Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer worldwide and one of the most fatal ones, causing
nearly 600,000 mortalities yearly (1,2).
Hepatitis B virus (HBV) infection is one of the major causes of HCC
worldwide; epidemiological studies show that 80% of all HCC occurs
in HBV-infected individuals. Hepatitis B virus X protein (HBx) is a
key multifunctional regulatory protein that participates in viral
pathogenesis and carcinogenesis (3) and transactivates numerous
transcription factors, including cyclic adenosine monophosphate
response element-binding protein, activating transcription factor 2
(4), TATA-binding protein
(5), activator protein 1 (AP-1)
(6) and nuclear factor kappa B
(7). Moreover, HBx has been
implicated in the alteration of numerous signal transduction
pathways, including the RAS/RAF/mitogen-activated protein kinase
(MAPK) (8), mitogen-activated
protein kinase kinase kinase 1 (MEKK1)/JNK (6), Janus kinase (JAK)/signal transducer
and activator of transcription (STAT) (9), phosphoinositide 3-kinase (PI3K)/AKT
(10) and Notch1 signaling
(11) pathways, resulting in tumor
cell growth and survival.
Transforming growth factor β (TGF-β) induces cell
senescence and growth arrest via the downstream SMAD signaling
pathway in well-differentiated HCC cells, acting as a tumor
suppressor. Activation of the TGF-β receptor resulted in the
phosphorylation of SMAD2 and SMAD3, which in turn form a
heterotrimer together with SMAD4 and translocate into the nucleus
(12,13). SMAD3 is a modular protein with
conserved Mad-homology 1, intermediate linker and Mad-homology 2
domains (14). The C-terminal
serine residue of SMAD3 is phosphorylated by activated TGF-β type I
receptor (TβRI), whereas the linker domain is phosphorylated by
other kinases, including MAPKs (15–18)
and cyclin-dependent kinases (19). In contrast to the tumor-suppressive
role of the C-terminal phosphorylated SMAD3 (pSMAD3C), SMAD3
phosphorylated at the linker region (pSMAD3L) correlates with
enhanced cell proliferation and invasion. The different
phosphorylated sites reversibly shift SMAD-dependent signaling
between tumor suppression and promotion (20,21).
During carcinogenesis, tumor cells acquire advantage through
selective reduction of the tumor-suppressive activity of TGF-β
together with augmentation of its oncogenic activity (22). The alterations in the TGF-β signal
transduction pathway may be involved in the development of HCC in
long-standing HBV infection. In the progression of HBx-associated
hepatocarcinogenesis, HBx shifts TGF-β signaling from the
TβRI/pSMAD3C tumor-suppressive pathway to the JNK/pSMAD3L oncogenic
pathway in early chronic hepatitis B (18).
Given the roles of HBx and TGF-β, the present study
hypothesized that inhibition of HBx-induced activation of
JNK/pSMAD3L sensitizes HCC cells to TGF-β and promotes the
anti-cancer activity of TGF-β. The effects of TGF-β and JNK
inhibitor SP600125 on cell growth in well-differentiated HCC cells
with forced HBx expression were investigated. The present study
provided an important molecular mechanism which may be utilized for
the treatment of HBV-associated hepatocarcinogenesis.
Materials and methods
Cell culture
Huh-7 and Hep3B cell lines (obtained from the Liver
Cancer Institute, Zhongshan Hospital, Fudan University, Shanghai,
China) and the lentivirus packaging cell line 293T (purchased from
China Center for Type Culture Collection, Wuhan, China) were
maintained in Dulbecco's Modified Eagle's Medium (DMEM) containing
10% fetal bovine serum (FBS; both from Hyclone, Thermo Fisher
Scientific, Waltham, MA, USA), 50 mg/ml penicillin-streptomycin and
0.1 mM non-essential amino acids (both from Hyclone, Thermo Fisher
Scientific) under a humidified 5% CO2 atmosphere at
37°C.
Reagents and antibodies
Trypsin-EDTA was purchased from Hyclone. opti-MEM
medium was from Gibco-BRL (Invitrogen Life Technologies, Carlsbad,
USA). Puromycin (Merck Calbiochem, Darmstadt, Germany), polybrene
(Sigma-Aldrich, St. Louis, MO, USA), penicillin-streptomycin,
Dual-Glo™ Luciferase Assay system (Promega, Madison, WI, USA),
Lipofectamine 2000 with Plus (Life Technologies, Carlsbad, CA, USA)
and recombinant human TGF-β1 (carrier-free; 580706; Biolegend, San
Diego, CA, USA) were used. Antibodies used in the present study are
listed in Table I.
 | Table IAntibodies used in the present
study. |
Table I
Antibodies used in the present
study.
| Antigen | Catalog number and
manufacturer | Application |
|---|
| HBx | sc-57760, Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA | 1:200 for WB |
| p-SMAD3C
(ser423/425) | 1880-1, Epitomics,
Burlingame, CA, USA | 1:1,000 for WB |
| p-SMAD3L
(ser213) | PA5-12694, Thermo
Fisher Scientific Inc., Waltham, MA, USA | 1:100 for WB |
| t-SMAD3 | 1735-1, Epitomics,
Burlingame, CA, USA | 1:2,000 for WB |
| SMAD2/3 | sc-133098, Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA | 1:500 for
WB
1:50 for IP |
| SMAD4 | 1676-1, Epitomics,
Burlingame, CA, USA | 1:2,000 for WB |
| SMAD4 | sc-7154, Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA | 1:50 for IP |
| p-JNK
(Thr183/Tyr185) | 4668, Cell
Signaling Technology, Inc., Danvers, MA, USA | 1:500 for WB |
| t-JNK | 9258, Cell
Signaling Technology, Inc., Danvers, MA, USA | 1:1,000 for WB |
| c-Myc | 1472-1, Epitomics,
Burlingame, CA, USA | 1:1,000 for WB |
| p21 | 2990-1, Epitomics,
Burlingame, CA, USA | 1:1,000 for WB |
| p15 | 4822, Cell
Signaling Technology, Inc., Danvers, MA, USA | 1:2,000 for WB |
| GAPDH | KC-5G4, KangChen
Bio-Tech, Shanghai, China | 1:10,000 for
WB |
| HRP-conjugated
anti-rabbit lgG | KangChen Bio-Tech,
Shanghai, China | 1:3,000 for WB |
| HRP-conjugated
anti-mouse lgG | KangChen Bio-Tech,
Shanghai, China | 1:3,000 for WB |
Plasmids
pLOV-cytomegalovirus (CMV)-enhanced green
fluorescent protein (eGFP)-HBx and pLOV-CMV-eGFP were constructed
by NeuronBiotech (Shanghai, China). Lentiviral gene expression
vector GV266-vec, lentiviral packaging vector pHelper 1.0 and
pHelper 2.0 were purchased from GeneChem Co, Ltd, (Shanghai,
China). The luciferase reporter assay driven by Smad binding
element 4 (SBE4-luc) SBE4-luc (Addgene plasmid no. 16495) was a
gift from Professor Bert Vogelstein (Department of Pathology,
Kimmel Cancer Center, Johns Hopkins University, Baltimore, USA)
(23). pRL-TK was purchased from
Promega (Madison, WI, USA).
Lentivirus production, viral infection
and establishment of stable clones
Lentiviral supernatants were produced as described
previously (24). Briefly, 293T
cells were transfected with pLOV-CMV-eGFP-HBx or pLOV-CMV-eGFP
plasmid by Lipofectamine 2000 and Plus, which were diluted with
opti-MEM (Gibco Life Technologies, Carlsbad, CA, USA), then
selected by 5 µg/ml puromycin for 2 weeks and finally used
to collect lentivirus-containing supernatants. The collected
retroviral and lentiviral supernatants were filtered through a
0.45-µm filter (PALL, Port Washington, NY, USA),
concentrated by Centricon Plus 70 (Merck Millipore, Darmstadt,
Germany) according to the manufacturer's instructions and used for
infection of HCC cells. 72 h following infection, cells were
selected with 2 µg/ml and 3 µg/ml puromycin for Hep3B
and Huh7 cells, respectively for 2 weeks. Selected pools of cells
were used for the subsequent experiments.
Cell proliferation assay
Cell proliferation was measured using the Cell
Counting Kit-8 (Beyotime Institute of Biotechnology, Shanghai,
China) according to the manufacturer's instructions. Hep3B and Huh7
(1.5×103) cells were treated with TGF-β1 (10 ng/ml) or
SP600125 (10 µM), which was replaced every 48 h for a total
of five days. At the indicated time-points, cells were incubated
with CCK-8 stain for 2 h according to the manufacturer's
instructions. The cell proliferation rate was assessed by measuring
the absorbance at 450 nm using a microplate reader (Elx 800; BioTek
Instruments, Inc., Winooski, VT, USA).
Soft agarose tumorigenicity assay
The soft agarose assay was performed as described
previously (11). Briefly, Hep3B
and Huh7 cells (1×104) were suspended in 1 ml 0.4% sea
plaque agarose (A9045; Sigma-Aldrich) containing 10% FBS and then
plated on top of 1 ml semisolid 0.8% agarose in six-well plates.
Cells were treated with TGF-β1 (10 ng/ml) or SP600125 (10
µM) very 48 h for two weeks. Colonies grown on soft agarose
were counted and pictures of colonies were captured using a
magnification of 100× using a phase-contrast microscope (Eclipse C1
System, Nikon, Tokyo, Japan).
Co-immunoprecipitation (co-IP) and
western blot analyses
Cells were lysed by sonication in
radioimmuno-precipitation assay lysis buffer (P0013D; Beyotime
Institute of Biotechnology) supplemented with Protease Inhibitor
Cocktail and Phosphatase Inhibitor PhosSTOP Tablets (Roche Applied
Science, Basel, Switzerland). Protein content was then measured
with the Bicinchoninic Acid kit (Beyotime Institute of
Biotechnology). The co-IP assays were performed as previous
described (25,26) with certain modifications.
Specifically, ~1×107 cells were lysed by sonication in 1
ml NP-40 lysis buffer (P0013F; Beyotime Institute of Biotechnology)
supplemented with the Protease Inhibitor Cocktail and PhosSTOP
Cocktail Tablets. Lysates were pre-cleared with purified mouse or
rabbit IgG (Wuhan Boster Biological Technology, Ltd., Wuhan, China)
antibodies for 15 min at 4°C on a rotating platform, then were
centrifuged at 15,000 × g for 15 min. Supernatants were incubated
with the corresponding antibodies overnight, then were incubated
with protein A/G beads (Abmart, Inc., Shanghai, China) for 4 h at
4°C on a rotating platform. Immunoprecipitates (IPs) and equal
quantities of cell lysates (40 µg) were then subjected to
western blot analyses. Samples of extract taken prior to IP were
processed in parallel with the IPs and were considered as the
''inputs''. Cell lysates or immunoprecipitates were separated with
SDS-PAGE (Wuhan Goodbio Technology Co., Ltd., Wuhan, China) and
transferred to polyvinylidene difluoride membranes (Roche Applied
Science). Nonspecific binding was blocked with Tris-buffered saline
(TBS) containing 5% non-fatty milk (Inner Mongolia Yili Industrial
Group Limited by Share Ltd, Hohhot, China) or bovine serum albumin
(Roche Applied Science) by incubating the membranes for 1 h at
37°C. The blots were then probed overnight at 4°C on a rotating
platform with primary antibodies in TBS with 0.1% Tween 20
(Sigma-Aldrich). Membranes were then washed three times with TBS
and incubated with horseradish peroxidase-conjugated secondary
antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove,
PA, USA). Immunoreactive bands were detected with chemiluminescence
(Life Technologies) and luminous signals from reactive bands were
assessed with the Alpha Innotech Fluorochem Imaging system
(Alphatron Asia Pte Ltd, Singapore, Singapore).
Transcriptional response assay
A transcriptional response assay was performed as
described previously (13).
Briefly, cells seeded in a 24-well plate were transiently
co-transfected with SBE4-Luc together with p-RL-TK reporter
constructs. Cells were treated with TGF-β (10 ng/ml) for 24 h (24 h
after transfection) and then luciferase activity was detected using
the Dual-Luciferase Reporter Assay System (Promega) according to
the manufacturer's instructions. The relative luciferase activity
was determined by a GloMax 20/20 Luminometer (Promega). Luciferase
activity was normalized to Renilla activity and presented as the
mean ± standard error of the mean (SEM) of triplicate
measurements.
Statistical analysis
Values are expressed as the mean ± SEM of triplicate
experiments. Statistical analyses were performed using SPSS 13.0
(SPSS, Inc., Chicago, IL, USA) software. One-way analysis of
variance or Student's t-test were used for analyzing
quantitative data between two groups. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
HBx promotes cell growth in
well-differentiated HCC cell lines
HBx has been reported to be implicated in
hepatocarcinogenesis (3) and the
present study was the first, to the best of our knowledge, to
investigate the effect of HBx on HCC cell growth. The
well-differentiated HCC cell lines Hep3B and Huh7 cells were
selected to establish stable clones with either forced expression
of HBx (Hep3B-HBx and Huh7-HBx) or which were transfected with the
empty vector as a negative control (Hep3B-vec and Huh7-vec). Stable
clones were confirmed by western blot analysis (Fig. 1A). The effects of altered
expression of HBx on cell growth were then assessed using the CCK8
cell proliferation assay. It was found that forced expression of
HBx resulted in an increase in the cell proliferation of Hep3B
(P<0.05) and Huh7 cells (P<0.01) (Fig. 1B). In addition, a soft agarose
assay was conducted, and a significant enhancement of Hep3B cell
growth (P<0.05) and Huh7 cell growth (P<0.05) was observed
(Fig. 1C).
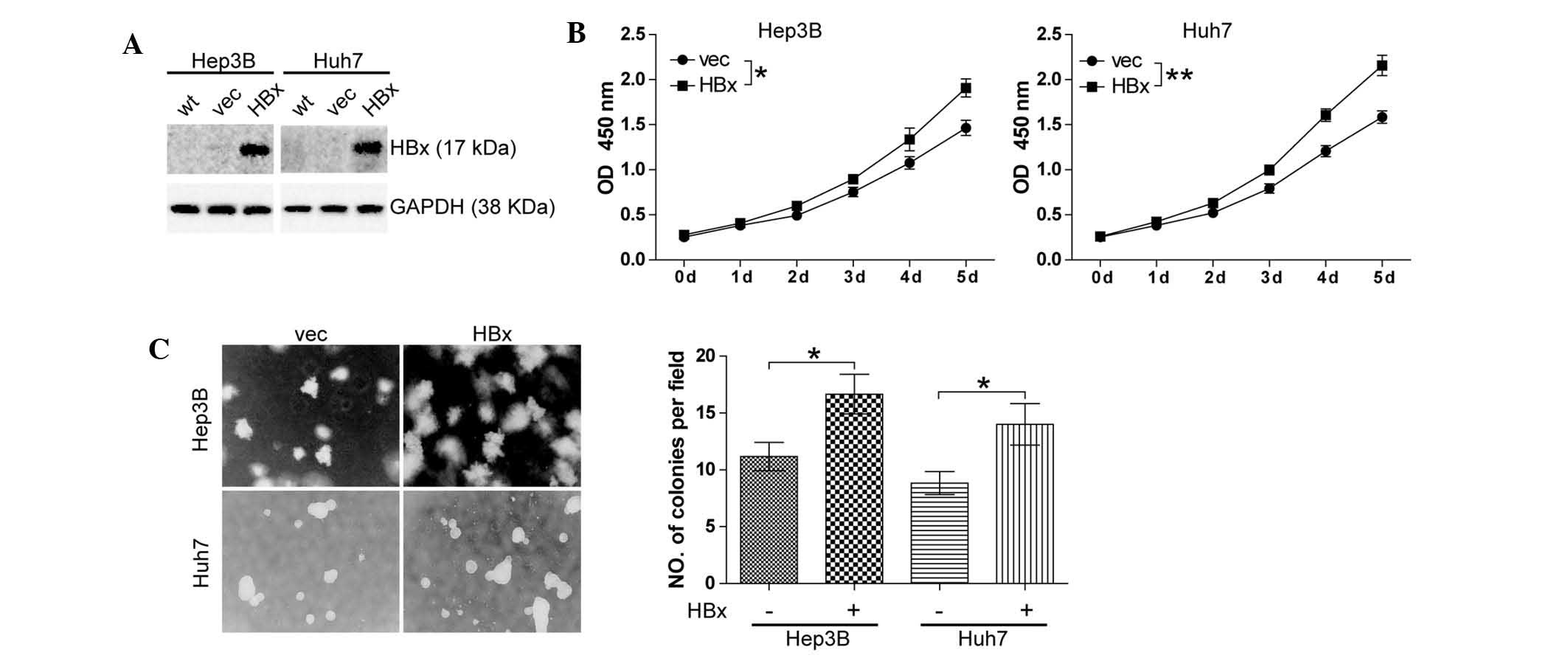 | Figure 1HBx promotes cell growth in
well-differentiated hepatocellular carcinoma cell lines. (A)
Expression of HBx in Hep3B and Huh7 stable clones generated by
lentiviral pLOV-CMV-eGFP vector or pLOV-CMV-eGFP-HBx transfection.
(B) A cell counting kit 8 cell proliferation assay was performed.
Cells (1.5×103) were cultured with complete culture
medium for five days. All experiments were conducted in triplicate.
*P<0.05, **P<0.01. (C) Soft agarose
assay was performed. Cells (1×104/ml) were cultured for
two weeks. Colonies grown on soft agarose were counted and pictures
of colonies were captured (magnification, ×100). Values are
expressed as the mean ± standard error of the mean of six fields,
with experiments conducted in triplicate. *P<0.05,
**P<0.01. HBx, hepatitis B virus X protein; eGFP,
enhanced green fluorescent protein; CMV, cytomegalovirus; vec,
vector; OD, optical density; wt, wild-type. |
TGF-β has no effect on cell growth in
well-differentiated HCC cell lines with forced HBx expression
Autocrine TGF-β signaling is commonly observed in
HCC cells and TGF-β signaling has a tumor-suppressive role in the
initial stage of HCC (13).
However, the effect of TGF-β signaling in HBx-expressing HCC cells
had not been elucidated thus far. The present study detected the
effect of TGF-β on cell growth in vector control cells (Hep3B-vec
and Huh7-vec) and cells with forced HBx expression (Hep3B-HBx and
Huh7-HBx) using the CCK8 cell proliferation assay. In the absence
of HBx, TGF-β significantly inhibited cell growth in Hep3B and Huh7
cell lines. By contrast, in the presence of HBx, Hep3B and Huh7
cells were significantly resistant to TGF-β-induced growth arrest
(Fig. 2A). The soft agarose assay
(Fig. 2B) produced similar
results. These results indicated that TGF-β-induced cell growth
arrest was blocked by HBx.
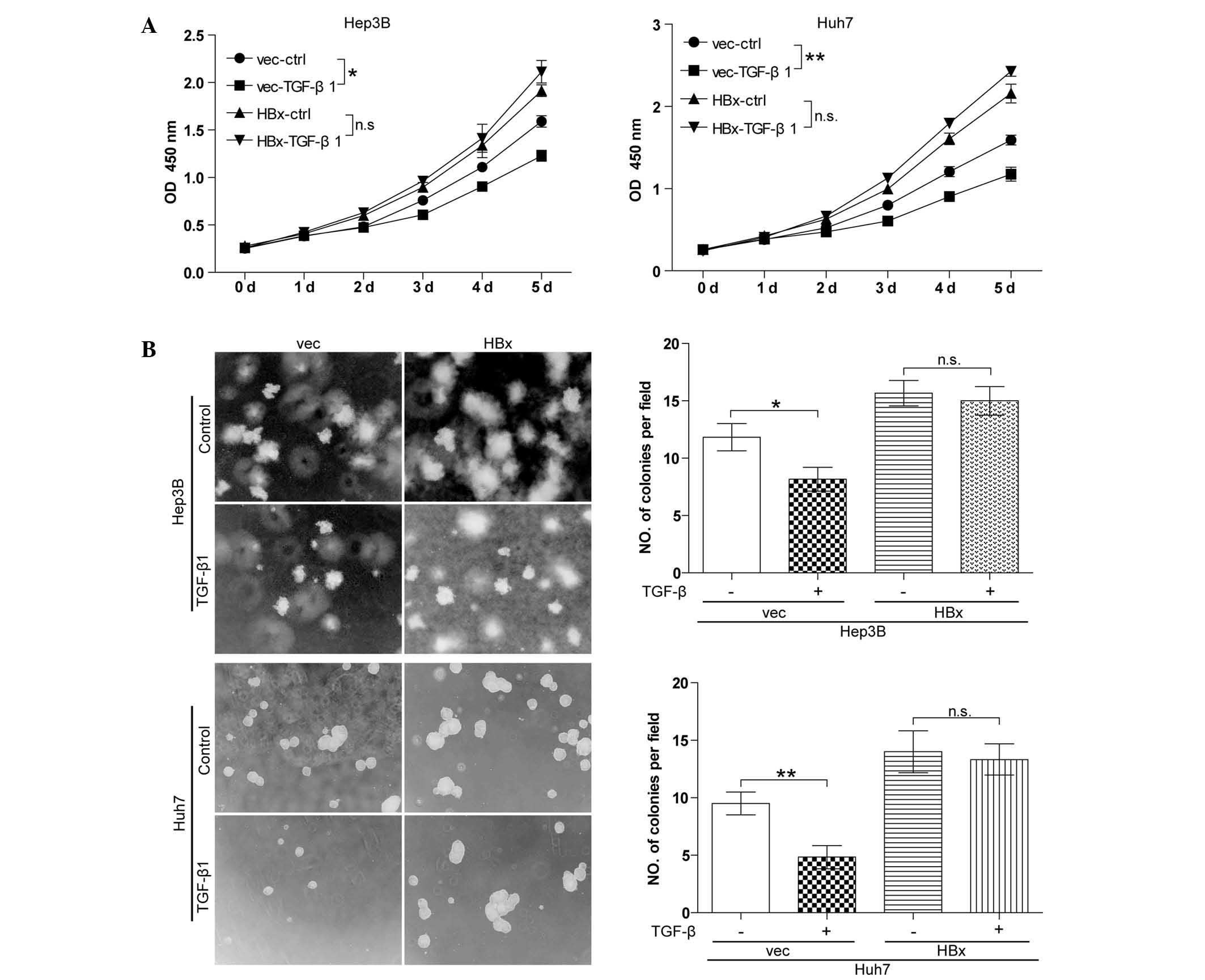 | Figure 2TGF-β has no effect on cell growth in
well-differentiated hepatocellular carcinoma cell lines with forced
HBx expression. (A) A cell counting kit 8 cell proliferation assay
was performed. Cells were treated with 10 ng/ml TGF-β1 every 48 h
for five days. All experiments were conducted in triplicate.
*P<0.05, **P<0.01. (B) A soft agarose
assay was performed. Cells were treated with 10 ng/ml TGF-β1 every
48 h for two weeks. Values are expressed as the mean ± standard
error of the mean of six fields, with experiments conducted in
triplicate. Magnification, ×100. *P<0.05,
**P<0.01. TGF, transforming growth factor; HBx,
hepatitis B virus X protein; vec, vector; ctrl, control; OD,
optical density; wt, wild-type. |
HBx activates JNK/pSMAD3L signaling and
promotes pSMAD3L/SMAD4 complex formation
To explore the mechanism by which HBx blocked
TGF-β1-induced cell growth arrest in well-differentiated HCC cells,
the present study detected the TGF-β/SMAD signaling activity in
response to exogenous TGF-β1 in the absence or presence of HBx with
a luciferase reporter assay using SBE4-luc. It was observed that
Hep3B and Huh7 displayed intact TGF-β signaling activity in the
absence of HBx. Forced expression of HBx enhanced SMAD signaling,
which was not affected by TGF-β (Fig.
3A). Next, western blot analysis was performed to evaluate the
activation of SMAD signaling. In the absence of HBx, TGF-β
predominantly phosphorylated SMAD3 at the C-terminal region
(pSMAD3C). In the presence of HBx, elevated JNK phosphorylation
(p-JNK) was found followed by elevated pSMAD3L together with
reduced pSMAD3C levels. The elevated p-JNK/pSMAD3L was independent
of TGF-β (Fig. 3B). Finally,
immunoprecipitation followed by western blotting was used to
examine the formation of the SMAD2/3/4 complex. In Hep3B cells,
TGF-β induced SMAD2/3/4 complex formation in the absence of HBx.
Forced expression of HBx resulted in an increased formation of
SMAD2/3/4 complex, which was not enhanced by TGF-β (Fig. 3C). In conclusion, TGF-β-induced
cell growth arrest was blocked by HBx via activation of JNK/pSMAD3L
signaling and an increase in pSMAD3L/SMAD4 complex formation.
 | Figure 3HBx activates JNK/pSMAD3L signaling
and promotes pSMAD3L/SMAD4 complex formation. (A) Hep3B and Huh7
cells were co-transfected with SBE4-luc and pRL-TK for 24 h and
then treated with TGF-β1 (10 ng/ml) for 24 h. Luciferase activity
was normalized to renilla luciferase activity and expressed as the
mean ± standard error of the mean of triplicate measurements.
***P<0.001. (B) Lysates from Hep3B and Huh7 cells
treated with 10 ng/ml TGF-β1 for 0.5 h were subjected to western
blot analysis to examine the indicated proteins. (C) Lysates from
Hep3B cells treated with 10 ng/ml TGF-β1 for 0.5 h were subjected
to IP of SMAD2/3 or SMAD4 followed by western blotting with SMAD4
or SMAD2/3 antibody. Input lysates were used for western blotting
with antibodies against indicated proteins. All experiments were
conducted in triplicate. TGF, transforming growth factor; Luc,
luciferase; OD, optical density; vec, vector; p, phosphorylated; t,
total; JNK, c-Jun N-terminal kinase; HBX, hepatitis B virus X
protein; IP, immunoprecipitation; IB, immunoblot. |
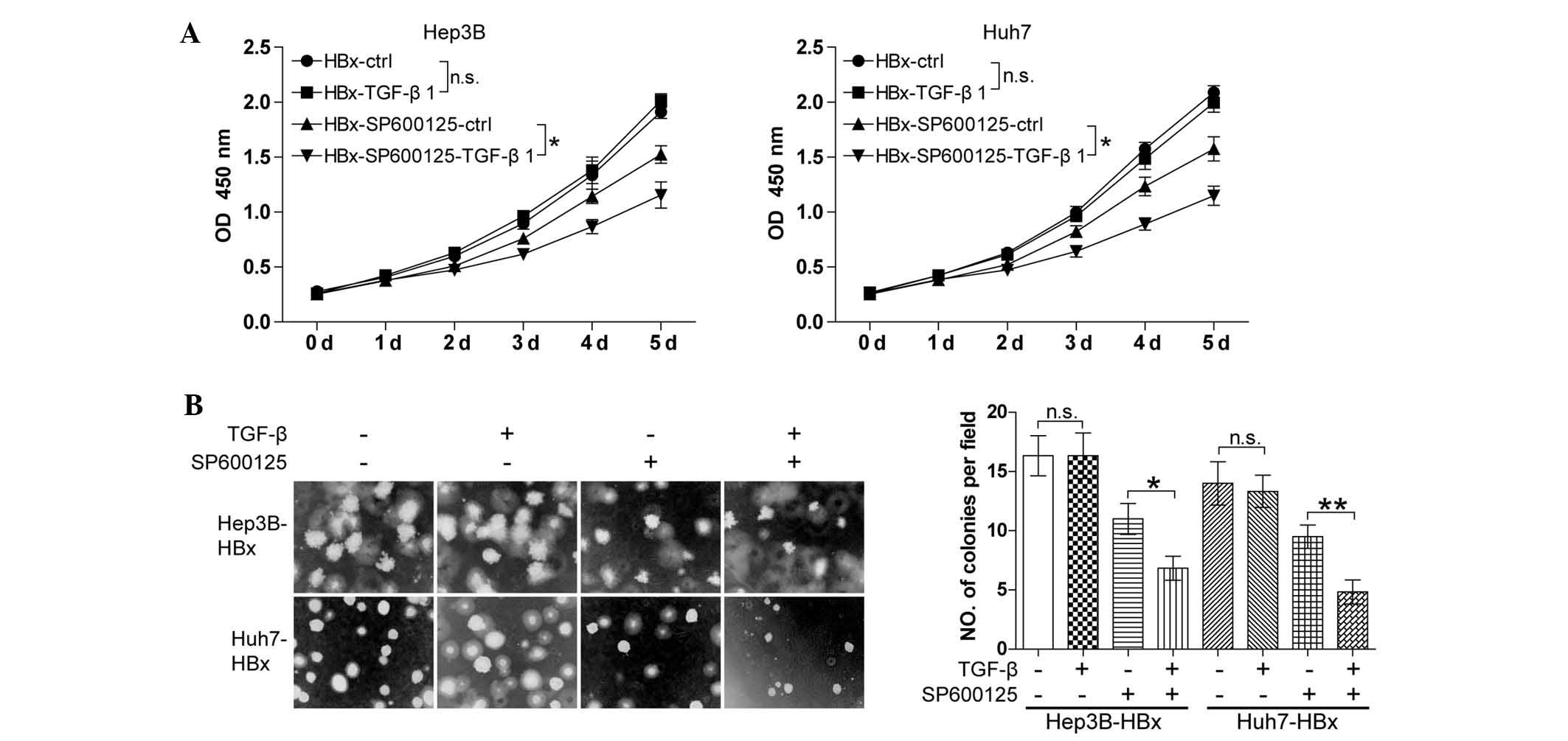
Targeting the JNK pathway favors TGF-β to
antagonize the oncogenic action of HBx
Elevated JNK phosphorylation induced by HBx is known
to be essential for the phosphorylation of SMAD3 at the linker
region for the restriction of the tumor-suppressive action of TGF-β
(18). The present study
hypothesized that inhibition of the JNK pathway may restore the
ability of TGF-β to induce cell growth arrest in HBx-expressing HCC
cells. To confirm this, the Hep3B-HBX and Huh7-HBX cell lines were
treated with TGF-β together with or without JNK inhibitor SP600125.
The CCK8 cell proliferation assay showed that TGF-β had no
significant effect on cell growth in the absence of SP600125.
SP600125 significantly inhibited the cell growth and the
combination of SP600125 and TGF-β resulted in an increased
reduction in cell growth compared with that of SP600125 alone
(Fig. 4A). In addition, a similar
result was observed using the soft agarose assay (Fig. 4B). The results showed that JNK
inhibitor SP600125 restored the capacity of TGF-β to induce cell
growth arrest in HBx-expressing well-differentiated HCC cells.
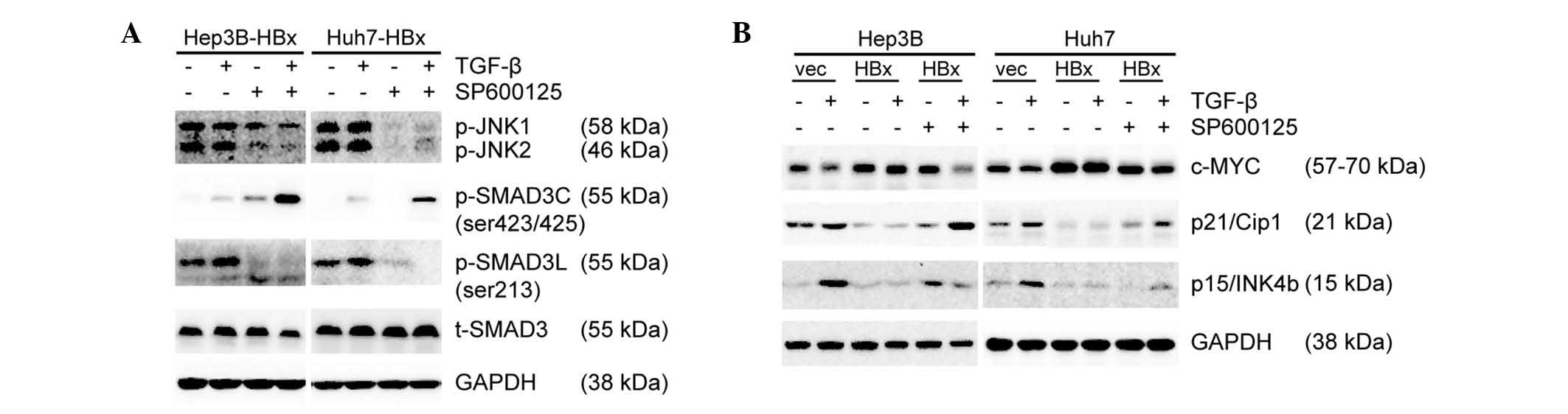
JNK inhibitor SP600125 promotes
pSMAD3C/SMAD4 complexe-induced reduction of c-Myc expression
To explore the underlying mechanism by which JNK
inhibitor SP600125 favored TGF-β to antagonize the oncogenic action
of HBx, the phosphorylation levels of JNK and SMAD3 were examined.
SP600125 significantly inhibited the phosphorylation of JNK and
SMAD3L and promoted the phosphorylation of SMAD3C induced by TGF-β
(Fig. 5A). c-Myc is implicated in
HCC cell senescence and cell cycle progression, and its targeting
of the SMAD2/3/4 transcription complex leads to the inhibition of
p15 and p21 (13,27). Therefore, the present study
assessed the protein expression of c-Myc, p21 and p15 by western
blot analysis. In the absence of HBx, TGF-β induced reduction of
c-Myc expression followed by upregulation of p21 and p15
expression. By contrast, forced expression of HBx resulted in an
opposite effect. In the presence of HBx, inhibition of JNK/Psmad3L
signaling with SP600125 resulted in reduced c-Myc expression and
upregulation of p21 and p15 expression, while TGF-β had no
significant effect on c-Myc, p15 and p21 expression. In combination
with SP600125, TGF-β regained the ability to inhibit c-Myc
expression followed by the upregulation of p21 and p15 expression
(Fig. 5B). In conclusion,
pSMAD3L/SMAD4 and pSMAD3C/SMAD4 complexes antagonize each other to
regulate c-Myc expression. SP600125 significantly inhibited the
phosphorylation of JNK and SMAD3L and promoted pSMAD3C/SMAD4
complex-induced reduction of c-Myc expression.
Discussion
HBx has a major role in the association between
chronic HBV infection and the development of HCC (28). HBx acts as a transactivator to
activate numerous key pathways dependent on various cell contexts,
including the RAS/RAF/MAPK (8),
MEKK1/JNK (6), JAK/STAT (9), PI3K/AKT (10) and Notch1 signaling (11) pathways, resulting in tumor cell
growth and survival. HBx also acts as a transcriptional factor that
stimulates the expression of proto-oncogenes, which control
hepatocellular proliferation, transformation, apoptosis and DNA
repair (29). In the present
study, it was found that HBx promoted cell growth via activation of
the JNK/pSMAD3L pathway in well-differentiated HCC cell lines,
which was consistent with the results of previous studies (10,18).
Even though HBx may transactivate certain other signaling pathways,
demonstration of the activation of the JNK/pSMAD3L pathway
sufficiently illustrates the oncogenic action of HBx.
Autocrine TGF-β is commonly observed in HCC cells
and its expression pattern is closely correlated with SA-β-Gal
activities in normal liver, cirrhosis and HCC (13). These findings indicated that TGF-β
may be a crucial factor to induce cell senescence and restrain the
progression of HCC in its initial stages. Combined with its
potential to induce growth arrest and apoptosis (13,30),
TGF-β treatment may be an attractive therapeutic option for the
prevention of HCC. However, this strategy is counteracted by fully
established HCC tumor cells, which reverse the actions of TGF-β
with the help of other factors (31). In the present study, HBx was
identified to be one such factor. In the absence of HBx, TGF-β
induced cell growth arrest in well-differentiated HCC cell lines.
By contrast, TGF-β had no effect on cell growth in the presence of
HBx. This result, together with those of previous studies (10,18),
indicated that blocking the transaction of HBx is an effective
method to restore the tumor-suppressive function of TGF-β. In other
words, inhibition of JNK/pSMAD3L signaling may maintain the
tumor-suppressive action of TGF-β. The present study found that a
JNK inhibitor, SP600125, significantly inhibited the cell growth in
well-differentiated HCC cell lines. In the presence of SP600125,
TGF-β regained its ability to induce cell growth arrest in HCC cell
lines with forced expression of HBx. These results suggested that
targeting JNK/pSMAD3L signaling is a potential therapy for
HBV-infected patients with HCC.
TGF-β/SMAD signaling regulates transcription of
numerous targeted genes, including plasminogen activator inhibitor
1 (31) and c-Jun (32). In this process, numerous
co-activators, including Cited2 (33), nuclear factor of activated T-cells
(NFAT) (34), AP-1 (32) and CREB binding protein/P300
(35), or co-suppressors,
including c-Ski, SnoN (36) and
mSin3A (37), have a key role. The
profile of SMAD-binding cofactors during development or under
various growth conditions determines cellular responses to TGF-β.
The proto-oncogene c-Myc is one target of TGF-β/SMAD signaling. The
transcriptional repression of c-Myc is dependent on direct SMAD3
binding to a novel SMAD binding site, termed a repressive SMAD
binding element, within the TGF-β inhibitory element of the c-Myc
promoter (38). Consistent with a
previous study (38), it was shown
that the pSMAD3C/SMAD4 transcriptional complex reduced the
expression of c-Myc. In addition, the pSMAD3L/SMAD4 transcriptional
complex had an opposite effect. pSMAD3C/SMAD4 complex recruits
co-suppressors, including p107, to the c-Myc promoter, inhibiting
the transcription of c-Myc (38).
These results indicated that the pSMAD3L/SMAD4 transcriptional
complex promoted c-Myc transcription via recruitment of
co-activators of c-Myc transcription, including Cited2 (33) and NFAT (34).
In conclusion, the present study revealed a dual
role of TGF-β on cell growth in well-differentiated HCC, which is
dependent on HBx. HBx shifts TGF-β from a tumor suppressor to a
tumor promoter. Therefore, anti-HBV therapy is very important to
improve HCC prognosis. Besides anti-viral therapy, inhibition of
activated JNK/pSMAD3L signaling caused by HBx is an important and
potential therapy for HBV-infected patients with HCC.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81372327 and
81072001), the State Key Project on Infection Disease of China
(nos. 2012ZX10002016-004 and 2012ZX10002010-001-004), and the
Chinese Ministry of Public Health for Key Clinical Project [no.
(2010) 493-51] to XP Chen. The authors are very grateful to Dr
Arian Laurence (Molecular Immunology and Inflammation Branch,
National Institute of Arthritis and Musculoskeletal and Skin
Diseases, National Institutes of Health, Bethesda, MD, USA) for
revising the manuscript.
References
|
1
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bouchard MJ and Schneider RJ: The
enigmatic X gene of hepatitis B virus. J Virol. 78:12725–12734.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maguire HF, Hoeffler JP and Siddiqui A:
HBV X protein alters the DNA binding specificity of CREB and ATF-2
by protein-protein interactions. Science. 252:842–844. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qadri I, Maguire HF and Siddiqui A:
Hepatitis B virus transactivator protein X interacts with the
TATA-binding protein. Proc Natl Acad Sci USA. 92:1003–1007. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Benn J, Su F, Doria M and Schneider RJ:
Hepatitis B virus HBx protein induces transcription factor AP-1 by
activation of extracellular signal-regulated and c-Jun N-terminal
mitogen-activated protein kinases. J Virol. 70:4978–4985.
1996.PubMed/NCBI
|
|
7
|
Su F and Schneider RJ: Hepatitis B virus
HBx protein activates transcription factor NF-kappaB by acting on
multiple cytoplasmic inhibitors of rel-related proteins. J Virol.
70:4558–4566. 1996.PubMed/NCBI
|
|
8
|
Benn J and Schneider RJ: Hepatitis B virus
HBx protein activates Ras-GTP complex formation and establishes a
Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci USA.
91:10350–10354. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee YH and Yun Y: HBx protein of hepatitis
B virus activates Jak1-STAT signaling. J Biol Chem.
273:25510–25515. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shih WL, Kuo ML, Chuang SE, Cheng AL and
Doong SL: Hepatitis B virus X protein inhibits transforming growth
factor-β-induced apoptosis through the activation of
phosphatidylinositol 3-kinase pathway. J Biol Chem.
275:25858–25864. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu J, Yun X, Jiang J, et al: Hepatitis B
virus X protein blunts senescence-like growth arrest of human
hepatocellular carcinoma by reducing Notch1 cleavage. Hepatology.
52:142–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Senturk S, Mumcuoglu M, Gursoy-Yuzugullu
O, Cingoz B, Akcali KC and Ozturk M: Transforming growth
factor-beta induces senescence in hepatocellular carcinoma cells
and inhibits tumor growth. Hepatology. 52:966–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Massagué J: TGF-beta signal transduction.
Ann Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kretzschmar M, Doody J, Timokhina I and
Massagué J: A mechanism of repression of TGFbeta/Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mori S, Matsuzaki K, Yoshida K, et al:
TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3
phosphorylation at the linker regions. Oncogene. 23:7416–7429.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Furukawa F, Matsuzaki K, Mori S, et al:
p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation
in rat myofibroblasts. Hepatology. 38:879–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murata M, Matsuzaki K, Yoshida K, et al:
Hepatitis B virus X protein shifts human hepatic transforming
growth factor (TGF)-beta signaling from tumor suppression to
oncogenesis in early chronic hepatitis B. Hepatology. 49:1203–1217.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsuura I, Denissova NG, Wang G, He D,
Long J and Liu F: Cyclin-dependent kinases regulate the
antiproliferative function of Smads. Nature. 430:226–231. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamagata H, Matsuzaki K, Mori S, et al:
Acceleration of Smad2 and Smad3 phosphorylation via c-Jun
NH(2)-terminal kinase during human colorectal carcinogenesis.
Cancer Res. 65:157–165. 2005.PubMed/NCBI
|
|
21
|
Sekimoto G, Matsuzaki K, Yoshida K, et al:
Reversible Smad-dependent signaling between tumor suppression and
oncogenesis. Cancer Res. 67:5090–5096. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roberts AB and Sporn MB: The Transforming
Growth Factor-βs. Peptide Growth Factors And Their Receptors.
Springer; pp. 419–472. 1990, View Article : Google Scholar
|
|
23
|
Zawel L, Dai JL, Buckhaults P, et al:
Human Smad3 and Smad4 are sequence-specific transcription
activators. Mol Cell. 1:611–617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ding ZY, Jin GN, Liang HF, et al:
Transforming growth factor β induces expression of connective
tissue growth factor in hepatic progenitor cells through Smad
independent signalling. Cell Signal. 25:1981–1992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xue J, Lin X, Chiu WT, et al: Sustained
activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer
metastasis. J Clin Invest. 124:564–579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dupont S, Mamidi A, Cordenonsi M, et al:
FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta
signaling, controls Smad4 monoubiquitination. Cell. 136:123–135.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Siegel PM and Massagué J: Cytostatic and
apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev
Cancer. 3:807–821. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Block TM, Mehta AS, Fimmel CJ and Jordan
R: Molecular viral oncology of hepatocellular carcinoma. Oncogene.
22:5093–5107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Zhang H and Ye L: Effects of
hepatitis B virus X protein on the development of liver cancer. J
Lab Clin Med. 147:58–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dzieran J, Fabian J, Feng T, et al:
Comparative analysis of TGF-β/Smad signaling dependent cytostasis
in human hepatocellular carcinoma cell lines. PloS one.
8:e722522013. View Article : Google Scholar
|
|
31
|
Dennler S, Itoh S, Vivien D, ten Dijke P,
Huet S and Gauthier JM: Direct binding of Smad3 and Smad4 to
critical TGFβ-inducible elements in the promoter of human
plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wong C, Rougier-Chapman EM, Frederick JP,
et al: Smad3-Smad4 and AP-1 complexes synergize in transcriptional
activation of the c-Jun promoter by transforming growth factor
beta. Mol Cell Biol. 19:1821–1830. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chou YT, Wang H, Chen Y, Danielpour D and
Yang YC: Cited2 modulates TGF-beta-mediated upregulation of MMP9.
Oncogene. 25:5547–5560. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Singh G, Singh SK, König A, et al:
Sequential activation of NFAT and c-Myc transcription factors
mediates the TGF-beta switch from a suppressor to a promoter of
cancer cell proliferation. J Biol Chem. 285:27241–27250. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng XH, Zhang Y, Wu RY and Derynck R: The
tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300
are coactivators for Smad3 in TGF-beta-induced transcriptional
activation. Genes Dev. 12:2153–2163. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koinuma D, Shinozaki M, Nagano Y, et al:
RB1CC1 protein positively regulates transforming growth factor-beta
signaling through the modulation of Arkadia E3 ubiquitin ligase
activity. J Biol Chem. 286:32502–32512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liao M, Zhang Y, Kang JH and Dufau ML:
Coactivator function of positive cofactor 4 (PC4) in Sp1-directed
luteinizing hormone receptor (LHR) gene transcription. J Biol Chem.
286:7681–7691. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Frederick JP, Liberati NT, Waddell DS, Shi
Y and Wang XF: Transforming growth factor beta-mediated
transcriptional repression of c-myc is dependent on direct binding
of Smad3 to a novel repressive Smad binding element. Mol Cell Biol.
24:2546–2559. 2004. View Article : Google Scholar : PubMed/NCBI
|