463–472. 1990.PubMed/NCBI
|
|
9
|
Evans NT and Naylor PF: The systemic
oxygen supply to the surface of human skin. Respir Physiol.
3:21–37. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stewart FA, Denekamp J and Randhawa VS:
Skin sensitization by misonidazole: a demonstration of uniform mild
hypoxia. Br J Cancer. 45:869–877. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stucker M, Struk A, Altmeyer P, Herde M,
Baumgärtl H and Lübbers DW: The cutaneous uptake of atmospheric
oxygen contributes significantly to the oxygen supply of human
dermis and epidermis. J Physiol. 538:985–994. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pursiheimo JP, Rantanen K, Heikkinen PT,
Johansen T and Jaakkola PM: Hypoxia-activated autophagy accelerates
degradation of SQSTM1/p62. Oncogene. 28:334–344. 2009. View Article : Google Scholar
|
|
13
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delgado MA, Elmaoued RA, Davis AS, Kyei G
and Deretic V: Toll-like receptors control autophagy. EMBO J.
27:1110–1121. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hussey S, Travassos LH and Jones NL:
Autophagy as an emerging dimension to adaptive and innate immunity.
Semin Immunol. 21:233–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao Y, Zhang CF, Rossiter H, Eckhart L,
König U, Karner S, Mildner M, Bochkov VN, Tschachler E and Gruber
F: Autophagy is induced by UVA and promotes removal of oxidized
phospho-lipids and protein aggregates in epidermal keratinocytes. J
Invest Dermatol. 133:1629–1637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee HM, Shin DM, Yuk JM, Shi G, Choi DK,
Lee SH, Huang SM, Kim JM, Kim CD, Lee JH and Jo EK: Autophagy
negatively regulates keratinocyte inflammatory responses via
scaffolding protein p62/SQSTM1. J Immunol. 186:1248–1258. 2011.
View Article : Google Scholar
|
|
18
|
Seo JS, Seol JW, Moon MH, Jeong JK, Lee YJ
and Park SY: Hypoxia protects neuronal cells from human prion
protein fragment-induced apoptosis. J Neurochem. 112:715–722. 2010.
View Article : Google Scholar
|
|
19
|
Candi E, Rufini A, Terrinoni A, Dinsdale
D, Ranalli M, Paradisi A, De Laurenzi V, Spagnoli LG, Catani MV,
Ramadan S, et al: Differential roles of p63 isoforms in epidermal
development: selective genetic complementation in p63 null mice.
Cell Death Differ. 13:1037–1047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eberle J, Fecker LF, Forschner T, Ulrich
C, Röwert-Huber J and Stockfleth E: Apoptosis pathways as promising
targets for skin cancer therapy. Br J Dermatol. 156(Suppl 3):
18–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mora R, Abschuetz A, Kees T, Dokic I,
Joschko N, Kleber S, Geibig R, Mosconi E, Zentgraf H,
Martin-Villalba A and Régnier-Vigouroux A: TNF-alpha- and
TRAIL-resistant glioma cells undergo autophagy-dependent cell death
induced by activated microglia. Glia. 57:561–581. 2009. View Article : Google Scholar
|
|
22
|
Herrero-Martín G, Høyer-Hansen M,
García-García C, Fumarola C, Farkas T, López-Rivas A and Jäättelä
M: TAK1 activates AMPK-dependent cytoprotective autophagy in
TRAIL-treated epithelial cells. EMBO J. 28:677–685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Itakura E and Mizushima N: p62 Targeting
to the autophagosome formation site requires self-oligomerization
but not LC3 binding. J Cell Biol. 192:17–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh K, Sharma A, Mir MC, Drazba JA,
Heston WD, Magi-Galluzzi C, Hansel D, Rubin BP, Klein EA and
Almasan A: Autophagic flux determines cell death and survival in
response to Apo2L/TRAIL (dulanermin). Mol Cancer. 13:702014.
View Article : Google Scholar : PubMed/NCBI
|