Introduction
The incidence and prevalence of diabetes mellitus
(DM) are increasing globally (1).
Cardiovascular complications are the leading causes of morbidity
and mortality in patients with diabetes. Among them, diabetic
cardiomyopathy (DCM) is defined as a distinct primary disease
process, independent of coronary artery disease, hypertension or
any other cardiovascular diseases, which leads to ventricular
dysfunction and heart failure (2).
However, the pathogenesis of DCM remains to be elucidated. To date,
certain cellular and molecular defects are believed to be
responsible for the pathogenesis of DCM, including glucotoxicity,
altered lipid metabolism, lipotoxicity, oxidative stress, abnormal
calcium handling, protein kinase C signaling,
apoptosis/inflammation/fibrosis and mitochondrial dysfunction
(3). In particular, growing
evidence has demonstrated that apoptosis of cardiomyocytes is vital
in the development of DCM (4,5).
Therefore, inhibition of cardiac apoptosis is of great importance
in the prevention and treatment of DCM (6).
Advanced oxidative protein products (AOPPs) are the
dityrosine-containing and cross-linking protein products, which are
formed during oxidative stress by the reaction between proteins and
chlorinated oxidants (7). AOPPs
were initially discovered in the plasma of patients with uremia and
undergoing dialysis, and are considered as a novel marker of
oxidative stress. Increased levels of plasma AOPPs were
demonstrated in patients with DM, coronary artery disease and
metabolic syndrome (8–10). In addition, AOPPs were
significantly higher in the diabetic group with vascular
complications compared with the group without complications
(11). In vitro, incubation
of AOPPs with human umbilical vein endothelial cells induced
superoxide generation, activated NAD(P)H oxidase,
extracellular-signal regulated kinase (ERK)1/2 and p38, and
promoted nuclear translocation of NF-κB, by bonding to the receptor
for advanced glycation end products (RAGEs) (12). In experimental models, accumulation
of AOPPs activated the p53/Bax/caspase-dependent proapoptotic
pathway via RAGE and resulted in podocyte apoptosis (13). However, the exact role of AOPPs in
the myocardial cells of DCM remains to be elucidated.
Glucagon-like peptide-1 (GLP-1) is a multifunctional
hormone secreted from intestinal endocrine L cells and is widely
used for the treatment of diabetes. As well as its effects on
glucose control, GLP-1 reduces gastric emptying, inhibits appetite
and exhibits a protective effect on cardiovascular diseases
(14,15). The GLP-1 receptor (GLP-1R) was
identified in the heart and vascular tissue, in addition to
pancreatic α and β cells (16,17).
A previous study demonstrated that the GLP-1R agonist, liraglutide,
inhibits endothelial cell dysfunction and alleviates
atherosclerotic injury (14). In
rats with chronic heart failure, subcutaneous infusion with GLP-1
and its analogue, exenatide, improves cardiac function, cardiac
remodeling and survival (18).
Furthermore, GLP-1 directly protects cardiomyocytes from
hypoxia/reoxygenation injury, predominantly by inhibiting their
apoptosis (19) and protects
vascular endothelial cells against AGE-induced apoptosis (20). However, whether GLP-1 has a
protective effect on AOPP-treated cardiomyocytes remains to be
elucidated.
Therefore, the present study aimed to investigate
whether AOPPs have a detrimental role in the survival of
cardiomyocytes and if GLP-1 exerts cardioprotective effects under
certain circumstances. The present study focussed on the effect of
GLP-1 on the apoptosis of H9c2 myocardial cells induced by AOPPs
and its underlying mechanism. It was demonstrated that GLP-1
alleviates AOPP-induced apoptosis via the PI3K/Akt/Bad pathway in
H9c2 cells. In conclusion, these data presented a theoretical
foundation and novel insight into the pathogenic and therapeutic
strategy of DCM.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (high glucose)
and phosphate-buffered saline (PBS) were purchased from Hyclone
(Logan, UT, USA). Fetal bovine serum (FBS), penicillin-streptomycin
solution and 0.25% Trypsin-EDTA were purchased from (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Dimethyl sulfoxide, diethyl
pyrocarbonate, rat serum albumin (RSA), bovine serum albumin (BSA),
paraformaldehyde, Triton X-100, 4′,6-diamidino-2-phenylindole
(DAPI), GLP-1 (7–36 amide) and the amebocyte lysate assay kit were
all purchased from Sigma-Aldrich (St Louis, MO, USA). The following
antibodies were purchased from Cell Signaling Technologies (Boston,
MA, USA): Rabbit monoclonal phosphorylated (p)-Akt (Ser473; cat.
no. CST4060P), rabbit monoclonal anti-Akt (cat. no. CST4695),
rabbit anti-B-cell lymphoma (Bcl)-2 (cat. no. CST2876) rabbit
monoclonal p-Bcl-2-associated death promoter (Bad; Ser136; cat. no.
CST4366P), rabbit anti-Bcl-2 associated X protein (Bax; cat. no.
CST2772) and rabbit anti-caspase-3 (cat. no. CST9662). Mouse
anti-RAGE antibody (cat. no. SC33662) was provided by Santa Cruz
Biotechnology, Inc. (Heidelberg, Germany). Rabbit anti-GLP1R
antibody (cat. no. ab39072) was purchased from Abcam (Cambridge,
MA, USA). A Cell Counting kit (CCK)-8 was purchased from Dojindo
Laboratories (Kumamoto, Japan). LY294002 and a Hoechst 33258
Staining kit were purchased from Beyotime Institute of
Biotechnology (Haimen, China). RNAiso plus, SYBR Premix EX Taq II
kit and PrimeScript™ RT reagent kit were purchased from Takara
Biotechnology Co., Ltd. (Dalian, China). Hypochlorous acid (HOCl)
was purchased from Fluke (Buchs, Swizerland). An annexin
V-fluorescein isothiocyanate (FITC) apoptosis detection kit was
purchased from eBioscience (San Diego, CA, USA). A bicinchoninic
assay protein assay kit was purchased from Keygen Biotech Co., Ltd.
(Nanjing, China). Rabbit anti-GAPDH antibody, anti-rabbit or
anti-mouse horseradish peroxidase-conjugated secondary
immunoglobulin (Ig)G were obtained from Wuhan Boster Biological
Technology, Ltd. (Wuhan, China). FITC-labeled anti-mouse IgG and
anti-rabbit IgG were purchased from EarthOx, LLC (San Francisco,
CA, USA).
Cell culture
The H9c2 (2-1) cells (21) were a generous gift from the
Department of Cardiology, Nanfang Hospital (Guangzhou, China). The
H9c2 cells were cultured in Dulbecco's modified Eagle's medium
(high glucose), containing 10% FBS and 1% penicillin-streptomycin
solution in a 5% carbon dioxide atmosphere at 37°C. The cells were
subcultured every 2–3 days at a ratio of 1:2 or 1:3.
AOPPs preparation and determination
AOPPs-RSA was prepared in vitro, as
previously described (7,13). Briefly, 20 mg/ml RSA solution was
mixed and placed with 40 mmol/l HOCl in PBS for 30 min at 37°C. The
prepared samples were dialyzed against PBS for 24 h to remove free
HOCl and passed through a Detoxi-Gel column (Pierce, Rockford, IL,
USA) to remove contaminated endotoxin. A total of 20 mg/ml RSA
solution was mixed with isometric PBS as a comparison. The content
of AOPPs in the AOPPs-RSA and unmodified RSA were 70.7 and 0.11
nmol/mg protein, respectively. Endotoxin levels in the AOPPs-RSA
and unmodified RSA were measured with the amebocyte lysate assay
kit and were determined to be >0.25 EU/ml.
Cell viability
The H9c2 cells were seeded into 96-well plates at a
density of 2.5×103 and were treated with the indicated
concentrations of AOPPs-RSA and GLP-1. Following incubation for 24
h, the medium was replaced with 10% CCK-8 solution for 2 h at 37°C.
The absorbance was measured at 450 nm with a microplate reader
(EXL808; BioTek, Winooski, VT, USA), according to the
manufacturer's instructions.
Cell apoptosis
Each group of cells were detected by double staining
with FITC-conjugated annexin V and propidium iodide (PI). The
collected cells were washed twice with PBS and stained with 5
µl annexin V-FITC for 15 min in the dark in binding buffer
(eBioscience, San Diego, CA, USA). Subsequently, 10 µl PI
was added at room temperature for 5 mins. Cell apoptosis was
determined using a FACSVerse flow cytometer (BD Biosciences, San
Jose, CA, USA) using the FACSuite program (BD Biosciences).
Hoechst 33258 staining
Following treatment, the H9c2 cells were fixed
overnight at 4°C, and were subsequently permeabilized and stained
with 10 µg/ml Hoechst 33258 for 5 min at room temperature.
The morphological changes were observed under a fluorescence
microscope (Axio Scope A1; Carl Zeiss, Jena, Germany) at a
wavelength of 350 nm.
Immunofluorescence assay
Following treatment, the cells were fixed in 4%
paraformaldehyde for 15 min and permeabilized with Triton X-100 for
15 min at room temperature. Following washing the cells twice with
PBS, the cells were blocked in 1% BSA for 30 min at room
temperature. The cells were subsequently incubated overnight at 4°C
with primary antibody (RAGE, 1:100; GLP-1, 1:100) in 1% BSA.
Following washing twice with PBS, the cells were incubated with
FITC-labeled anti-mouse or anti-rabbit IgG secondary antibodies
(1:200) for 1 h at room temperature. Finally, the cells were
stained with 100 ng/ml DAPI for 2 min at room temperature and
immunofluorescent images were captured using a fluorescence
microscope (Axio Scope A1; Carl Zeiss).
Western blot analysis
Following treatment, the cellular proteins were
extracted using radioimmunoprecipitation lysis buffer (Sangon
Biotech Co., Ltd,, Shanghai, China) for 30 min at 4°C and
centrifuged at 250 × g for 20 min at 4°C. The protein concentration
was measured using a bicinchoninic assay kit on a EXL808 microplate
reader. The proteins (50 µg) were separated on 12%
SDS-polyacrylamide gels (Whiga, Guangdong, China) and were
transferred onto polyvinylidene fluoride membranes (Bio-Rad
Laboratories, Hercules, CA, USA). The membranes were blocked in 5%
non-fat milk in Tris-buffered saline, containing 0.1% Tween-20
(TBST), for 2 h at room temperature and were subsequently
immunoblotted with primary antibodies for 2 h at room temperature.
Antibodies against p-Akt (1:2,000), Akt (1:1,000) p-Bad (1:1,000),
Bax (1:1,000), Bcl-2 (1:1,000), caspase-3 (1:1,000), RAGE (1:100)
and GLP-1R (1:1,000) were used. Following washing three times with
TBST, the membranes were incubated with anti-rabbit or anti-mouse
horseradish peroxidase-conjugated secondary IgG (1:2,500) at room
temperature for 1 h. The protein bands were visualized using an
enhanced chemiluminescence kit (EMD Millipore, Billerica, MA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted using RNAiso Plus,
according to the manufacturer's instructions. The purity of each
RNA sample was measured using a Nanodrop 2000 spectrophotometer
(Thermo Fisher Scientific, Inc.). To remove genomic DNA
contamination, the RNA samples were treated with gDNA Eraser
(Takara Biotechnology Co., Ltd.) at 42°C for 2 min. The
complementary DNA was synthesized in a 20 µl reaction
system, using the PrimeScript RT reagent kit at 37°C for 15 min and
85°C for 5 sec. The primers used were as follows: Rat RAGE,
forward: 5′-GGG ACA GTG TGG CTC GAATC-3′ and reverse: 5′-TCC CAA
GCC TGT TAG TTG CC-3′; rat GLP-1R, forward: 5′-AAT GCA GAC TCG CGA
AGTCC-3′ and reverse: 5′-TGA CGA AGC GTA GGG TTCCT-3′; GAPDH
(internal control), forward: 5′-AGGGCT GCCTTCTCTTGTGA-3′ and
reverse 5′-AAC TTG CCG TGG GTA GAG TCA-3′. RT-qPCR was performed
using the SYBR Premix EX Taq II kit on an ABI 7300 system (ABI,
Foster City, CA, USA). The cycling conditions were 93°C for 15 min,
followed by 40 cycles of 93°C for 15 sec, 55°C for 25 sec and 72°C
for 25 min. The data were normalized against GAPDH and the negative
control. Data analysis was performed using the 2−ΔΔCq
method.
Statistical analysis
All data are expressed as the mean ± standard
deviation. Differences between groups were assessed by one-way or
Welch analysis of variances (equal variances not assumed) once
normality was confirmed. For pairwise comparison, a least
significant difference test or Dunnett's T3 test was used when the
equal variances were not assumed. Spearman's rank correlation was
used for correlation analysis. P<0.05 was considered to indicate
a statistically significant difference. All analyses were performed
with SPSS 13.0 software (SPSS Inc., Chicago, IL, USA).
Results
GLP-1 attenuated AOPP-induced toxicity in
H9c2 cells
To confirm if AOPPs are toxic on cell viability, a
CCK-8 assay was performed in AOPP-treated H9c2 cells. As shown in
Fig. 1A, incubation with AOPPs-RSA
for 24 h significantly inhibited cell viability (84.2 to 64.5%) in
a dose-dependent manner (r=−0.797, P=0.000) while RSA caused no
effect. In addition, H9c2 cells were incubated with 1 µg/ml
AOPP-RSA for 24 h in the presence or absence of different
concentrations of GLP-1. Compared with AOPP-RSA, GLP-1 partly
restored cell viability in a dose-dependent manner (r=0.698,
P=0.004), with a peak at 50 nmol/l (97.9%; Fig. 1B). Therefore, subsequent
experiments were performed using 1 µg/ml AOPPs-RSA and 50
nmol/l GLP-1. These results demonstrated that GLP-1 protects
AOPPs-RSA-induced toxicity in H9c2 cells.
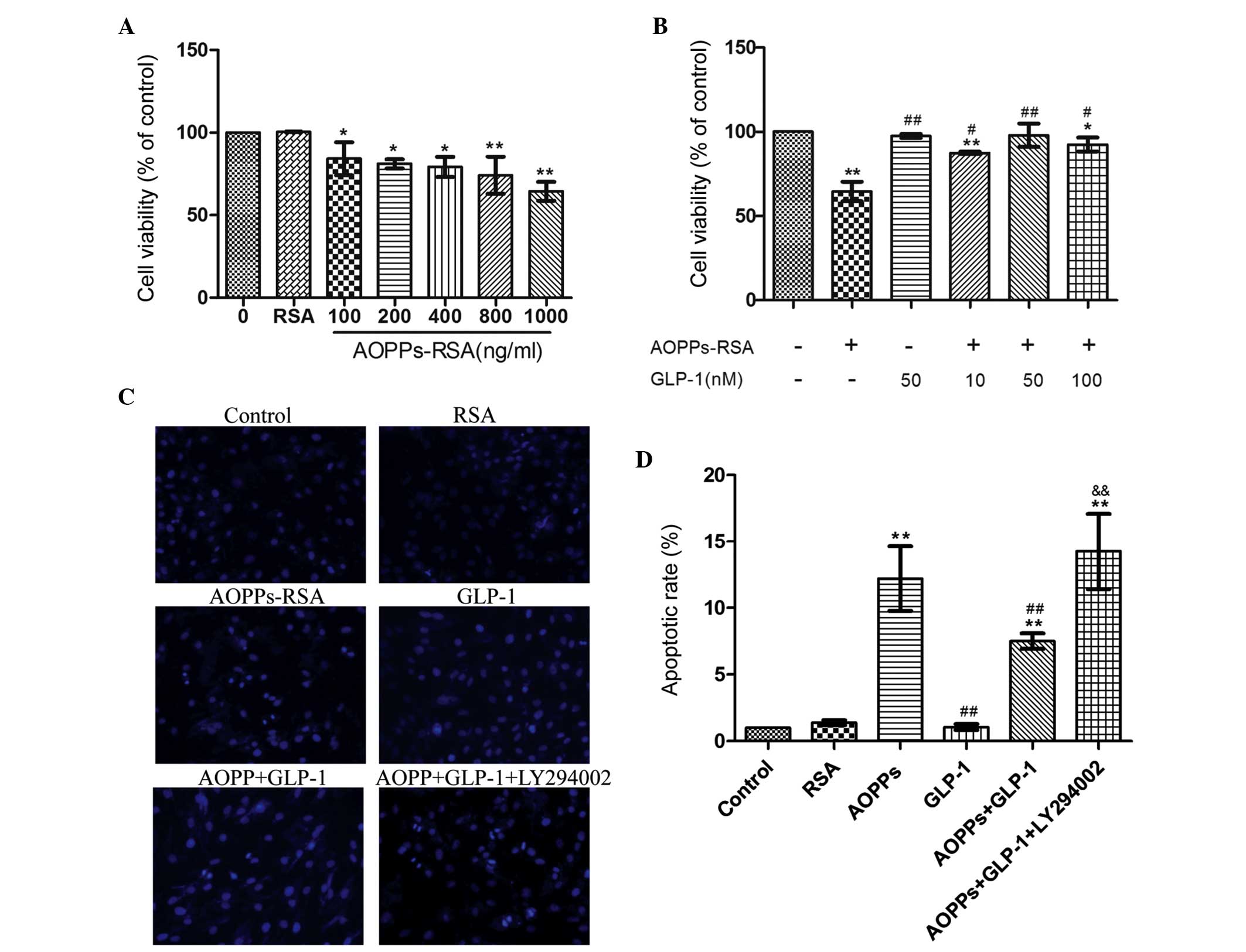 | Figure 1GLP-1 attenuated AOPPs-RSA-induced
toxicity and apoptosis in H9c2 cells. The cell viability,
morphological changes and apoptotic rates were assessed using a
cell counting kit-8 assay, Hoechst 33258 staining and flow
cytometry, respectively. (A) The cells were incubated with RSA (1
µg/ml), or increasing concentrations of AOPPs-RSA for 24 h.
AOPPs-RSA inhibited cell viability in a dose-dependent manner. (B)
The cells were incubated with 1 µg/ml AOPPs-RSA for 24 h in
the presence or absence of different concentrations of GLP-1. GLP-1
improved cell viability in a dose-dependent manner. (C) The
morphological changes in H9c2 cells were detected using a
fluorescence microscope. Chromatin condensation and nuclear
fragmentation represented apoptotic cells, while the diffuse blue
fluorescence represented normal cells (magnification, ×200). (D)
The apoptotic rate of each treatment was determined. The data are
expressed as the mean ± standard deviation of three independent
experiments (*P<0.05 and **P<0.01, vs.
control; #P<0.05 and ##P<0.01, vs.
AOPPs-treated cells; &&P<0.01, vs. AOPPs +
GLP-1-treated group). GLP-1, Glucagon-like peptide-1; AOPPs,
advanced oxidation protein products; RSA, rat serum albumin. |
GLP-1 attenuates AOPP-induced apoptosis
in H9c2 cells
The proapoptotic effect of AOPPs on H9c2 cells was
assessed. The morphological changes were detected by staining cells
with Hoechst 33258 (Fig. 1C). An
increase in the number of apoptotic cells was observed in the
AOPP-treated cells and a decrease in the AOPPs- and GLP-1-treated
cells. Following co-incubation with LY294002, the number of
apoptotic cells increased. As shown in Fig. 1D, cells incubated with AOPPs-RSA
exhibited a significant increase in the apoptotic rate (15.8%)
compared with the control group (1.3%; P<0.01). This increment
was blunted in the presence of GLP-1 (9.7%, P<0.01, vs. the
AOPPs group). The PI3K inhibitor, LY294002, was used to determine
the mechanisms behind the antiapoptotic effects of GLP-1. The
apoptotic rate of this group was significantly increased (18.4%,
P<0.01, vs. the AOPPs + GLP-1 group). These data indicated that
GLP-1 attenuates AOPP-RSA-induced apoptosis in H9c2 cells and this
effect may be associated with the PI3K pathway.
GLP-1 may protect AOPP-induced apoptosis
by down- regulating the expression of RAGE
Previously, AOPPs were confirmed to exert their
proapoptotic effect via RAGE (22). It was confirmed that the expression
of RAGE was upregulated by exposure to AOPP-RSA in a dose-dependent
manner (Fig. 2A) in H9c2 cells. In
addition, GLP-1 (50 nM for 24 h) significantly downregulated the
protein and mRNA expression levels of RAGE (Fig. 2B and C). Therefore, GLP-1 may
protect AOPP-induced apoptosis by downregulating the expression of
RAGE.
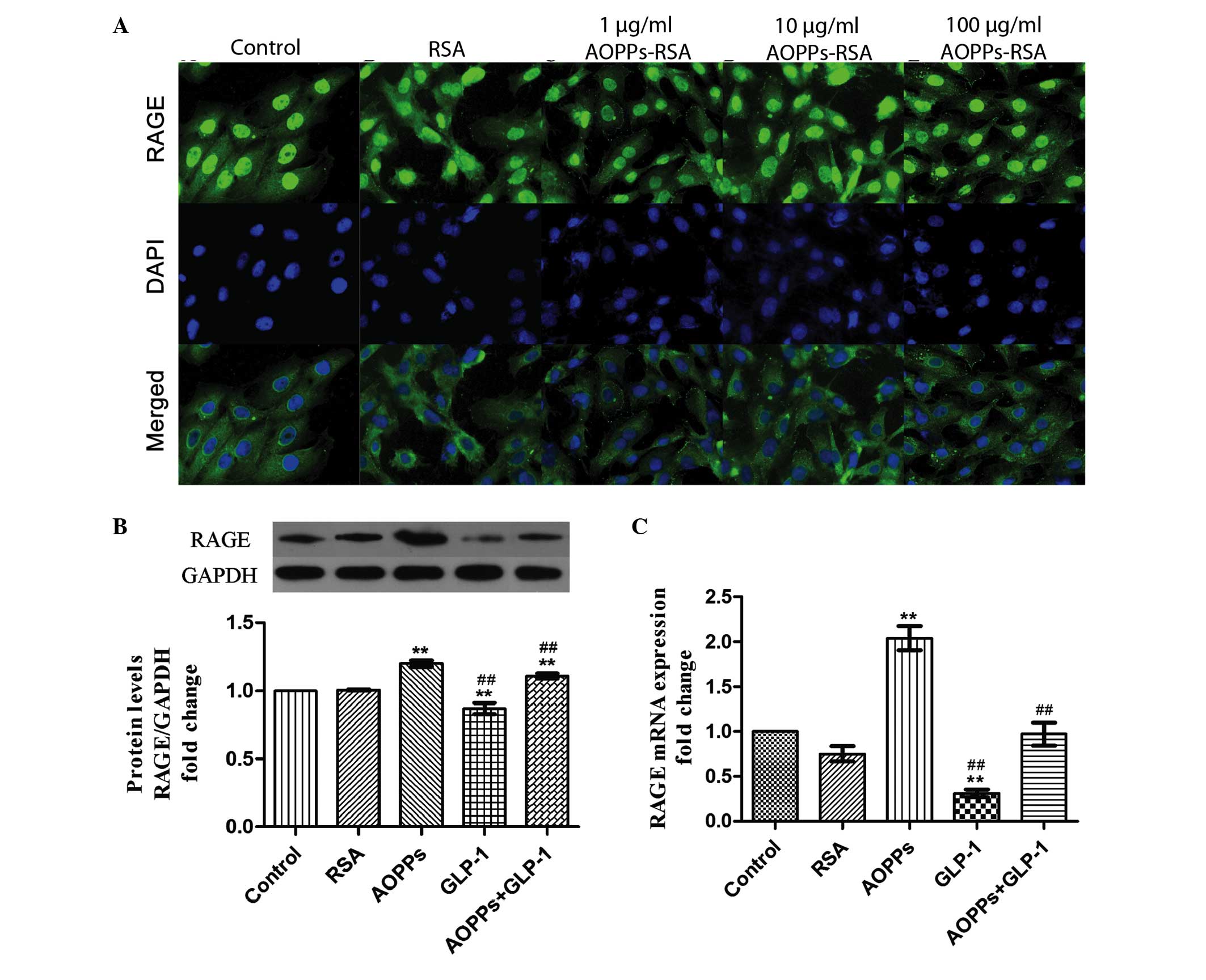 | Figure 2GLP-1 downregulates the expression of
RAGE in H9c2 cells. (A) H9c2 cells were immunostained with
anti-mouse RAGE primary antibody and fluorescein
isothiocyanate-conjugated secondary antibody (green), and were
counterstained with DAPI (blue). The expression of RAGE was
upregulated by exposure of increasing concentrations of AOPPs-RSA
for 24 h (magnification, ×400). (B) GLP-1 significantly decreased
the protein expression of RAGE, as determined by western blotting.
(C) GLP-1 significantly decreased the mRNA expression of RAGE as
determined by reverse transcription-quantitative polymerase chain
reaction. The data are expressed as the mean ± standard deviation
of three independent experiments (**P<0.01, vs.
control; ##P<0.01, vs. AOPPs-RSA-treated cells).
GLP-1, Glucagon-like peptide-1; AOPPs, advanced oxidation protein
products; RSA, rat serum albumin; RAGE, receptor for advanced
glycation end product; DAPI, 4′,6-diamidino-2-phenylindole. |
GLP-1 exerts its protective function via
the GLP-1R in H9c2 cells
The presence of the GLP-1R has been identified in
different cells and the interaction of GLP-1 with the GLP-1R has
been determined to be responsible for multiple function of GLP-1
(23). Therefore, the present
study confirmed that H9c2 cells express the GLP-1R (Fig. 3A). In addition, the mRNA and
protein expression levels of GLP-1R were upregulated by exposure to
GLP-1 itself (Fig. 3B and C).
Therefore, it was speculated that GLP-1 may exert its protective
function via the GLP-1R in H9c2 cells.
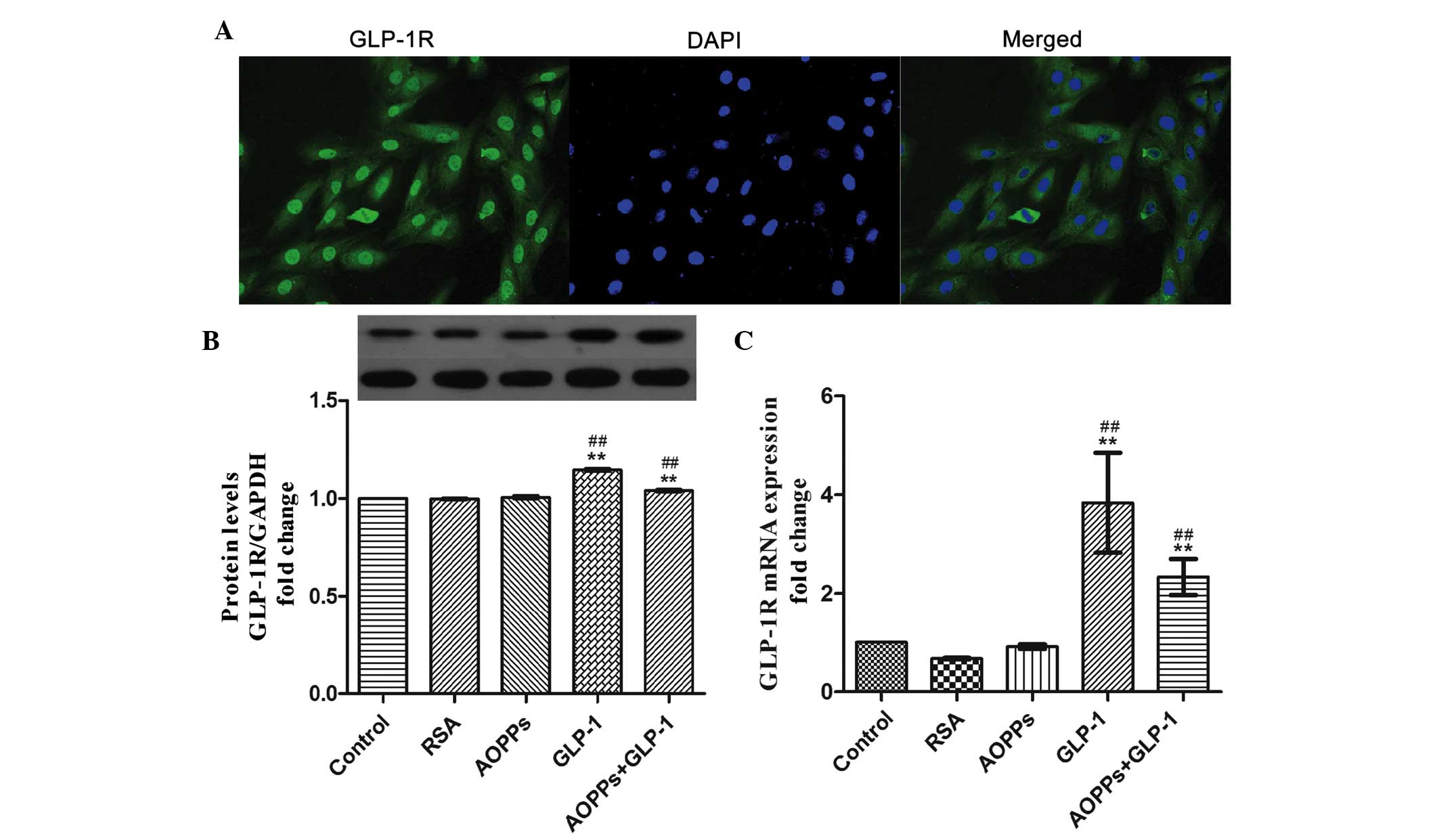 | Figure 3GLP-1 exerts its protective function
via the GLP-1 receptor in H9c2 cells. (A) H9c2 cells were
immunostained with anti-rabbit GLP-1R primary antibody and
fluorescein isothiocyanate-conjugated secondary antibody (green),
and counterstained with DAPI (blue; magnification, ×200). (B) GLP-1
significantly increased the expression of the GLP-1R, as determined
by western blot analysis. (C) The mRNA expression of GLP-1R was
increased following exposure to GLP-1 (50 nM) for 24 h, as measured
by reverse transcription-quantitative polymerase chain reaction.
The data are expressed as the mean ± standard deviation of three
independent experiments (**P<0.01, vs. control;
##P<0.01, vs. AOPPs-treated cells). GLP-1,
Glucagon-like peptide-1; GLP-1R, GLP-1 receptor; AOPPs, advanced
oxidation protein products; RSA, rat serum albumin; DAPI,
4′,6-diamidino-2-phenylindole. |
GLP-1 prevents AOPP-induced apoptosis via
the PI3K/Akt/Bad pathway in H9c2 cells
The role of the PI3K/Akt/Bad pathway and GLP-1 in
preventing AOPP-induced apoptosis in H9c2 cells was investigated.
Firstly, AOPP-RSA significantly inactivated Akt phosphorylation at
Ser473 (Fig. 4A) and subsequently
reduced Bad phosphorylation at Ser136 (Fig. 4B) compared with the control and RSA
group. In addition, a decrease of the anti-apoptotic protein, Bcl-2
(Fig. 4C), and an increase of the
proapoptotic protein, Bax (Fig.
4D), was demonstrated in AOPPs-RSA treated cells. In addition,
the elevated expression of caspase-3 was demonstrated by western
blotting (Fig. 4E).
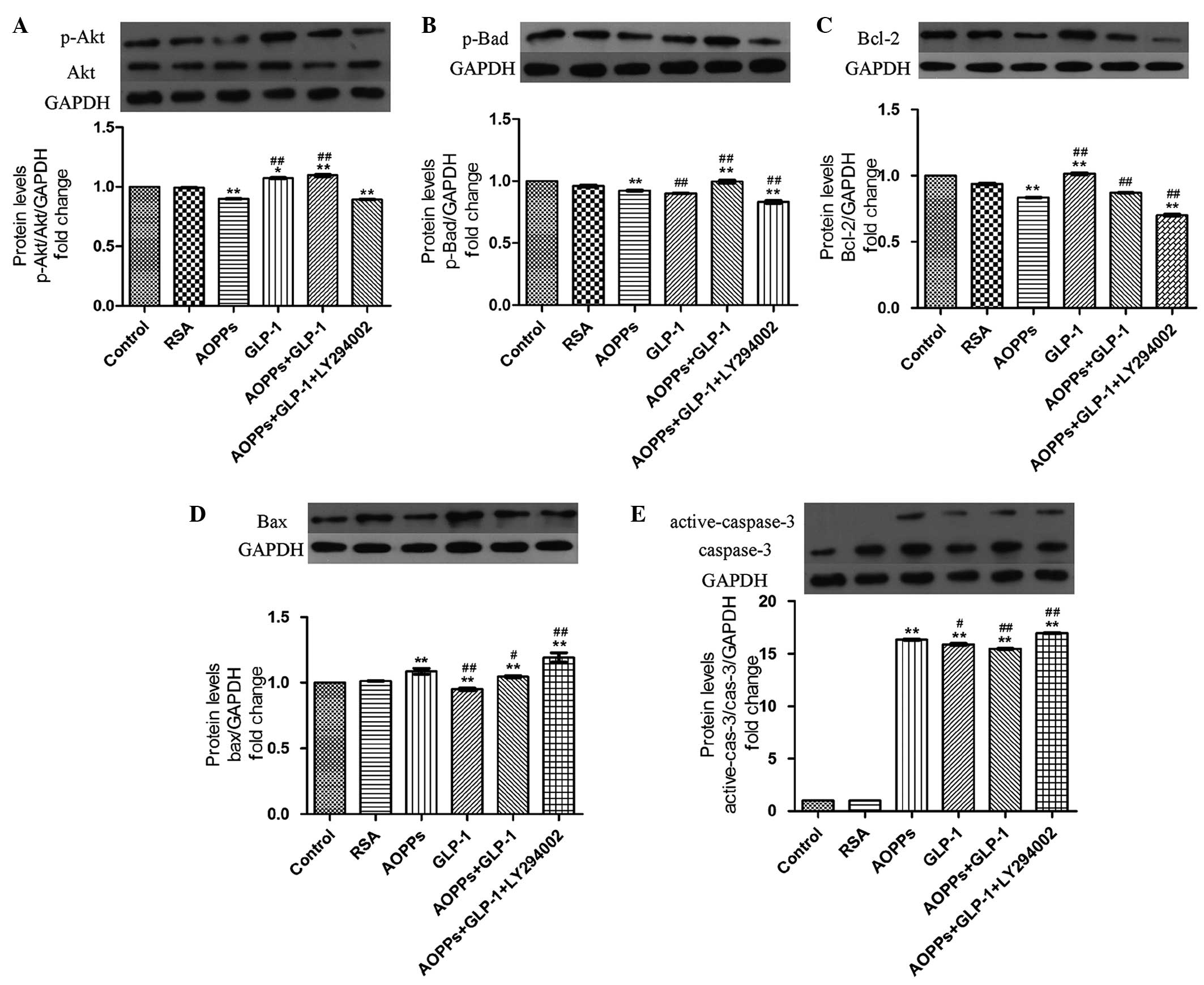 | Figure 4GLP-1 prevents AOPPs-induced
apoptosis via the PI3K/Akt/BAD pathway in H9c2 cells. The cells
were exposed to AOPPs-RSA (1 µg/ml), GLP-1 (50 nM) or
LY294002 (10 µM) for 24 h. The protein expression levels
were detected by western blotting. GLP-1 reversed AOPPs-RSA-induced
(A) p-Akt and (B) p-bad inactivation. LY294002 significantly
inhibited the effect of GLP-1. AOPPs-RSA decreased the expression
of (C) Bcl-2 and increased the expression of (D) Bax and (E)
activated caspase-3. GLP-1 reversed these effects, however, was
suppressed by LY294002. The data are expressed as the mean ±
standard deviation of three independent experiments
(*P<0.05 and **P<0.01, vs. control;
#P<0.05 and ##P<0.01, vs. AOPPSs-RSA
treated cells). GLP-1, Glucagon-like peptide-1; AOPPs, advanced
oxidation protein products; RSA, rat serum albumin; p-,
phosphorylated; Bcl, B-cell lymphoma; Bax, Bcl-2-associated X
protein; Bad, Bcl-2-associated death promoter. |
However, these changes were reversed in the presence
of GLP-1 (Fig. 4). Furthermore,
the effect of GLP-1 was abrogated following treatment of the H9c2
cells with a PI3K inhibitor, LY294002 (Fig. 4). These results indicated that
GLP-1 prevented AOPP-induced apoptosis via the PI3K/Akt/Bad pathway
in H9c2 cells.
Discussion
The major findings of the present study were as
follows: i) AOPPs induced apoptosis in H9c2 cells; ii) GLP-1
prevented AOPP-induced cell apoptosis; iii) GLP-1 exerted these
cardioprotective effects predominantly via the PI3K/Akt/Bad
pathway. This is the first study, to the best of our knowledge, to
confirm that GLP-1 protected against AOPP-induced apoptosis in H9c2
cells.
AOPPs, as novel markers of oxidative stress, have
been explored in several cell types. Previous studies have
investigated the proapoptotic effect of AOPPs on vascular
endothelial cells (12), podocytes
(13), intestine epithelial cells
(24) and rat osteoblast-like
cells (25). However, previous
studies, which focus on the effect of AOPPs in cardiomyocytes are
rare. Valente et al (26)
demonstrated that AOPPs induce neonatal and adult mouse
cardiomyocyte death via Nox2/Rac1/super-oxide-dependent
TRAF3IP2/JNK signaling. In the present study, it was revealed that
AOPP-treatment significantly increased the apoptotic rate of a rat
ventricular myoblast cell line, H9c2. In addition, AOPPs decreased
the phosphorylation of Akt and subsequently reduced the
phosphorylation of Bad. This data suggested that AOPPs induce H9c2
cell apoptosis and that the Akt/Bad pathway may be involved.
Cardiomyocyte apoptosis is a major event in the pathogenesis of DCM
(27,28). In addition, DM is associated with
an increased production of AOPPs (8,11,29,30).
In this regard, the present study hypothesized that AOPP-induced
apoptosis may be involved in the development of DCM. Since adult
cardiomyocytes possess a finite capacity to proliferate, the loss
of cardiomyocytes results in cardiac dysfunction and heart failure
(31). Therefore, suppression of
cardiomyocyte apoptosis is a crucial strategy for the prevention of
DCM.
GLP-1 has been widely investigated in recent years.
Numerous studies have confirmed that GLP-1 has anti-apoptotic
functions in different types of cell, including pancreatic β cells
(32), cholangiocytes (33), neurons (34) and cardiomyocytes (35). Consistent with previous reports,
the present study indicated that GLP-1 (7–36)
inhibits AOPP-induced toxicity and apoptosis in H9c2 cells. The
present study next investigated the mechanisms of this
antiapoptotic effect of GLP-1.
Cytoprotective actions of GLP-1 have been
substantiated in the heart. Notably, GLP-1 directly interacts with
the myocardium due to the presence of the receptor of the GLP-1R
(36,37). The present results demonstrated
that the GLP-1R was expressed in H9c2 cells. Therefore, it is
possible that GLP-1 can exhibit protective effect on H9c2 cells.
AOPPs and AGEs are similar in structure and biological activity.
AOPPs are demonstrated to induce cardiomyocyte death by interacting
with a member of the immunoglobulin superfamily of cell surface
molecules, RAGE (26). In the
present study, it was confirmed that the expression of RAGE
occurred in H9c2 cells and was upregulated following exposure to
increasing concentration of AOPPs-RSA. In addition, it was revealed
that GLP-1 downregulated not only the mRNA expression of RAGE, but
also the protein expression. These data may partly explain why
GLP-1 can inhibit AOPP-induced apoptosis in H9c2 cells.
The mechanisms underlying the anti-apoptotic effect
of GLP-1 on the heart have been investigated by in vivo and
in vitro experiments. For instance, in murine HL-1
cardiomyocytes, GLP-1 prevents the exposure of phosphatidylserine,
the increase of the Bax:Bcl-2 ratio, the activation of Bad,
mitochondrial membrane depolarization, the release of cytochrome
c, the activation of caspase-3 and DNA fragmentation,
induced by staurosporine (35).
GLP-1R agonist, exendin-4, attenuates high glucose-induced
cardiomyocyte apoptosis in association with decreased endoplasmic
reticulum stress and markers of enhanced SERCA2a activity (38). Furthermore, exenatide increased the
expression levels of p-Akt and Bcl-2, decreased the activity of
caspase-3, reduced infarct size, and prevented deterioration of
systolic and diastolic cardiac function in a porcine model of
myocardial ischaemia/reperfusion (39). However, the mechanisms of GLP-1 on
the AOPP-induced apoptosis remain to be elucidated. Therefore, the
present study investigated the PI3K/Akt pathway as a candidate for
the underlying mechanisms.
It has been reported that GLP-1 and its analogues
inhibit cardiomyocyte apoptosis by regulating numerous
apoptotic-associated pathways, including the PI3K (40), cAMP (23) and ERK1/2 pathways (41). The present study demonstrated that
when treated with a PI3K inhibitor, LY294002, the apoptotic rate of
GLP-1 and AOPPs co-treated cells was increased, indicating that
GLP-1 requires activation of the PI3K pathway to prevent
AOPP-induced apoptosis in H9c2 cells. PI3K is a type of cellular
protein kinase, which is involved in cell survival, growth and
proliferation. Activated PI3K subsequently phosphorylated its
downstream Ser/Thr protein kinase, Akt, also known as protein
kinase B. The activation of the Akt pathway provides cells with a
survival signal, which allows them to withstand apoptotic stimuli
(42). Numerous reports elucidated
the important roles of activating the PI3K/Akt signaling pathway in
competing cell apoptosis in the heart (43–45).
Bad, a Bcl-2 family member, can combine with antiapoptotic factor
Bcl-2 or Bcl-xL to form a proapoptotic complex. However, when Bad
is phosphorylated at Ser136 by Akt, it is released from the
proapoptotic complex and forms a complex with 14-3-3 proteins in
the cytosol, therefore inactivating its proapoptotic function
(46). Bad is hypothesized to be a
downstream target of Akt in promoting cell survival (47). The present data revealed that
AOPP-treatment inactivated Akt and Bad phosphorylation, while GLP-1
restored this suppression. These effects of GLP-1 were correlated
with the attenuation of cell apoptosis. However, LY294002 abolished
these effects of GLP-1. Therefore, the antiapoptotic effects of
GLP-1 were associated with, at least in part, activation of the
PI3K/Akt/Bad pathway.
Apoptosis is the process of programmed cell death,
which is regulated through the balance between proapoptotic and
antiapoptotic proteins. The Bcl-2 family, as the key regulators of
apoptosis, consists of both proapoptotic proteins, including Bax
and Bad, and antiapoptotic members, including Bcl-2 and Bcl-xL. The
proapoptotic protein Bax is necessary for mitochondrial outer
membrane permeabilization, inducing cytochrome c release and
leading to the activation of caspases (48). However, Bcl-2 inhibits this process
by suppressing the translocation of Bax and therefore, reducing the
activity of the caspases (49).
Caspase-3 is a type of proteinase, which has a central role in the
execution-phase of cell apoptosis only when it has been activated.
Activation of caspase-3 requires proteolytic processing of its
inactive zymogen into activated p17 and p12 fragments. Consistent
with previous studies (35,50),
the present study demonstrated that AOPPs decreased the expression
of Bcl-2, and increased the expression of Bax and activation of
caspase-3 in H9c2 cells. GLP-1 reversed these changes.
Nevertheless, the effects of GLP-1 were partly abrogated by
co-incubation with the PI3K inhibitor, LY294002. These findings
suggested that the mechanism by which GLP-1 protects H9c2 cells
against the apoptotic effects of AOPPs is by shifting the balance
between proapoptotic and antiapoptotic proteins via the
PI3K/Akt/Bad pathway.
The prominent finding of the present study was that
GLP-1 protected cardiomyocytes against AOPP-induced apoptosis. The
possible mechanism may be associated with the PI3K/Akt/Bad survival
pathway. Currently, no single effective treatment exists for DCM.
The present findings provided a conceivable mechanism of the
development of DCM and rendered a novel application of GLP-1
exerting favorable cardiac effects for the treatment of DCM.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81270966) and the
Natural Science Foundation of Guangdong Province, China (no.
S2012010009494). The authors would like to thank Dr Yulin Liao
(Department of Cardiology, Nanfang Hospital, Guangdong, P.R. China)
for generously providing the H9c2 cells. The authors appreciate the
technical support from Guangdong Provincial Key Laboratory of
Malignant Tumor Epigenetics and Gene regulation (Sun Yat-Sen
Memorial Hospital, Sun Yat-Sen University, Guangdong, China).
References
|
1
|
Chen L, Magliano DJ and Zimmet PZ: The
worldwide epidemiology of type 2 diabetes mellitus-present and
future perspectives. Nat Rev Endocrinol. 8:228–236. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Battiprolu PK, Lopez-Crisosto C, Wang ZV,
Nemchenko A, Lavandero S and Hill JA: Diabetic cardiomyopathy and
metabolic remodeling of the heart. Life Sci. 92:609–615. 2013.
View Article : Google Scholar :
|
|
3
|
Schilling JD and Mann DL: Diabetic
cardiomyopathy: Bench to bedside. Heart Fail Clin. 8:619–631. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuethe F, Sigusch HH, Bornstein SR, Hilbig
K, Kamvissi V and Figulla HR: Apoptosis in patients with dilated
cardiomyopathy and diabetes: A feature of diabetic cardiomyopathy?
Horm Metab Res. 39:672–676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fiordaliso F, Li B, Latini R, Sonnenblick
EH, Anversa P, Leri A and Kajstura J: Myocyte death in
streptozotocin-induced diabetes in rats in angiotensin
II-dependent. Lab Invest. 80:513–527. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baraka A and AbdelGawad H: Targeting
apoptosis in the heart of streptozotocin-induced diabetic rats. J
Cardiovasc Pharmacol Ther. 15:175–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Witko-Sarsat V, Friedlander M,
Capeillère-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers
P and Descamps-Latscha B: Advanced oxidation protein products as a
novel marker of oxidative stress in uremia. Kidney Int.
49:1304–1313. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalousová M, Skrha J and Zima T: Advanced
glycation end-products and advanced oxidation protein products in
patients with diabetes mellitus. Physiol Res. 51:597–604. 2002.
|
|
9
|
Kaneda H, Taguchi J, Ogasawara K, Aizawa T
and Ohno M: Increased level of advanced oxidation protein products
in patients with coronary artery disease. Atherosclerosis.
162:221–225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Atabek ME, Keskin M, Yazici C, Kendirci M,
Hatipoglu N, Koklu E and Kurtoglu S: Protein oxidation in obesity
and insulin resistance. Eur J Pediatr. 165:753–756. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tabak O, Gelisgen R, Erman H, Erdenen F,
Muderrisoglu C, Aral H and Uzun H: Oxidative lipid, protein and DNA
damage as oxidative stress markers in vascular complications of
diabetes mellitus. Clin Invest Med. 34:E163–E171. 2011.PubMed/NCBI
|
|
12
|
Guo ZJ, Niu HX, Hou FF, Zhang L, Fu N,
Nagai R, Lu X, Chen BH, Shan YX and Tian JW: Advanced oxidation
protein products activate vascular endothelial cells via a
RAGE-mediated signaling pathway. Antioxid Redox Signal.
10:1699–1712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou LL, Hou FF, Wang GB, Yang F, Xie D,
Wang YP and Tian JW: Accumulation of advanced oxidation protein
products induces podocyte apoptosis and deletion through
NADPH-dependent mechanisms. Kidney Int. 76:1148–1160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gaspari T, Liu H, Welungoda I, Hu Y,
Widdop RE, Knudsen LB, Simpson RW and Dear AE: A GLP-1 receptor
agonist liraglutide inhibits endothelial cell dysfunction and
vascular adhesion molecule expression in an ApoE-/-mouse model.
Diab Vasc Dis Res. 8:117–124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lønborg J, Vejlstrup N, Kelbæk H, Bøtker
HE, Kim WY, Mathiasen AB, Jørgensen E, Helqvist S, Saunamäki K,
Clemmensen P, et al: Exenatide reduces reperfusion injury in
patients with ST-segment elevation myocardial infarction. Eur Heart
J. 33:1491–1499. 2012. View Article : Google Scholar
|
|
16
|
Kieffer TJ and Habener JF: The
glucagon-like peptides. Endocr Rev. 20:876–913. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tomas E and Habener JF: Insulin-like
actions of glucagon-like peptide-1: A dual receptor hypothesis.
Trends Endocrinol Metab. 21:59–67. 2010. View Article : Google Scholar
|
|
18
|
Liu Q, Anderson C, Broyde A, Polizzi C,
Fernandez R, Baron A and Parkes DG: Glucagon-like peptide-1 and the
exenatide analogue AC3174 improve cardiac function, cardiac
remodeling, and survival in rats with chronic heart failure.
Cardiovasc Diabetol. 9:762010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie Y, Wang SX, Sha WW, Zhou X, Wang WL,
Han LP, Li DQ and Yu DM: Effects and mechanism of glucagon-like
peptide-1 on injury of rats cardiomyocytes induced by
hypoxia-reoxygenation. Chin Med J (Engl). 121:2134–2138. 2008.
|
|
20
|
Zhan Y, Sun HL, Chen H, Zhang H, Sun J,
Zhang Z and Cai DH: Glucagon-like peptide-1 (GLP-1) protects
vascular endothelial cells against advanced glycation end products
(AGEs)-induced apoptosis. Med Sci Monit. 18:BR286–BR291. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Newman RA, Hacker MP and Krakoff IH:
Amelioration of adriamycin and daunorubicin myocardial toxicity by
adenosine. Cancer Res. 41:3483–3488. 1981.PubMed/NCBI
|
|
22
|
Zhou LL, Cao W, Xie C, Tian J, Zhou Z,
Zhou Q, Zhu P, Li A, Liu Y, Miyata T, et al: The receptor of
advanced glycation end products plays a central role in advanced
oxidation protein products-induced podocyte apoptosis. Kidney Int.
82:759–770. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ravassa S, Zudaire A and Díez J: GLP-1 and
cardioprotection: From bench to bedside. Cardiovasc Res.
94:316–323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie F, Sun S, Xu A, Zheng S, Xue M, Wu P,
Zeng JH and Bai L: Advanced oxidation protein products induce
intestine epithelial cell death through a redox-dependent, c-jun
N-terminal kinase and poly (ADP-ribose) polymerase-1-mediated
pathway. Cell Death Dis. 5:e10062014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhong ZM, Bai L and Chen JT: Advanced
oxidation protein products inhibit proliferation and
differentiation of rat osteoblast-like cells via NF-kappaB pathway.
Cell Physiol Biochem. 24:105–114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Valente AJ, Yoshida T, Clark RA,
Delafontaine P, Siebenlist U and Chandrasekar B: Advanced oxidation
protein products induce cardiomyocyte death via
Nox2/Rac1/superoxide-dependent TRAF3IP2/JNK signaling. Free Radic
Biol Med. 60:125–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cai L, Wang Y, Zhou G, Chen T, Song Y, Li
X and Kang YJ: Attenuation by metallothionein of early cardiac cell
death via suppression of mitochondrial oxidative stress results in
a prevention of diabetic cardiomyopathy. J Am Coll Cardiol.
48:1688–1697. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuethe F, Sigusch HH, Bornstein SR, Hilbig
K, Kamvissi V and Figulla HR: Apoptosis in patients with dilated
cardiomyopathy and diabetes: a feature of diabetic cardiomyopathy?
Horm Metab Res. 39:672–676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Piwowar A, Knapik-Kordecka M and Warwas M:
Markers of oxidative protein damage in plasma and urine of type 2
diabetic patients. Br J Biomed Sci. 66:194–199. 2009.
|
|
30
|
Piwowar A, Knapik-Kordecka M and Warwas M:
AOPP and its relations with selected markers of
oxidative/antioxidative system in type 2 diabetes mellitus.
Diabetes Res Clin Pract. 77:188–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Narula J, Arbustini E, Chandrashekhar Y
and Schwaiger M: Apoptosis and the systolic dysfunction in
congestive heart failure. Story of apoptosis interruptus and zombie
myocytes. Cardiol Clin. 19:113–126. 2001. View Article : Google Scholar
|
|
32
|
Puddu A, Storace D, Durante A, Odetti P
and Viviani GL: Glucagon-like peptide-1 counteracts the detrimental
effects of advanced glycation end-products in the pancreatic beta
cell line HIT-T 15. Biochem Biophys Res Commun. 398:462–466. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marzioni M, Alpini G, Saccomanno S,
Candelaresi C, Venter J, Rychlicki C, Fava G, Francis H, Trozzi L
and Benedetti A: Exendin-4, a glucagon-like peptide 1 receptor
agonist, protects cholangiocytes from apoptosis. Gut. 58:990–997.
2009. View Article : Google Scholar :
|
|
34
|
Harkavyi A and Whitton PS: Glucagon-like
peptide 1 receptor stimulation as a means of neuroprotection. Br J
Pharmacol. 159:495–501. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ravassa S, Zudaire A, Carr RD and Diez J:
Antiapoptotic effects of GLP-1 in murine HL-1 cardiomyocytes. Am J
Physiol Heart Circ Physiol. 300:H1361–H1372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS,
Drucker DJ and Husain M: Cardioprotective and vasodilatory actions
of glucagon-like peptide 1 receptor are mediated through both
glucagon-like peptide 1 receptor-dependent and -independent
pathways. Circulation. 117:2340–2350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bullock BP, Heller RS and Habener JF:
Tissue distribution of messenger ribonucleic acid encoding the rat
glucagon-like peptide-1 receptor. Endocrinology. 137:2968–2978.
1996.PubMed/NCBI
|
|
38
|
Younce CW, Burmeister MA and Ayala JE:
Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis
via inhibition of endoplasmic reticulum stress and activation of
SERCA2a. Am J Physiol Cell Physiol. 304:C508–C518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Timmers L, Henriques JP, de Kleijn DP,
Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek
JJ, Doevendans PA, et al: Exenatide reduces infarct size and
improves cardiac function in a porcine model of ischemia and
reperfusion injury. J Am Coll Cardiol. 53:501–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ban K, Kim KH, Cho CK, Sauvé M, Diamandis
EP, Backx PH, Drucker DJ and Husain M: Glucagon-like peptide
(GLP)-1 (9-36) amide-mediated cytoprotection is blocked by exendin
(9-39) yet does not require the known GLP-1 receptor.
Endocrinology. 151:1520–1531. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huisamen B, Genis A, Marais E and Lochner
A: Pre-treatment with a DPP-4 inhibitor is infarct sparing in
hearts from obese, pre-diabetic rats. Cardiovasc Drugs Ther.
25:13–20. 2011. View Article : Google Scholar
|
|
42
|
Yao R and Cooper GM: Requirement for
phosphati-dylinositol-3 kinase in the prevention of apoptosis by
nerve growth factor. Science. 267:2003–2006. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mullonkal CJ and Toledo-Pereyra LH: Akt in
ischemia and reperfusion. J Invest Surg. 20:195–203. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shao S, Nie M, Chen C, Chen X, Zhang M,
Yuan G, Yu X and Yang Y: Protective action of liraglutide in beta
cells under lipotoxic stress via PI3K/Akt/FoxO1 pathway. J Cell
Biochem. 115:1166–1175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chang G, Zhang P, Ye L, Lu K, Wang Y, Duan
Q, Zheng A, Qin S and Zhang D: Protective effects of sitagliptin on
myocardial injury and cardiac function in an ischemia/reperfusion
rat model. Eur J Pharmacol. 718:105–113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chipuk JE and Green DR: How do BCL-2
proteins induce mitochondrial outer membrane permeabilization?
Trends Cell Biol. 18:157–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dejean LM, Ryu SY, Martinez-Caballero S,
Teijido O, Peixoto PM and Kinnally KW: MAC and Bcl-2 family
proteins conspire in a deadly plot. Biochim Biophys Acta.
1797:1231–1238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cunha DA, Ladrière L, Ortis F,
Igoillo-Esteve M, Gurzov EN, Lupi R, Marchetti P, Eizirik DL and
Cnop M: Glucagon-like peptide-1 agonists protect pancreatic
beta-cells from lipotoxic endoplasmic reticulum stress through
upregulation of BiP and JunB. Diabetes. 58:2851–2862. 2009.
View Article : Google Scholar : PubMed/NCBI
|














