Introduction
Indoleamine 2,3-dioxygenase (IDO) degrades
L-tryptophan via the kynurenine pathway. L-tryptophan depletion
activates general control non-derepressible (GCN)2 kinase, which
phosphorylates eukaryotic initiation factor 2α, altering the
translation program of T-cells and leading to the inhibition of
cellular proliferation and anergy (1). An additional pathway able to sense
amino acid deprivation is the mammalian target of rapamycin complex
(mTORC)1 pathway (2).
Under inflammatory conditions IDO is upregulated in
antigen presenting cells (APCs), including monocytes, macrophages
and dendritic cells, and restricts the T-cell response (3,4).
Expression of IDO in APCs reduces graft rejection (5–7) and
ameliorates autoimmune diseases (8–10).
In addition, IDO is expressed in certain non-immune cells.
Expression of IDO in a paternally derived placental trophoblast is
required for a successful semi-allogenic pregnancy (11,12),
while expression in tumor cells contributes to their escape from
immunosurveillance (13). Patients
on hemodialysis are characterized by impaired adaptive immunity and
exhibit increased expression of IDO, further enhanced in those who
are non-responders to vaccination against hepatitis B virus
(14). In these patients, plasma
IDO levels are negatively associated with the T-cell count
(15). Therefore, IDO is an
enzyme, which serves an important role in immune system homeostasis
and clarification of its mechanism of action may contribute to an
improved understanding of immune system physiology, potentially
leading to novel means of pharmaceutical intervention.
In previous studies, IDO-induced L-tryptophan
depletion was demonstrated to activate the GCN2 kinase, whilst
mTORC1 was unaffected in human alloreactive T-cells. In addition,
in parallel with a reduction in T-cell proliferation, IDO reduced
glucose consumption and lactate production by T-cells (16,17).
This indicates that IDO suppresses aerobic glycolysis in activated
T-cells. The majority of rapidly proliferating cancer cells are
characterized by an increased ratio of cytoplasmic glycolysis to
mitochondrial glucose oxidation, a phenomenon first described by
Otto Warburg, and hence termed the Warburg effect or aerobic
glycolysis (18). Rapidly
proliferating activated T-cells reprogram their metabolic pathways
from pyruvate oxidation via the Krebs cycle to the glycolytic,
pentose-phosphate and glutaminolytic pathways in order to fulfill
the bioenergetic and biosynthetic demands of proliferation
(19). Notably, IDO reveals no
effect on the levels of activated pyruvate dehydrogenase or its
inactive phosphorylated-Ser393 form, which controls the influx of
pyruvate into the Krebs cycle (16).
A previous study demonstrated that in alloreactive
T-cells, IDO increases the levels of p53, which contributes to the
suppression of proliferation and aerobic glycolysis. The
IDO-induced increase in p53 upregulated the expression levels of
cyclin-dependent kinase inhibitor p21. IDO and p53 both reduced
glucose consumption. The IDO-induced increase in p53 levels reduced
the expression of glucose transporter-1, and increased the
expression of TP53-induced glycolysis and apoptosis regulator,
inhibiting glucose influx into T-cells and reducing glycolysis.
However, IDO downregulated lactate dehydrogenase-A (LDH-A) and
glutaminase (GLS)2, which are key enzymes in aerobic glycolysis and
glutaminolysis, respectively, in a p53-independent manner. In
addition, IDO and not p53 reduced lactate production (20).
As T-cell activation increases the transcription
factor c-Myc, which subsequently upregulates LDH-A and GLS2
(19), in the present study the
effect of IDO or direct GCN2 kinase activation on the expression
levels of c-Myc, LDH-A and GLS2 in T-cells was investigated. In
addition, the effect of IDO or direct GCN2 kinase activation on the
expression of T-cell receptor (TCR)-complex ζ-chain was
investigated, as downregulation of this key molecule has been
previously demonstrated to reduce the expression of c-Myc and
T-cell proliferation (21).
Materials and methods
Subjects
Blood samples were collected from 10 non-related
healthy volunteers (5 males and 5 females; age, 27–49 years).
Informed consent was obtained from each individual enrolled in the
study and the study protocol was approved by the by the ethics
committee of the University Hospital of Larissa, Medical School,
University of Thessaly (Larissa, Greece).
Peripheral blood mononuclear cell (PBMC),
and T-cell isolation and culture
PBMCs were isolated from whole blood by
Ficoll-Hypaque density gradient centrifugation (Histopaque 1077;
Sigma-Aldrich, St. Louis, MO, USA) and quantified using an optical
microscope (Axiovert 40 C; Carl Zeiss AG, Oberkochen, Germany) and
a Neubauer chamber (Paul Marienfeld GmbH, Lauda-Königshofen,
Germany). Cell viability was assessed by trypan blue staining
(Sigma-Aldrich).
PBMCs were resuspended in RPMI-1640 medium
containing L-glutamine and 10 mM 4-(2-hydroxyethyl)-1-pip
erazineethanesulfonic acid, and supplemented with 10% fetal bovine
serum and antibiotic-antimycotic solution (dilution, 1:100) (all
from Sigma-Aldrich).
For the experiments with the GCN2 kinase activator,
tryptophanol (TRP; Sigma-Aldrich), T-cells were isolated from PBMCs
using a Pan T-cell Isolation kit (Miltenyi Biotec GmbH, Bergisch
Gladbach, Germany). Non-T-cells were indirectly magnetically
labeled and were depleted from the PBMC samples. Isolated T-cells
were cultured in the same medium as the PBMCs. All cultures were
incubated at 37°C in a humidified atmosphere containing 5%
CO2.
Assessment of cell proliferation in
two-way mixed lymphocyte reactions
Two-way mixed lymphocyte reactions (MLRs) were
performed in 96-well plates for 7 days in the presence or absence
of 100 µΜ IDO inhibitor, 1-methyl-DL-tryptophan (1-MT;
Sigma-Aldrich) or 30 µM p53 inhibitor, pifithrin-α (PFT;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The
concentrations of 1-MT and PFT were selected, according to previous
experiments that demonstrated efficacy without toxicity (1,16,20,22).
Pifithrin-α was refreshed in the cell cultures at day 4. A total of
5×104 PBMCs from each member of the MLR couple were
used, with a total of 1×105 PBMCs in each well. Cultures
of resting PMBCs with a population of 1×105 cells/well
were used as the control.
At the end of the 7-day period, cell proliferation
was assessed using a cell proliferation enzyme-linked immunosorbent
assay (ELISA; Roche Diagnostics, Basel, Switzerland) using
bromodeoxyuridine labeling and immunoenzymatic detection according
to the manufacturer's protocol. The proliferation index was
calculated as the ratio of the optical density (OD) derived from
each MLR to the mean of the ODs derived from the control resting
PBMC cultures of the two subjects that constituted the specific
MLR. The following formula was used: Proliferative index = OD of
the MLR from subjects A and B/{[(OD of resting PBMCs from subject A
+ OD of resting PBMCs from subject B)]/2}. A total of 10 MLRs were
performed. All experiments were performed in triplicate, and the
results presented are the mean of the three measurements.
Isolation of T-cells from MLRs and
assessment of ζ-chain, c-Myc, p53, p21, LDH-A and GLS2 levels
A total of 10 MLRs were performed in 12-well plates
for 7 days. The number of PBMCs from each member of the MLR couple
was 5×105, with a total of 1×106 PBMCs/well.
The expression levels of ζ-chain, c-Myc, p53, p21, LDH-A and GLS2
were assessed in the presence or absence of 100 µΜ 1-MT or
30 µΜ PFT. PFT was refreshed in the cell cultures on day 4.
Following the 7 day culture period, the T-cells were isolated by
negative selection using the Pan T-cell Isolation kit (Miltenyi
Biotec GmbH).
Isolated T-cells were counted using an optical
microscope and a Neubauer chamber, and cell viability was
determined by trypan blue staining (Sigma-Aldrich). Equal numbers
of T-cells from each MLR were lyzed using the T-PER tissue protein
extraction reagent (Thermo Fisher Scientific, Inc., Waltham, MA,
USA), supplemented with protease and phosphatase inhibitors
(Sigma-Aldrich and Roche Diagnostics, respectively). The protein
was quantified using a Bradford assay (Sigma-Aldrich) and western
blotting was performed. Equal quantities of protein extracts (50
µg) from each sample were loaded for electrophoresis in
precast 4–12% gradient bis-tris polyacrylamide gels (Invitrogen;
Thermo Fisher Scientific, Inc.). Subsequently, the proteins were
transferred onto polyvinylidene difluoride (PVDF) membranes
(Invitrogen; Thermo Fisher Scientific, Inc.). Blots were blocked in
5% w/v non-fat dry milk (Regilait, Saint Martin Belle Roche,
France) diluted in 1X Tris-buffered saline (Thermo Fisher
Scientific, Inc.) supplemented with 0.1% Tween-20 (Sigma-Aldrich).
The blots were then incubated with the primary antibodies at 4°C
for 16 h, followed by secondary antibody incubation (anti-rabbit
immunoglobulin G, horseradish peroxidase-linked antibody; Cell
Signaling Technology, Inc., Danvers, MA, USA) for 30 min at room
temperature. A pre-stained protein ladder (Invitrogen; Thermo
Fisher Scientific, Inc.) was used as a marker. The bands were
visualized by enhanced chemiluminescent detection using the
LumiSensor Plus Chemiluminescent Horseradish Peroxidase Substrate
kit (GenScript, Piscataway, NJ, USA) and analysis was performed
using Image J software v 1.49 (National Institute of Health,
Bethesda, MD, USA). For the reprobing of PVDF blots, the previous
primary and secondary antibodies were removed using Restore Western
Blot Stripping Buffer (Thermo Fisher Scientific, Inc.), according
to the manufacturer's instructions. The PVDF membrane was then
reused and western blotting resumed as described, using a different
primary antibody.
The following primary antibodies, all raised in
rabbits with specificity for humans, were used for western
blotting: Anti-ζ-chain (cat. no. sc-20919; dilution, 1/100; Santa
Cruz Biotechnology, Inc.), anti-c-Myc (cat. no. 5605; dilution,
1/500; Cell Signaling Technology, Inc.), anti-p53 (cat. no. 9282;
dilution, 1/500; Cell Signaling Technology, Inc.), anti-p21 (cat.
no. 2947; dilution, 1/500; Cell Signaling Technology, Inc.),
anti-LDH-A (cat no. 2012; dilution, 1/1,000; Cell Signaling
Technology, Inc.), anti-GLS2 (cat no. AP17426PU-N; dilution, 1/100;
Acris Antibodies, San Diego, CA, USA) and anti-β-actin (cat no.
4967; dilution, 1/2,500; Cell Signaling Technology, Inc.).
Stimulation of isolated T-cells with
TRP
T-cells were isolated from PBMCs using the Pan
T-cell Isolation kit (Miltenyi Biotec GmbH). Isolated T-cells were
counted using an optical microscope and a Neubauer chamber. Cell
viability was assessed by trypan blue staining (Sigma-Aldrich).
T-cells were cultured in the presence or absence of
anti-CD2, anti-CD3 and anti-CD28 conjugated beads, using the T-Cell
activation/expansion kit (Miltenyi Biotec GmbH) at a bead to cell
ratio of 1:2. Stimulated T-cells were cultured in the presence or
absence of TRP (0.25 mM). The concentration of TRP was selected
according to previous studies that demonstrated efficacy without
toxicity (1,20).
Investigation of the effect of TRP on the
proliferation of T-cells
T-cell proliferation was assessed using a Cell
Proliferation ELISA (Roche Diagnostics). Resting, stimulated or
stimulated in the presence of 0.25 mM TRP T-cells were cultured in
96-well plates (1×105cells/well) for 72 h. All
experiments were performed in T-cells derived from the blood of 10
individuals in triplicate, and the results are presented as the
mean of the three measurements.
Assessment of the effect of TRP on
ζ-chain, c-Myc, p53, p21, LDH-A and GLS2 levels in T-cells
The proteins were extracted from resting, stimulated
or stimulated TRP-treated T-cells cultured in 12-well plates
(1×106 cells/well) for 12 h in order to measure the
expression levels of ζ-chain, c-Myc, p53, p21, LDH-A and GLS2 by
western blotting. The primary antibodies were anti-ζ-chain (Santa
Cruz Biotechnology, Inc.), anti-c-Myc (Cell Signaling Technology,
Inc.), anti-p53 (Cell Signaling Technology, Inc.), anti-p21 (Cell
Signaling Technology, Inc.), anti-LDH-A (Cell Signaling Technology,
Inc.), anti-GLS2 (Acris Antibodies) and anti-β-actin (Cell
Signaling Technology, Inc.). Experiments were performed in T-cells
derived from the blood of 10 individuals.
Statistical analysis
Normality of the evaluated variables was assessed
and confirmed by one-sample Kolmogorov-Smirnov test. For comparison
of means, the sphericity assumption was evaluated by Mauchly's test
and if it failed, degrees of freedom were corrected using
Greenhouse-Geisser or Huynh-Feldt estimates of sphericity.
Comparison of means was performed by one-way repeated-measures
analysis of variance followed by Bonferroni's correction test. The
values were normalized against the control group, and are presented
as the mean ± standard deviation. SPSS 13.0 for Windows (SPSS Inc.,
Chicago, IL, USA) was used for all statistical analyses. P<0.05
was considered to indicate a statistically significant
difference.
Results
IDO and p53 reduce T-cell
proliferation
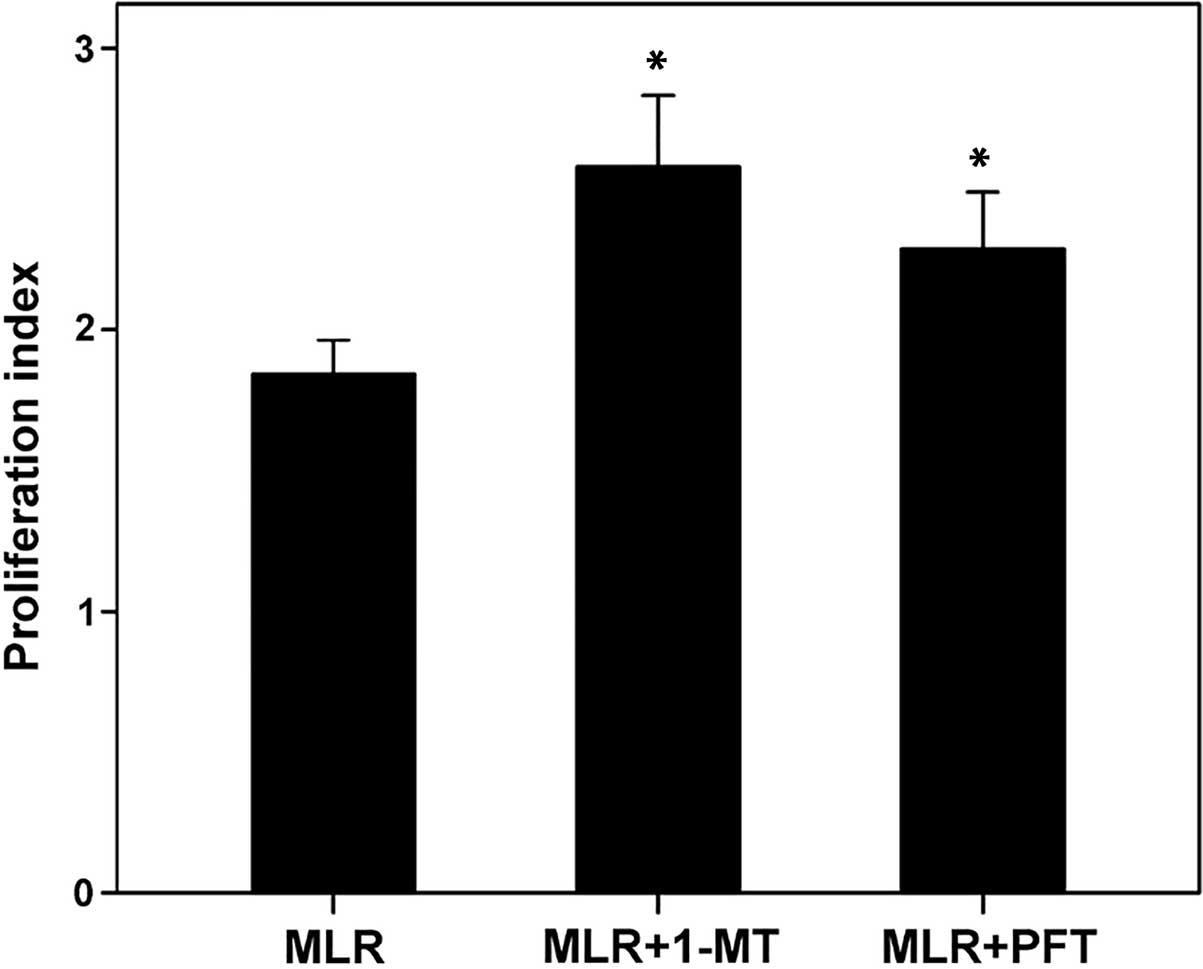
In MLRs, the inhibition of IDO by 1-MT enhanced the
T-cell proliferation index from 1.84±0.15 to 2.57±0.30
(P<0.001). In addition, p53 inhibition by PFT enhanced the
T-cell proliferation index to 2.29±0.24 (P<0.001). The
proliferation index was not significantly different between the
1-MT or PFT treatment groups (P=0.55; Fig. 1).
IDO induces the expression levels of p53
and p21, however IDO alone reduces the expression levels of
ζ-chain, c-Myc, LDH-A and GLS2
In MLRs, IDO inhibition reduced the expression
levels of p53 and p21 in T-cells. In T-cells derived from
1-MT-treated MLRs, the expression levels of p53 were reduced to
0.70±0.15, compared with the level in untreated MLRs (P=0.004;
Figs. 2 and 3). Similarly, following treatment with
1-MT, the expression of p21 was significantly reduced compared with
the untreated MLRs (0.41±0.12; P<0.001). In addition, PFT
reduced the expression levels of p53 and p21 to 0.54±0.15 (P=0.001)
and 0.49±0.16 (P<0.001), respectively (Figs. 2 and 3).
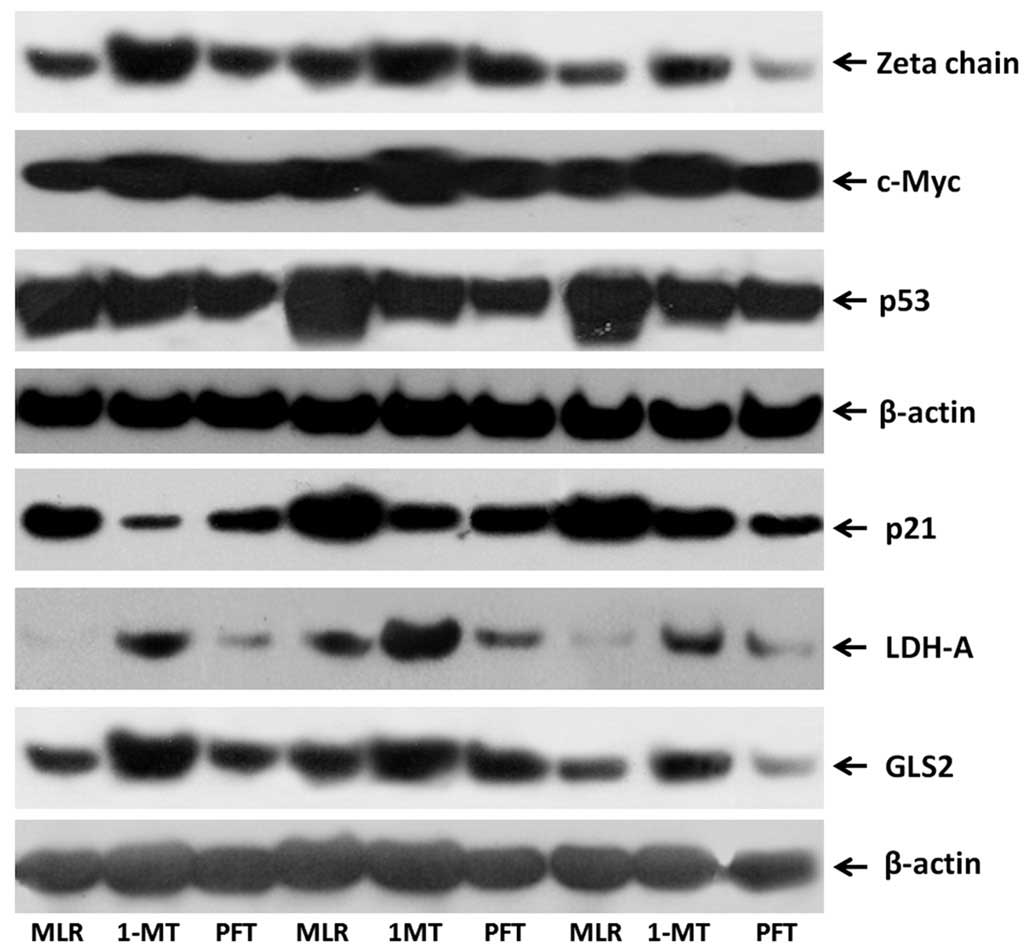 | Figure 2Western blot images presenting the
effect of 1-MT or PFT treatment of MLRs on the expression levels of
TCR-complex ζ-chain, c-Myc, p53, p21, LDH-A and GLS2 in
alloreactive T-cells. Ten MLRs were conducted in the presence or
absence of the indoleamine 2,3-dioxygenase inhibitor 1-MT and the
p53 inhibitor PFT, following which the T-cells were isolated and
western blotting conducted. The western blotting lanes correspond
to three representative experiments of the ten conducted. 1-MT,
1-methyl-DL-tryptophan; PFT, pifithrin-α; MLRs, mixed lymphocyte
reactions; TCR, T-cell receptor; LDH-A, lactose dehydrogenase A;
GLS2, glutaminase 2. |
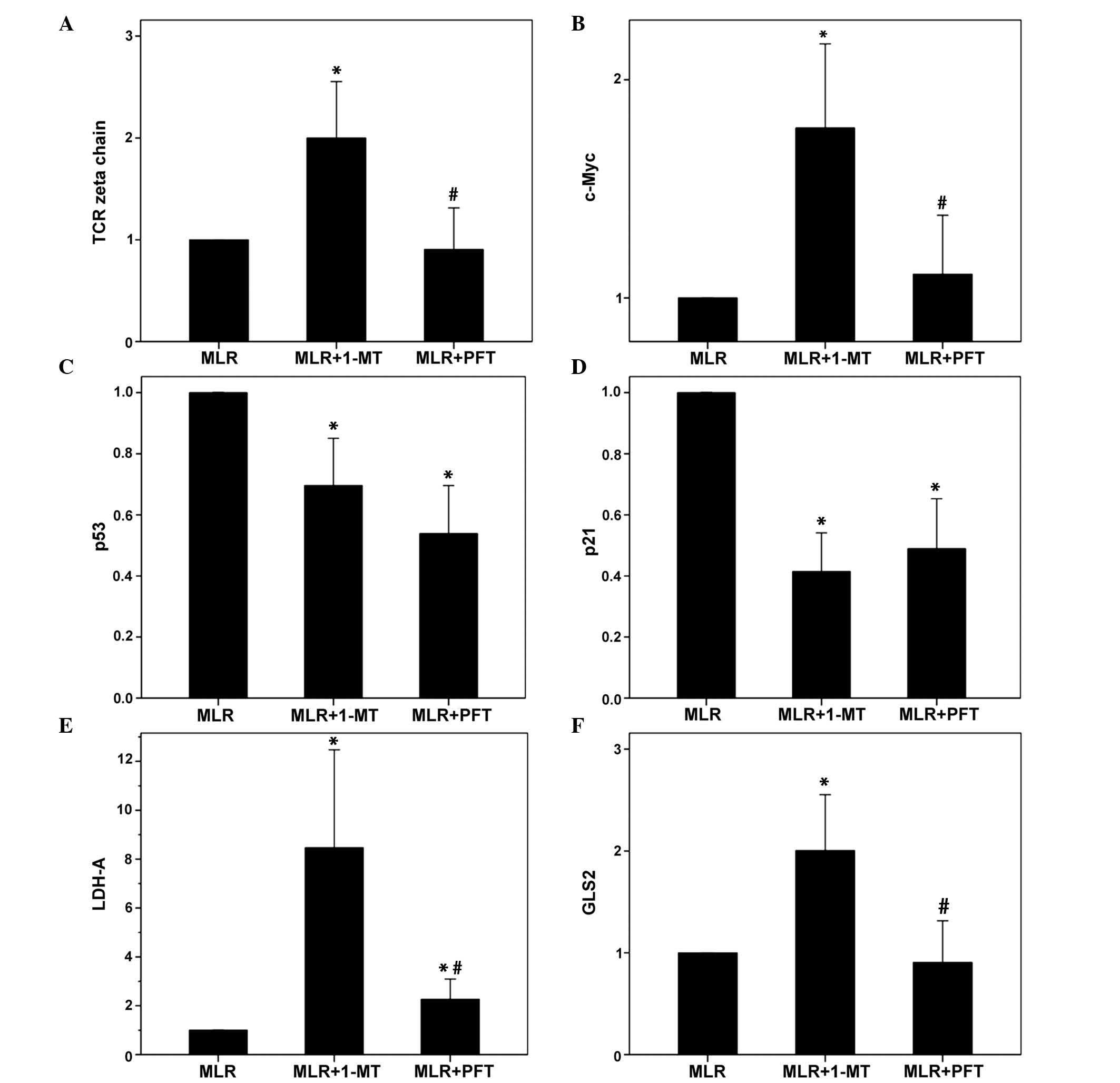 | Figure 3The effect of 1-MT or PFT treatment
of MLRs on the expression levels of TCR-complex ζ-chain, c-Myc,
p53, p21, LDH-A and GLS2 in alloreactive T-cells. Ten MLRs were
conducted in the presence or absence of the indoleamine
2,3-dioxygenase inhibitor 1-MT or the p53 inhibitor PFT, following
which the T-cells were isolated and western blotting conducted. (A)
1-MT however not PFT increased TCR-complex ζ-chain expression. (B)
1-MT however not PFT increased c-Myc expression. 1-MT and PFT
reduced (C) p53 expression and (D) p21 expression. (E) 1-MT
markedly induced LDH-A expression, whereas PFT increased it to a
lesser extent. (F) 1-MT significantly increased GLS2 expression,
while PFT did not affect the expression levels. Values are
presented as the mean ± 95% confidence intervals.
*P<0.05 vs. untreated MLR; #P<0.05 vs.
1-MT-treated MLR. 1-MT, 1-methyl-DL-tryptophan; PFT, pifithrin-α;
MLRs, mixed lymphocyte reactions; TCR, T-cell receptor; LDH-A,
lactose dehydrogenase A; GLS2, glutaminase 2. |
In MLRs, IDO, however not p53, reduced the
expression levels of ζ-chain, c-Myc, LDH-A and GLS2. Compared with
untreated MLRs, in 1-MT-treated T-cells, the expression of ζ-chain
increased to 2.0±0.53 (P=0.006), c-Myc to 1.78±0.37 (P=0.003),
LDH-A to 8.47±8.05 (P=0.001) and GLS2 to 2.0±0.52 (P=0.005;
Figs. 2 and 3).
Compared with untreated MLRs, in the PFT-treated
T-cells the expression levels of ζ-chain, c-Myc and GLS2 were
unaltered, with levels of 0.90±0.16 (P=0.573), 1.11±0.26 (P=0.348)
and 0.90±0.16 (P=0.577), respectively. PFT increased the expression
of LDH-A to 2.27±1.67 (P=0.05), however, to a lesser extent
compared with 1-MT, which increased LDH-A to 8.47±8.05 (P=0.001;
Figs. 2 and 3).
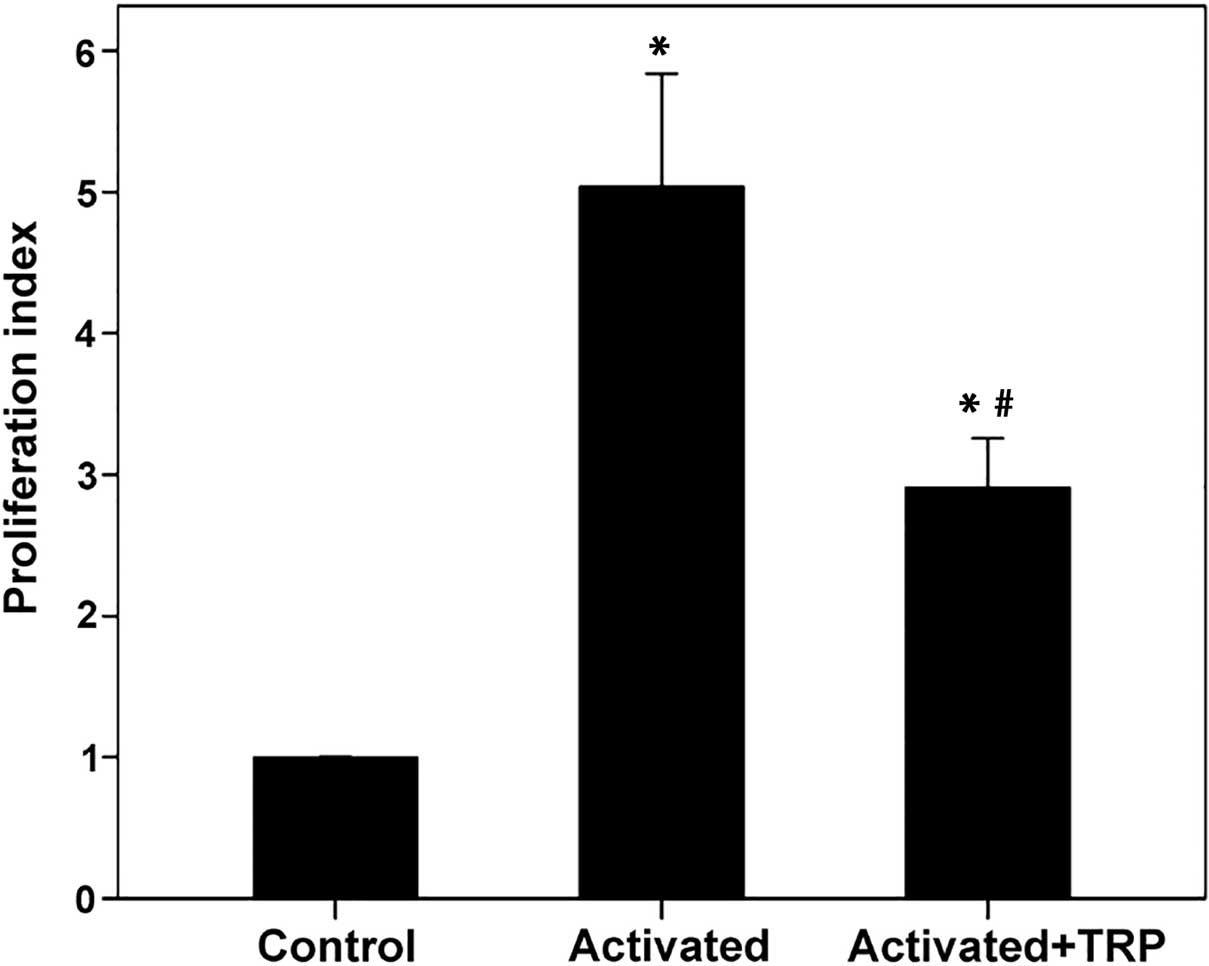
In activated T-cells, TRP reduces T-cell
proliferation
In T-cells activated with anti-CD2, anti-CD3 and
anti-CD28, TRP reduced proliferation. The proliferation index was
5.04±0.96 in activated T-cells and 2.91±0.42 in activated T-cells
treated with TRP (P<0.001; Fig.
4).
In activated T-cells, TRP induces the
expression levels of p53 and p21, while reducing the expression
levels of ζ-chain, c-Myc, LDH-A and GLS2
Direct activation of the GCN2 kinase by TRP induced
the expression levels of p53 and p21 in activated T-cells. Compared
with the unactivated control T-cells, p53 levels were increased in
activated T-cells to 3.56±1.83 (P<0.001), with TRP-treated
activated T-cells exhibiting a further increase to 5.2±2.24
(P<0.001). It is noteworthy that in the absence of TRP,
activation of T-cells increased the levels p53, however, to a
significantly lesser extent (P<0.001; Figs. 5 and 6).
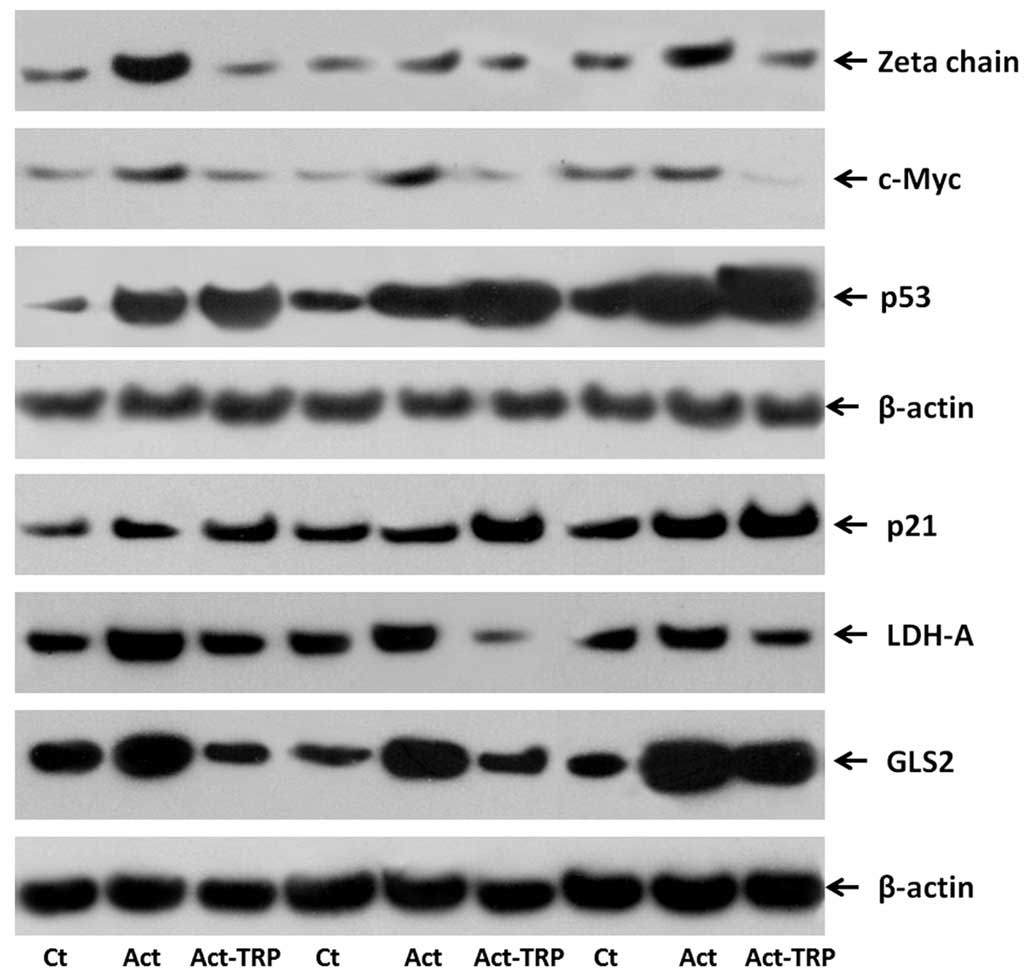 | Figure 5Western blot images presenting the
effect of TRP treatment on the expression levels of TCR-complex
ζ-chain, c-Myc, p53, p21, LDH-A and GLS2 in activated T-cells.
Isolated T-cells were resting or activated with anti-CD2, anti-CD3
and anti-CD28 in the presence or absence of the GCN2 kinase
activator, TRP. The western blotting lanes correspond to three
representative experiments of the ten conducted. TRP, tryptophanol;
TCR, T-cell receptor; LDH-A, lactose dehydrogenase A; GLS2,
glutaminase 2; CD, cluster of differentiation; GCN2, general
control nonderepressible 2; Ct, control; Act, activated. |
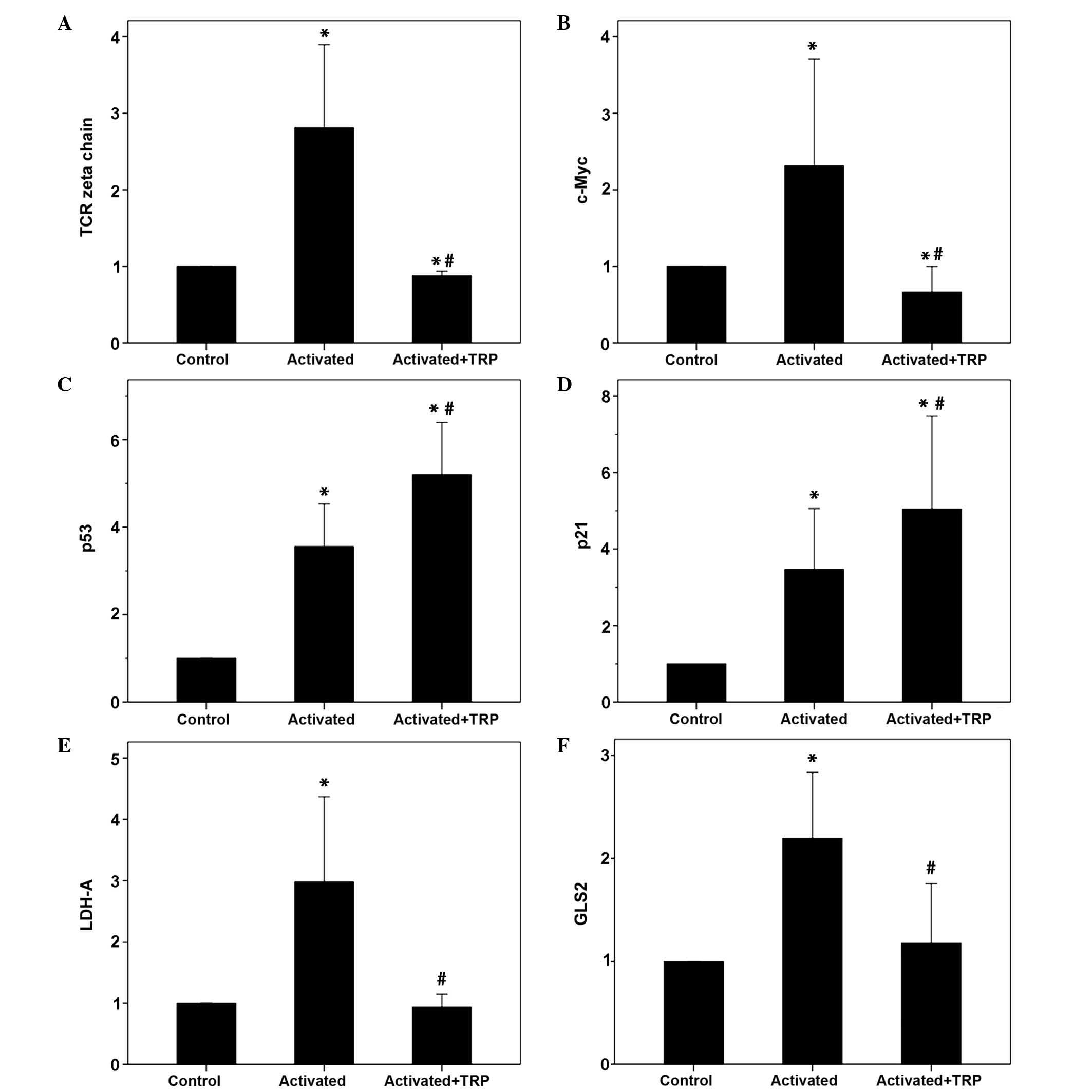 | Figure 6The effect of TRP treatment on the
expression levels of TCR-complex ζ-chain, c-Myc, p53, p21, LDH-A
and GLS2 in activated T-cells. Isolated T-cells were resting or
activated with anti-CD2, anti-CD3 and anti-CD28 in the presence or
absence of the GCN2 kinase activator TRP. Ten experiments were
conducted. (A) T-cell activation increased TCR-complex ζ-chain
expression, whereas treatment with TRP reduced expression. (B)
T-cell activation increased c-Myc expression, whereas treatment
with TRP reduced expression. (C) T-cell activation increased p53
expression levels, which were further increased following TRP
treatment. (D) T-cell activation increased the expression of p21,
which was further increased by TRP treatment. (E) Activation of
T-cells increased LDH-A expression, whereas treatment with TRP
reduced expression. (F) The expression levels of GLS2 were
increased in activated T-cells however were reduced following TRP
treatment. Values are presented as the mean ± 95% confidence
intervals. *P<0.05 vs. unactivated control T-cells;
#P<0.05 vs. activated T-cells. TRP, tryptophanol;
TCR, T-cell receptor; LDH-A, lactose dehydrogenase A; GLS2,
glutaminase 2; CD, cluster of differentiation; GCN2, general
control nonderepressible 2. |
Similar results were observed regarding the
expression of p21. Compared with the unactivated T-cells,
TRP-treated activated T-cells exhibited an increase in the
expression of p21 to 5.05±5.76 (P=0.002), whereas in untreated
activated T-cells, p21 levels were increased to 3.47±3.78
(P=0.004). It is noteworthy that in the absence of TRP, activation
of T-cells increased the expression of p21 (P=0.001) however, to a
significantly lesser extent (Figs.
5 and 6).
Activation of T-cells resulted in an almost 3-fold
increase in ζ-chain expression (2.81±1.30; P=0.06). However,
treatment of activated T-cells with TRP reduced the levels of
ζ-chain to 0.88±0.07 (P=0.02; Figs.
5 and 6).
Similar results were observed regarding the
expression of c-Myc. Activation of T-cells resulted in an increase
in c-Myc expression to 2.32±1.60 (P=0.045). However, treatment of
activated T-cells with TRP reduced the levels of c-Myc to 0.67±0.40
(P=0.040; Figs. 5 and 6).
Compared with unactivated T-cells, T-cell activation
with anti-CD2, anti-CD3 and anti-CD28 increased the expression of
LDH-A 3-fold (2.98±2.60; P=0.008) and GLS2 2-fold (2.19±0.77;
P=0.003). Concurrent treatment with TRP abolished these alterations
in the expression of LDH-A and GLS2, with levels of 0.94±0.39
(P=0.523) and 1.18±0.69 (P=0.482), respectively (Figs. 5 and 6).
Discussion
IDO suppresses T-cell proliferation and concurrently
inhibits aerobic glycolysis and glutaminolysis (16,20).
In addition, IDO increases the levels of p53, which has been
suggested to contribute to reduced T-cell proliferation and
downregulation of various factors involved in aerobic glycolysis.
Notably, IDO-induced increases in the levels of p53 do not alter
LDH-A and GLS2 levels (20). In
addition, IDO downregulates TCR-complex ζ-chain (23). Of the ten immunoreceptor tyrosine
activation motifs (ITAMs) of the TCR-complex, six are within the
ζ-chain dimer. Reduced phosphorylation of TCR-complex ITAMs results
in reduced c-Myc expression and subsequently reduced T-cell
proliferation (21). In addition,
upon T-cell activation, c-Myc is upregulated and induces the
expression levels of LDH-A and GLS2 (19). In the current study, it was
investigated whether IDO-induced L-tryptophan depletion or direct
GCN2 kinase activation promote the following sequence of events:
TCR-complex ζ-chain downregulation, reduced c-Myc expression,
reduced T-cell proliferation and downregulation of LDH-A and GLS2
levels. In addition, the effect of IDO or direct GCN2 kinase
activation on p53 expression was investigated.
For the purposes of the present study, the MLR as a
model of alloreactivity was used (24), in addition to the specific IDO
inhibitor 1-MT. 1-MT is a competitive, non-toxic IDO inhibitor
(25), which has been successfully
used to breach the immune privilege of the placenta and tolerance
against grafts (5,11). In addition, the p53 inhibitor PFT
was used to investigate the association between p53 and c-Myc, and
to confirm the p53-independent effects of IDO on the expression
levels of LDH-A and GLS2. PFT acts downstream of p53 and reversibly
inhibits p53-dependent transcriptional activation (22). Furthermore, a system lacking
IDO-bearing APCs was used in order to distinguish the effect of
GCN2 kinase activation from a possible effect of kynurenine, and to
investigate whether this activation is adequate to induce the
observed alterations by IDO, independently of mTORC1. Isolated
T-cells were activated with anti-CD2, anti-CD3 and anti-CD28
antibodies in the presence or absence of TRP. TRP is a competitive
inhibitor of the tryptophanyl-tRNA synthetase. By raising the pool
of uncharged tRNA, TRP acts as a pharmacologic activator of the
GCN2 kinase (26). Notably,
halofuginone, which activates the GCN2 kinase, exerts its
immunomodulatory properties without altering signaling through the
mTORC1 (27).
The present study demonstrated that in MLRs, the IDO
inhibitor, 1-MT, increased T-cell proliferation, indicating that
IDO reduces proliferation. Furthermore, the p53 inhibitor, PFT,
increased proliferation, which indicated that p53 inhibits
proliferation in MLRs. In addition to the activation of the GCN2
kinase, the immunomodulatory effects of IDO have been attributed to
kynurenine, the first breakdown product in the IDO-dependent
tryptophan degradation pathway. Kynurenine is able to affect
T-cells by activating the aryl hydrocarbon receptor (28,29).
In order to elucidate whether GCN2 activation alone is adequate for
suppressing T-cell proliferation, the current study used a
kynurenine free, APC-free system of isolated T-cell activation to
investigate the effects of the GCN2 kinase activator, TRP. This
demonstrated that in activated T-cells, TRP inhibited T-cell
proliferation.
As observed in a previous study (30), IDO induced the expression levels of
p53 and p21 in MLR-derived T-cells, contributing to reduced T-cell
proliferation due to the p53-mediated upregulation of p21, the
latter being a potent cyclin-dependent kinase inhibitor, which
induces G1-phase cell-cycle arrest (31). The reduction in the expression
levels of p53 and p21 in PFT-treated alloreactive T-cells indicates
that PFT potentially downregulates a positive feedback loop that
controls the expression of p53 in these cells (32).
The present study demonstrated that direct
activation of the GCN2 kinase by TRP in T-cells activated with
anti-CD2, anti-CD3 and anti-CD28 markedly increased p53 and p21
expression, suggesting that activation of this kinase alone is
sufficient for these alterations. Notably, compared with the
resting control T-cells, in activated T-cells p53 and p21 levels
were increased, however to a lesser extent compared with in the
TRP-treated activated T-cells. This may be an intrinsic cell
mechanism for controlling proliferation. For instance, in primary
embryonic fibroblasts, c-Myc, a transcription factor that is
required for cell proliferation, activates the
p19ARF-mouse double minute 2 homolog-p53 tumor
suppressor pathway (33).
Furthermore, in accordance with a previous study
(30), IDO reduced LDH-A and GLS2
levels in MLR-derived T-cells. The p53 inhibitor, PFT, revealed no
effect on GLS2 expression and exerted a relatively minor effect on
the LDH-A levels. In the present study, direct GCN2 kinase
activation by TRP in T-cells activated with anti-CD2, anti-CD3 and
anti-CD28 reduced LDH-A and GLS2 levels indicating that GCN2 kinase
activation alone is sufficient to induce downregulation of the
enzymes involved in aerobic glycolysis and glutaminolysis,
respectively. Since, upon activation, rapidly proliferating T cells
rely on aerobic glycolysis and glutaminolysis in order to fulfill
their bioenergetic and biosynthetic demands (19), this p53- and kynurenine-independent
downregulation of LDH-A and GLS2, respectively, by IDO may
contribute to its immunosuppressive effects.
In MLRs, IDO reduced the levels of TCR-complex
ζ-chain and c-Myc, whereas PFT had no effect, indicating that p53
does not effect the levels of ζ-chain and c-Myc. The reduction in
c-Myc may be attributed to the reduced levels of ζ-chain. It is
known that reduced phosphorylation of the TCR complex ITAMs
downregulates c-Myc expression and inhibits T-cell proliferation
(21). Therefore, beyond the
IDO-induced increase in p53, the IDO-induced decrease in c-Myc may
additionally contribute to the IDO-induced inhibition of T-cell
proliferation. Similar results were obtained by TRP treatment of
isolated activated T-cells, indicating that the GCN2 kinase alone
is sufficient for the downregulation of TCR-complex ζ-chain and
c-Myc. It is noteworthy that the levels of TCR-complex ζ-chain were
increased in activated T-cells in contrast with resting T-cells,
although the responsible mechanisms remain to be elucidated.
The results of the present study supported the
notion that in primary human T-cells, IDO reduces LDH-A levels
through the downregulation of c-Myc. LDH-A, which converts pyruvate
to lactate in the last step of aerobic glycolysis, is a putative
c-Myc target gene. Transgenic mice overexpressing c-Myc in the
liver exhibit increased hepatic glycolytic enzyme activity and
overproduce lactate (34). In
addition, transfected rodent fibroblasts overexpressing LDH-A
alone, or those transformed by c-Myc overproduce lactate (35). Furthermore, in activated murine
T-cells, c-Myc is upregulated and induces LDH-A expression
(19).
Upregulation of c-Myc in activated mouse T-cells has
been demonstrated to increase the levels of GLS2 (19). In addition to rapidly proliferating
T-cells, numerous rapidly proliferating cells, notably cancer
cells, reprogram their mitochondrial metabolism to depend on
glutaminolysis to sustain cellular viability and Krebs cycle
anapleurosis. In transformed cells, overexpression of c-Myc results
in the concurrent conversion of glucose to lactate and the
oxidation of glutamine via the Krebs cycle (36). In this study, in human T-cells,
IDO-induced GLS2 downregulation may be mediated through the
reduction in c-Myc expression.
Thus, in the present study, by using a single
experimental model it was possible to confirm the observations of
previous studies, which used diverse experimental models. In
addition, the present study provided further insight into the
mechanism potentially responsible for the immunosuppressive effects
of IDO. More precisely, these data demonstrated that IDO, through
GCN2 kinase activation, downregulates TCR-complex ζ-chain and
c-Myc, resulting in the suppression of T-cell proliferation and
reduction in the levels of LDH-A and GLS2, which are key enzymes
involved in aerobic glycolysis and glutaminolysis,
respectively.
References
|
1
|
Munn DH, Sharma MD, Baban B, Harding HP,
Zhang Y, Ron D and Mellor AL: GCN2 kinase in T cells mediates
proliferative arrest and anergy induction in response to
indoleamine 2,3-dioxygenase. Immunity. 22:633–642. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cobbold SP, Adams E, Farquhar CA, Nolan
KF, Howie D, Lui KO, Fairchild PJ, Mellor AL, Ron D and Waldmann H:
Infectious tolerance via the consumption of essential amino acids
and mTOR signaling. Proc Natl Acad Sci USA. 106:12055–12060. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
King NJ and Thomas SR: Molecules in focus:
Indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol. 39:2167–2172.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Curti A, Trabanelli S, Salvestrini V,
Baccarani M and Lemoli RM: The role of indoleamine 2,3-dioxygenase
in the induction of immune tolerance: Focus on hematology. Blood.
113:2394–2401. 2009. View Article : Google Scholar
|
|
5
|
Alexander AM, Crawford M, Bertera S,
Rudert WA, Takikawa O, Robbins PD and Trucco M: Indoleamine
2,3-dioxygenase expression in transplanted NOD Islets prolongs
graft survival after adoptive transfer of diabetogenic splenocytes.
Diabetes. 51:356–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beutelspacher SC, Pillai R, Watson MP, Tan
PH, Tsang J, McClure MO, George AJ and Larkin DF: Function of
indoleamine 2,3-dioxygenase in corneal allograft rejection and
prolongation of allograft survival by over-expression. Eur J
Immunol. 36:690–700. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Tredget EE, Ghaffari A, Lin X,
Kilani RT and Ghahary A: Local expression of indoleamine
2,3-dioxygenase protects engraftment of xenogeneic skin substitute.
J Invest Dermatol. 126:128–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seo SK, Choi JH, Kim YH, Kang WJ, Park HY,
Suh JH, Choi BK, Vinay DS and Kwon BS: 4-1BB-mediated immunotherapy
of rheumatoid arthritis. Nat Med. 10:1088–1094. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gurtner GJ, Newberry RD, Schloemann SR,
McDonald KG and Stenson WF: Inhibition of indoleamine
2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in
mice. Gastroenterology. 125:1762–1773. 2003. View Article : Google Scholar
|
|
10
|
Kwidzinski E, Bunse J, Aktas O, Richter D,
Mutlu L, Zipp F, Nitsch R and Bechmann I: Indolamine
2,3-dioxygenase is expressed in the CNS and down- regulates
autoimmune inflammation. FASEB J. 19:1347–1349. 2005.PubMed/NCBI
|
|
11
|
Munn DH, Zhou M, Attwood JT, Bondarev I,
Conway SJ, Marshall B, Brown C and Mellor AL: Prevention of
allogeneic fetal rejection by tryptophan catabolism. Science.
281:1191–1193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mellor AL, Sivakumar J, Chandler P, Smith
K, Molina H, Mao D and Munn DH: Prevention of T cell-driven
complement activation and inflammation by tryptophan catabolism
during pregnancy. Nat Immunol. 2:64–68. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Munn DH and Mellor AL: Indoleamine
2,3-dioxygenase and tumor-induced tolerance. J Clin Invest.
117:1147–1154. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eleftheriadis T, Liakopoulos V, Antoniadi
G, Stefanidis I and Galaktidou G: Indoleamine 2,3-dioxygenase is
increased in hemodialysis patients and affects immune response to
hepatitis B vaccination. Vaccine. 29:2242–2247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eleftheriadis T, Yiannaki E, Antoniadi G,
Liakopoulos V, Pissas G, Galaktidou G and Stefanidis I: Plasma
indoleamine 2,3-dioxygenase and arginase type I may contribute to
decreased blood T-cell count in hemodialysis patients. Ren Fail.
34:1118–1122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eleftheriadis T, Pissas G, Yiannaki E,
Markala D, Arampatzis S, Antoniadi G, Liakopoulos V and Stefanidis
I: Inhibition of indoleamine 2,3-dioxygenase in mixed lymphocyte
reaction affects glucose influx and enzymes involved in aerobic
glycolysis and glutaminolysis in alloreactive T-cells. Hum Immunol.
74:1501–1509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eleftheriadis T, Pissas G, Karioti A,
Antoniadi G, Liakopoulos V, Dafopoulou K, Pournaras S, Koukoulis G
and Stefanidis I: The indoleamine 2,3-dioxygenase inhibitor
1-methyl-tryptophan suppresses mitochondrial function, induces
aerobic glycolysis and decreases interleukin-10 production in human
lymphocytes. Immunol Invest. 41:507–520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J and Green DR: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eleftheriadis T, Pissas G, Antoniadi G,
Spanoulis A, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase increases p53 levels in alloreactive human T cells
and both indoleamine 2,3-dioxygenase and p53 suppress glucose
uptake, glycolysis and proliferation. Int Immunol. 26:673–684.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guy CS, Vignali KM, Temirov J, Bettini ML,
Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, et
al: Distinct TCR signaling pathways drive proliferation and
cytokine production in T cells. Nat Immunol. 14:262–270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Komarov PG, Komarova EA, Kondratov RV,
Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV: A chemical
inhibitor of p53 that protects mice from the side effects of cancer
therapy. Science. 285:1733–1737. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fallarino F, Grohmann U, You S, McGrath
BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi
C, et al: The combined effects of tryptophan starvation and
tryptophan catabolites down-regulate T cell receptor zeta-chain and
induce a regulatory phenotype in naive T cells. J Immunol.
176:6752–6761. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato T, Deiwick A, Raddatz G, Koyama K and
Schlitt HJ: Interactions of allogeneic human mononuclear cells in
the two-way mixed leucocyte culture (MLC): Influence of cell
numbers, subpopulations and cyclosporin. Clin Exp Immunol.
115:301–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jia L, Schweikart K, Tomaszewski J, Page
JG, Noker PE, Buhrow SA, Reid JM, Ames MM and Munn DH: Toxicology
and pharmacokinetics of 1-methyl-d-tryptophan: Absence of toxicity
due to saturating absorption. Food Chem Toxicol. 46:203–211. 2008.
View Article : Google Scholar
|
|
26
|
Jiang HY, Wek SA, McGrath BC, Scheuner D,
Kaufman RJ, Cavener DR and Wek RC: Phosphorylation of the alpha
subunit of eukaryotic initiation factor 2 is required for
activation of NF-kappaB in response to diverse cellular stresses.
Mol Cell Biol. 23:5651–5663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sundrud MS, Koralov SB, Feuerer M, Calado
DP, Kozhaya AE, Rhule-Smith A, Lefebvre RE, Unutmaz D, Mazitschek
R, Waldner H, et al: Halofuginone inhibits TH17 cell
differentiation by activating the amino acid starvation response.
Science. 324:1334–1338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mezrich JD, Fechner JH, Zhang X, Johnson
BP, Burlingham WJ and Bradfield CA: An interaction between
kynurenine and the aryl hydrocarbon receptor can generate
regulatory T cells. J Immunol. 185:3190–3198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Opitz CA, Litzenburger UM, Sahm F, Ott M,
Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller
M, et al: An endogenous tumour-promoting ligand of the human aryl
hydrocarbon receptor. Nature. 478:197–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Elefheriadis T, Pissas G, Antoniadi G,
Spanoulis A, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase increases p53 level in alloreactive human T-cells
and both indoleamine 2,3-dioxygenase and p53 suppress glucose
uptake, glycolysis and proliferation. Int Immunol. 26:673–684.
2014. View Article : Google Scholar
|
|
31
|
Brady CA and Attardi LD: p53 at a glance.
J Cell Sci. 123(Pt 15): 2527–2532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Harris SL and Levine AJ: The p53 pathway:
Positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eischen CM, Weber JD, Roussel MF, Sherr CJ
and Cleveland JL: Disruption of the ARF-Mdm2-p53 tumor suppressor
pathway in Myc-induced lymphomagenesis. Genes Dev. 13:2658–2669.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Valera A, Pujol A, Gregori X, Riu E, Visa
J and Bosch F: Evidence from transgenic mice that myc regulates
hepatic glycolysis. Faseb J. 9:1067–1078. 1995.PubMed/NCBI
|
|
35
|
Shim H, Dolde C, Lewis BC, Wu CS, Dang G,
Jungmann RA, Dalla-Favera R and Dang CV: c-Myc transactivation of
LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad
Sci USA. 94:6658–6663. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Le A, Lane AN, Hamaker M, Bose S, Gouw A,
Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al:
Glucose-independent glutamine metabolism via TCA cycling for
proliferation and survival in B cells. Cell Metab. 15:110–121.
2012. View Article : Google Scholar : PubMed/NCBI
|