Introduction
Hepatocellular carcinoma (HCC) represents the second
most common cause of cancer-associated mortality worldwide due to
late diagnosis and poor treatment options (1). Studies have suggested that aberrant
regulation of cell death signaling pathways and loss of several
tumor suppressors, including apoptotic pathways and tumor
suppressor miRNAs (miRNAs/miRs), are involved in the genesis of
HCC. However, the molecular pathogenesis of HCC has largely
remained elusive (2,3). Although surgery and liver
transplantation are able to cure patients with early-stage HCC,
chemotherapy is the main treatment option for patients with
advanced or unresectable liver cancers (4).
Doxorubcin (DOX), also known as adriamycin, is
widely used as an anti-tumor drug. Its mechanisms of action include
the induction of apoptosis of tumor cells by intercalating into
their DNA to inhibit its transcription and initiate a DNA damage
response (5). DOX-based
chemotherapy is used against a wide range of cancer types,
including ovarian (6), breast
(7) and bladder cancer (8) as well as HCC (9). However, the clinical application of
DOX is limited due to cardiac toxicity and drug resistance
(10). Therefore, it is urgently
required to develop novel and efficient approaches to improve the
curative effects of DOX for HCC treatment.
The relevance of miRNAs in tumorigenesis and drug
resistance of cancer types, including HCC, has become increasingly
known (11,12). miRNAs are a class of small,
endogenous, non-coding, single-stranded RNAs of 19–25 nucleotides,
which either induce mRNA degradation or inhibit mRNA translation
via binding with imperfect complementary sequences within the
3′-untranslated region (3′-UTR) of the target mRNA (13). Aberrant expression of miRNAs is
associated with tumor development through regulation of their
respective target genes. For example, miR-218 was reported to
suppress the growth of esophageal squamous cell carcinoma by
regulating the PI3K/AKT/mTOR pathway (14). miR-17 and miR-106b are frequently
downregulated in the serum of gastric cancer patients, which may
therefore be utilized as tumor markers for the early diagnosis of
gastric cancer (15). Although
miR-101 has been demonstrated to suppress the growth of multiple
cancer types, including HCC (16–18),
its utilization as a target in chemotherapy has remained to be
validated. The present study aimed to assess the role of miR-101 in
HCC as well its ability to enhance the chemotherapeutic efficacy of
DOX. Furthermore, the underlying mechanism was elucidated by
identifying a direct target of miR-101 using bioinformatics
analysis and a luciferase reporter assay. The present study
indicated that the combination of forced overexpression of miR-101
enhanced the apoptosis-inducing effects of doxorubicin via myeloid
cell leukemia 1 (Mcl-1), which may represent a novel approach for
the treatment of HCC.
Materials and methods
Cell culture
The HepG2, Hep3B, Huh7 and PLC human HCC cell lines
and the L-O2 normal liver cell line were obtained from the
Institute of Biochemistry and Cell Biology, Chinese Academy of
Sciences (Shanghai, China) and cultured in Dulbecco's modified
Eagle's medium (DMEM) (Gibco; Thermo Fisher Scientific, Waltham,
MA, USA) with 10% fetal bovine serum (FBS; Gibco) at 37°C in a
humidified atmosphere containing 5% CO2.
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) analysis of miR-101 and Mcl-1
mRNA expression
Total RNA was isolated from cultured cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific) following the
manufacturer's instructions. For analysis of mature miR-101
expression, stem-loop RT-qPCR (19) was employed using Moloney murine
leukemia virus Reverse Transcriptase (Invitrogen) following the
manufacturer's instructions. The miR-101 RT primer (Ribobio,
Guangzhou, China) had the following sequence: 5′-CTC AAC TGG TGT
CGT GGA GTC GGC AAT TCA GTT GAG ATG TCATG-3′. Relative expression
was calculated using the 2−ΔΔCq method (20) and normalized to the expression of
U6 small RNA. Mcl-1 mRNA was also quantified by RT-qPCR using the
PrimeScript RT reagent kit (Takara Bio Inc., Otsu, Japan) for
reverse transcription and SYBR® Premix Ex Taq II (Takara
Bio, Inc.) for PCR amplification on an Applied Biosystems 7900HT
thermocycler (Thermo Fisher Scientific). The PCR reaction mix
consisted of 12.5 µl 2X SYBR® Premix Ex Taq II, 1
µl forward primer (10 µM), 1 µl reverse primer
(10 µM), 2 µl cDNA and 8.5 µl H2O.
PCR was performed under the following thermal cycling conditions:
95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C
for 30 sec, and one cycle of 95°C for 15 sec, 60°C for 60 sec and
95°C for 15 sec for dissociation. PCR primers were obtained from
Ribobio and had the following sequences: Mcl-1 forward, 5′-TGG CTA
AAC ACT TGA AGACC-3′ and reverse, 5′-GGA AGA ACT CCA CAA ACCC-3′;
β-actin forward, 5′-CAG AGC CTC GCC TTT GCC-3′ and reverse, 5′-GTC
GCC CAC ATA GGA ATC-3′. After the products were purified using the
MiniBEST DNA Fragment Purification kit (cat. no. 9761; Takara Bio,
Inc.) according to the manufacturer's instructions, the PCR
products were quantified by reading the absorbance at 260 nm PCR
products. The expression levels were normalized to β-actin.
Plasmid construction
Bioinformatics analysis using TargetScan (http://www.targetscan.org/) indicated that Mcl-1
represents a target gene of miR-101. To verify this, luciferase
reporter vectors containing a wild-type (W) or mutated (M) fragment
of the 3′-UTR of Mcl-1 were constructed. The 3′-UTR of Mcl-1 was
amplified by PCR using cDNA from HepG2 cells as a template. The
Mcl-1 3′UTR and open reading frame fragment were amplified by PCR.
The following primers were utilized: Mcl-1 3′UTR:
Mcl-1-SpeI, 5′-CGA CTA GTG CAA CAA ACA AAC TTT GTT TG-3′ and
Mcl-1-HindIII, 5′-CGA AGC TTG CAA AGT TCA AAA GGG TAT GA-3′;
Mcl-1 open reading frame: Mcl-1-HindIII, 5′-CGA AGC TTA TGT
TTG GCC TCAAAA GAA AC-3′ and Mcl-1 EcoRI, 5′-CGG AAT TCT GTC
TTA TTA GAT ATG CCA AAC CAG-3′. The reaction conditions were as
follows: 95°C for 5 min; 35 cycles of 95°C for 45 sec, 58°C for 45
sec and 72°C for 60 sec; and 72°C for 5 min. PCR products and the
vector were then digested with SpeI/HindIII or
HindIII/EcoRI (Takara Bio, Inc.). To ligate the Mcl-1
fragment into the vector, the digestion products were incubated
with T4 DNA Ligase (Takara Bio, Inc.). The resulting recombinant
plasmid was transformed into E. coli (DH5 strain; Tiangen
Co., Ltd., Beijing, China) for replication and then extracted from
E. coli using an EndoFree Plasmid Maxi kit (Qiagen, Hilden,
Germany). Total RNA was isolated from cultured cells using TRIzol
reagent, and the cDNA library was synthesized by using the
PrimeScript RT reagent kit (Takara Bio, Inc.) according to the
manufacturer's instructions. The obtained nucleotides were cloned
into the SpeI/HindIII site of the pMIR vector
(pMIR-REPORT™ miRNA Expression Reporter Vector System; Ambion;
Thermo Fisher Scientific). For cloning, 1 µl restriction
enzymes (Takara Bio, Inc.), 2 µl 10X buffer (Takara Bio,
Inc.) and 1 µg DNA were used with H2O added for a
total volume of 20 µl. The recombinant plasmid was named
pMIR-Mcl-1-W. The mutant plasmid, pMIR-Mcl-1-M, was created by
mutating the seed regions of the miR-101 binding sites (CAG UAC
UGUA) by using a site-directed mutagenesis kit (Takara Bio,
Inc.).
To construct an Mcl-1 overexpression vector, the
Mcl-1 open reading frame without the 3′-UTR was amplified by PCR
with HepG2 cDNA as a template and cloned into the
HindIII/EcoRI site of the pcDNA3.1 vector
(Invitrogen) with the respective restriction enzymes (Takara Bio,
Inc.) according to the above-mentioned procedure; the resulting
recombinant plasmid was named pcDNA-Mcl-1.
Transient transfection
HepG2 cells were seeded into 12-well plates and
transfected at ~80% confluence. The Firefly luciferase reporters
(pMIR-Mcl-1-W or pMIR-Mcl-1-M; 2 µg/ml), Renilla luciferase
pRL-TK vector (100 ng/ml; Promega Corp., Madison, WI, USA),
eukaryotic expression vector (pcDNA3.1 or pcDNA-Mcl-1; 2
µg/ml), RNA oligonucleotides [negative control
oligonucleotide (NCO; 5′-GUA CUA ACC UAU GUG AAA UAG-3′; Genepharma
Co., Shanghai, China), miR-101 mimics (5′-UAC AGU ACU GUG AUA ACU
GAA-3′; Genepharma Co.) or multidrug restistance 1 (MDR1) small
interfering (si)RNA (5′-GGA AAA GAA ACC AAC UGU CUU-3′; 50 nM;
Ribobio)], were transiently transfected into the HepG2 cells with
Lipofectamine 2000 reagent (Invitrogen) following the
manufacturer's instructions.
Luciferase assay
HepG2 cells were seeded into 48-well plates at 80%
cell confluence, followed by transfection with the Firefly
luciferase reporters pMIR plus the Renilla luciferase pRL-TK vector
in combination with miR-101-mimics and NCO using Lipofectamine
2000. Luciferase activity was measured 48 h after transfection by
using the Dual-Luciferase Reporter assay system (Promega Corp.)
according to the manufacturer's instructions. Firefly luciferase
activity was normalized to Renilla luciferase activity for each
transfected well.
Measurement of cell viability by
3-(4,5-dimethylthi-azol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT)
The effect of miR-101, DOX (Sigma-Aldrich, St.
Louis, MO, USA) and their combination on the viability of HepG2
cells was measured using an MTT assay. HepG2 cells were seeded in
triplicate in a 96-well plate at a density of 5×103 per
well. Following culture for 12 h, cells were transfected with
miR-101 or NCO. Following incubation for 24 h, cells were treated
with various concentrations of DOX (0.1, 0.25, 0.5, 1, 1.5 and 2
µg/ml) for 48 h. The inhibition of cell viability was
detected using the MTT assay as described previously using MTT and
dimethyl sulfoxide (Sigma-Aldrich) (21). The absorbance values of wells from
the various treatment groups at 570 nm were determined using a
SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA) and compared
with the absorbance of the control cells to calculate the relative
cell viability.
Doxorubicin accumulation
HepG2 cells were transfected with miR-101 mimics,
NCO or MDR1 siRNA for 24 h, and the cells were then treated with
0.25 µg/ml doxorubicin. After 2 h of incubation, the cells
were washed three times with phosphate-buffered saline and the mean
fluorescence intensity of intracellular DOX was determined using
flow cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ,
USA) at an excitation wavelength of 488 nm and an emission
wavelength of 575 nm.
Western blot analysis
Total cellular extracts were prepared with
radioimmunoprecipitation assay lysis buffer (Cell Signaling
Technology, Inc., Danvers, MA, USA). The protein concentration was
determined using the bicinchoninic acid assay (Pierce
Biotechnology, Inc., Rockford, IL, USA) and equal amounts of
protein (50 µg) were separated by 12.5% sodium dodecyl
sulfate polyacrylamide gel electrophoresis (Sigma-Aldrich).
Proteins were then transferred onto polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA) by electroblotting.
Non-specific binding was blocked using 5% (w/v) skimmed milk in
Tris-buffered saline with 1% Tween-20 (TBST) for 2 h at room
temperature. The membranes were then incubated with the primary
antibodies rabbit anti-β-actin monoclonal antibody (mAb) (cat. no.
4970; 1:1,000 dilution; Cell Signaling Technology, Inc.) and Mcl-1
rabbit mAb (cat. no. 5453; 1:1,000 dilution; Cell Signaling
Technology, Inc.) overnight at 4°C. Following several washes with
TBST, the membranes were incubated in horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G (cat no.
7074; 1:2,000 dilution; Cell Signaling Technology, Inc.) for 2 h at
room temperature and were then washed five times with TBST for 10
min each. Blots were visualized using an enhanced chemiluminescence
detection kit (cat. no. 32106; Pierce Biotechnology, Inc.) and
images were captured using Gel Doc™ EZ (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Measurement of apoptosis
DOX was added to the cell medium at a final
concentration of 0.25 µg/ml at 24 h after transfection with
miR-101 mimics. After 24 h of incubation, cells were collected,
stained with Annexin V/propidium iodide (PI) (Sigma-Aldrich, St.
Louis, MO, USA) for 15 min at room temperature according to the
manufacturer's instructions and analyzed using flow cytometry
(FACSCalibur).
Statistical analysis
Statistical analysis was performed using SPSS 15.0
software (SPSS, Inc., Chicago, IL, USA). Student's t-test and
analysis of variance were used to compare mean values. Values are
expressed as the mean ± standard deviation and derived from at
least three independent experiments. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
miR-101 is downregulated and Mcl-1 is
upregulated in HCC
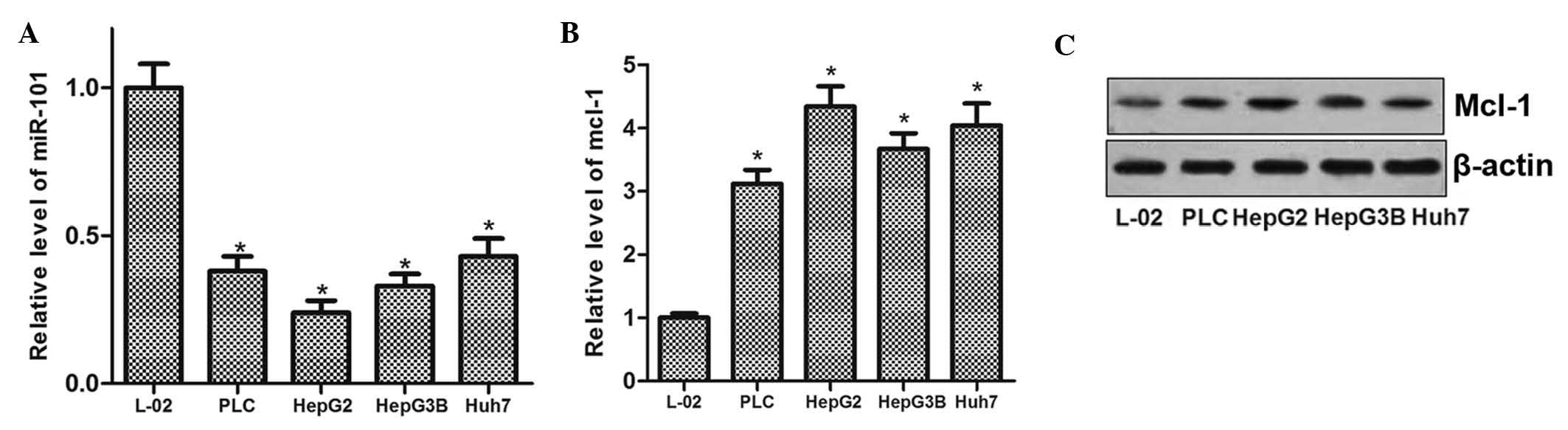
To evaluate the role of miR-101 in HCC cells as well
as any mechanistic association with Mcl-1, the expression levels of
miR-101 and Mcl-1 were determined in the L-O2 normal liver cell
line as well as in four HCC cell lines (PLC, HepG2, Hep3B and
Huh7). The results showed that miR-101 expression was reduced in
the human HCC cell lines compared with that in L-O2 cells (Fig. 1A). Conversely, Mcl-1 was
significantly upregulated in all four liver cancer cell lines at
the mRNA as well as at the protein level (Fig. 1B and C). These results suggested
that miR-101 exerts a tumor suppressor role in HCC, and that the
expression of Mcl-1 is involved in liver carcinogenesis.
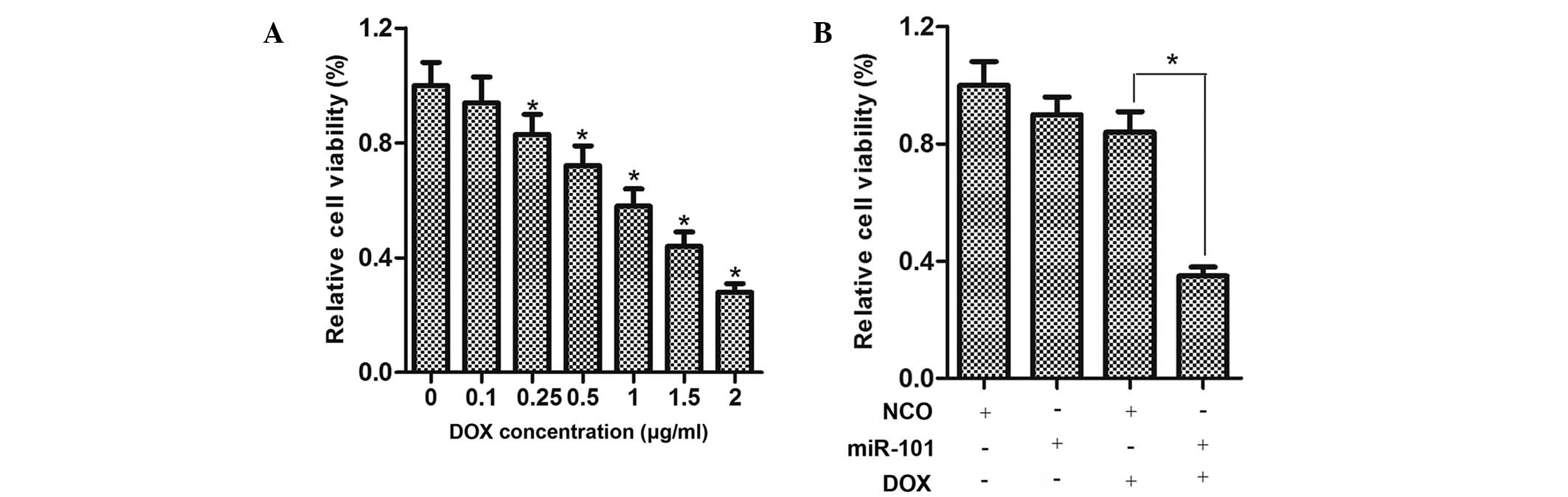
miR-101 sensitizes HCC cells to DOX
The viability of HepG2 cells treated with various
concentrations of DOX for 48 h was determined using an MTT assay,
demonstrating that DOX inhibited the cell viability in a
dose-dependent manner (Fig. 2A).
To examine the effects of miR-101 on the viability of HCC cells,
HepG2 cells were transfected with miR-101 mimics for 24 h and
treated with DOX at a moderate concentration (0.25 µg/ml)
for 48 h. As shown in Fig. 2B, the
viability of HepG2 cells treated with DOX plus miR-101 was
considerably lower compared to that of cells treated with DOX or
miR-101 mimics alone.
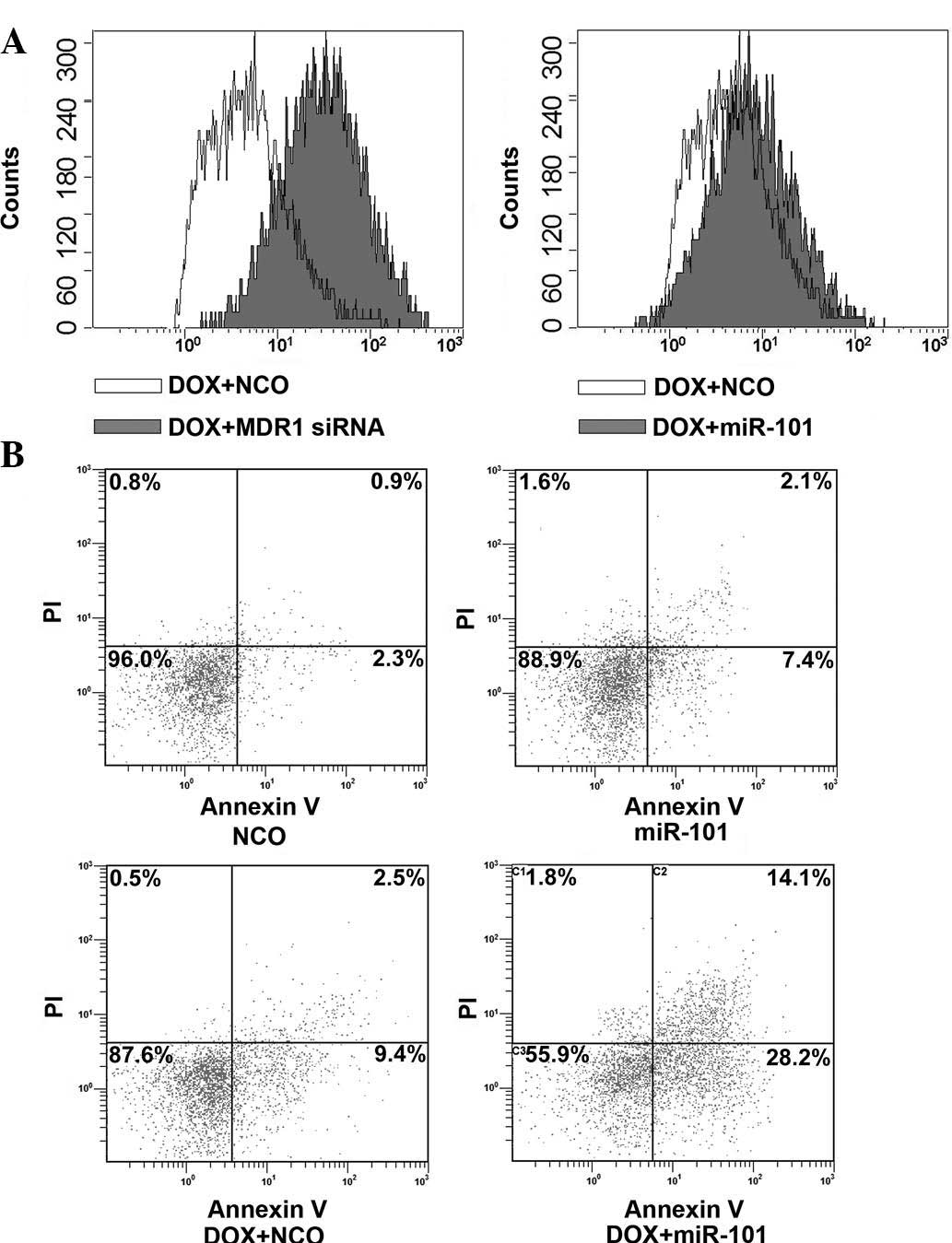
miR-101 does not affect the accumulation
of DOX in HCC cells
To determine the underlying mechanisms of
miR-101-mediated sensitization of HCC cells to DOX, the present
study assessed the effects of miR-101 mimics on DOX accumulation in
HepG2. Cells transfected with MDR1 (encoding P-glycoprotein) siRNA
to increase the accumulation of DOX (22) were used as the positive control. As
expected, transfection of MDR1 siRNA significantly increased the
uptake of DOX in HepG2 cells (Fig.
3A). However, the accumulation of DOX in HepG2 cells was not
influenced by transfection with miR-101 mimics, suggesting that a
mechanism other than the enhancement of drug accumulation is
responsible for the synergism of miR-101 and DOX.
miR-101 enhances apoptosis induction by
DOX
To further investigate the underlying mechanisms of
miR-101-mediated sensitization of HCC cells to DOX, the
apoptosis-inducing effects of miR-101, DOX and their combination
were assessed by Annexin V/PI staining and flow cytometric
analysis. As shown in Fig. 3B, a
marked increase of HepG2-cell apoptosis was observed upon combined
treatment with DOX and miR-101 compared with that in the single
treatment groups. These results suggested that the overexpression
of miR-101 sensitizes HCC cells to DOX through enhancing the
apoptotic rate.
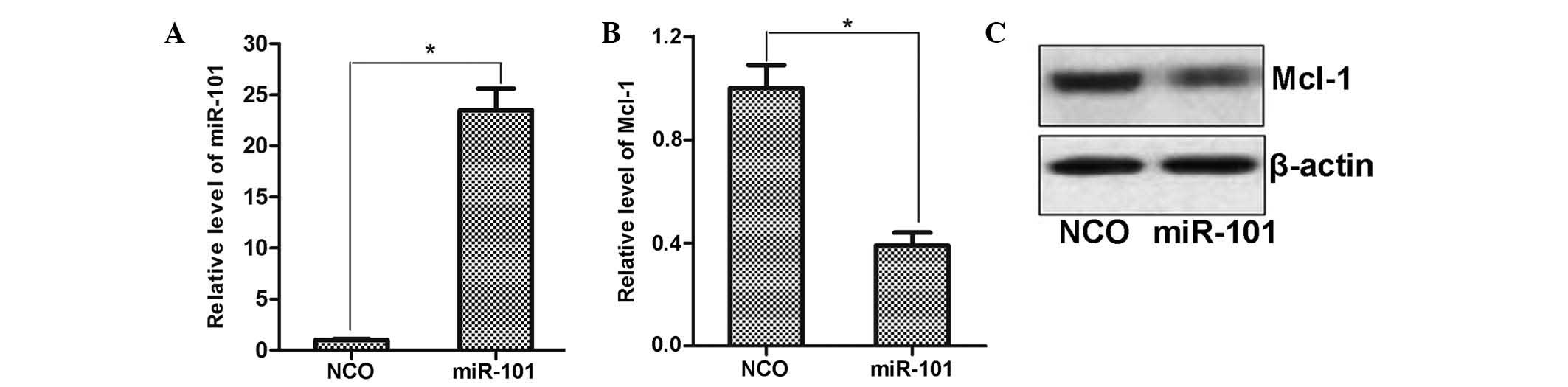
miR-101 negatively regulates Mcl-1
expression in HCC cells
To explore the regulatory mechanisms of miR-101, the
public miRNA database TargetScan was used for prediction of its
target genes, which suggested Mcl-1 as a possible target. To
determine the regulation of Mcl-1 expression by miR-101, miR-101
mimics were transfected into HepG2 cells and the effects on Mcl-1
expression were assessed by RT-qPCR and western blot analysis. As
shown in Fig. 4A, transfection
with miR-101 mimics significantly enhanced the levels of miR-101 in
HepG2 cells compared to those in the NCO-transfected group.
Furthermore, consistent with the bioinformatics prediction,
ectopically expressed miR-101 decreased the expression of Mcl-1 at
the mRNA as well as at the protein level (Fig. 4B and C). This result suggested that
Mcl-1 expression is regulated by miR-101 in HCC cells.
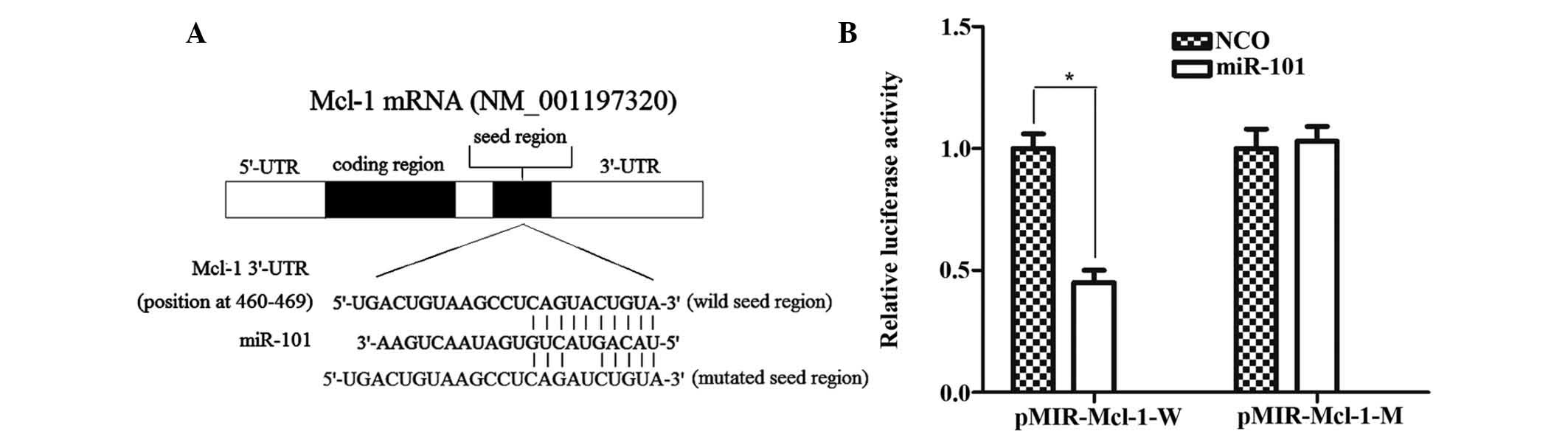
Mcl-1 is a direct target of miR-101
As illustrated in Fig.
5A, Mcl-1 contains a putative miR-101 target site in its 3′-UTR
(5′-CAG UAC UGUA-3′; nucleotides 460-469 of Mcl-1 mRNA 3′-UTR) and
is therefore a potential target of miR-101. To confirm whether
miR-101 directly regulates Mcl-1 expression, the target sequence of
the Mcl-1 3′-UTR or a mutant sequence was cloned into the pMIR
luciferase reporter vector, which was then respectively transfected
into HepG2 cells in combination with miR-101 mimics. The results
showed that co-transfection with miR-101 significantly suppressed
the luciferase activity of the reporter plasmid containing the
wild-type 3′-UTR of Mcl-1 compared with transfection with NCO,
while the luciferase activity of the reporter plasmid carrying the
mutant 3′-UTR remained unaffected by the miR-101 mimics (Fig. 5B). All these results confirmed that
Mcl-1 is a direct target of miR-101 in HepG2 cells.
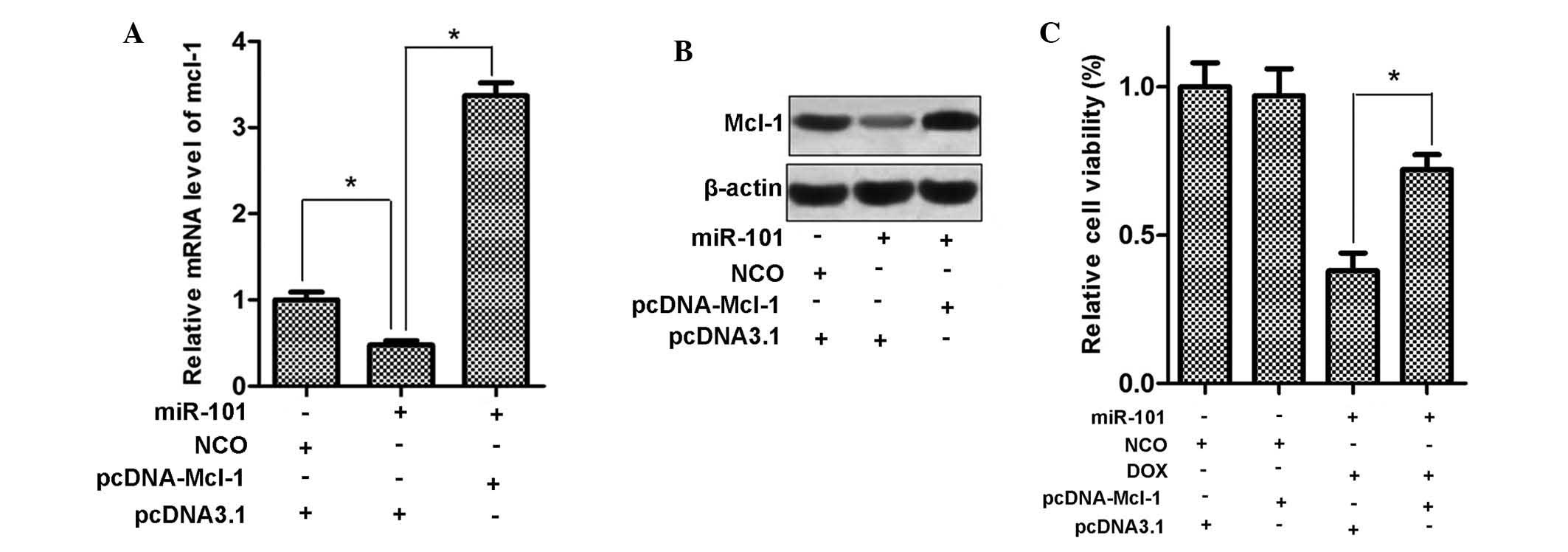
Mcl-1 mediates the sensitization of HCC
cells to DOX by miR-101
Since miR-101 sensitized HepG2 cells to DOX and
Mcl-1 is a direct target of miR-101, the present study examined
whether eukaryotic expression vector-mediated upregulation of Mcl-1
reverses the cytotoxic effects of DOX plus miR-101 in HepG2 cells.
First, HepG2 cells were transfected with pcDNA3.1 carrying with the
open reading frames of the Mcl-1 gene, and Mcl-1 expression was
evaluated at the mRNA and protein level. As shown in Fig. 6A and B, transfection with
pcDNA-Mcl-1 significantly increased the expression of Mcl-1 in
HepG2 cells, even in the presence of miR-101 mimics. Furthermore,
as expected, exogenous expression of Mcl-1 significantly decreased
the cytotoxicity of DOX combined with miR-101 in HepG2 cells
(Fig. 6C). These results supported
the notion that Mcl-1 is involved in the sensitization of HCC cells
to doxorubicin by miR-101.
Discussion
It has been demonstrated that miRNAs are involved in
carcinogenesis through regulating the expression of a large variety
of genes, including oncogenes and tumor suppressor genes (23). It has been reported that
dysregulation of miR-101 is associated with tumorigenesis,
development and invasion, which is essential in multiple cancer
types, including bladder cancer (24) and human hepatocellular carcinoma
(25,26). Furthermore, it has bee reported
that overexpression of miR-101 induced apoptosis in gastric cancer
cells (27). The present study
demonstrated that the expression of miR-101 was reduced in four
human HCC cell lines compared with that in a normal liver cell
line, illustrating that miR-101 acts as a tumor suppressor in liver
cancer. By contrast, Mcl-1 was significantly upregulated in all
four liver cancer cell lines. These results inferred that miR-101
and Mcl-1 are inversely correlated in HCC.
Mcl-1 is a key anti-apoptotic protein of the B-cell
lymphoma 2 family, which controls apoptosis (28). In addition, Mcl-1 has been
implicated in resistance to chemotherapy in multiple cancer types.
Sun et al (29)
demonstrated that siRNA-mediated inhibition of Mcl-1 effectively
increased the sensitivity of human colon cancer cells to
ABT-737-induced apoptosis, while apoptosis was significantly
attenuated by enforced expression of Mcl-1. As high expression of
Mcl-1 contributes to the ability of cancer cells to survive and
evade apoptosis induced by chemotherapeutic drugs, high levels of
Mcl-1 protein have been correlated with poor prognosis of various
cancer types (30,31).
The present study found that miR-101 significantly
increased the cytotoxic effects of DOX in HepG2 cells.
Flow-cytometric analysis revealed that the accumulation of DOX in
HepG2 cells was not influenced by miR-101, suggesting that other
pathways are responsible for the observed synergism of miR-101 and
DOX. It was then demonstrated that miR-101 enhanced the anti-cancer
effects of DOX by increasing its ability to induce apoptosis in
HepG2 cells. Bioinformatics analysis revealed that Mcl-1 was a
putative target of miR-101. Since downregulation of Mcl-1 is a
mechanism of apoptosis induction (32), the present study determined the
effects of miR-101 mimics on Mcl-1 in HepG2 cells, revealing that
upregulation of miR-101 expression significantly reduced Mcl-1
expression. Furthermore, a luciferase reporter assay proved that
miR-101 directly targeted the Mcl-1 3′-UTR. Of note, enforced
expression of Mcl-1 by transfection of pcDNA-Mcl-1 markedly reduced
the effects of miR-101 on the sensitivity of HCC cells to DOX,
suggesting that miR-101 enhances the sensitivity of HCC cells to
DOX via Mcl-1. It is therefore concluded that miR-101 is involved
in DOX resistance of HCC through regulating Mcl-1 expression.
In conclusion, the results of the present study
demonstrated that miR-101 was downregulated in HCC cell lines,
while its overexpression enhanced the sensitivity of HepG2 cells to
the chemotherapeutic agent DOX by facilitating apoptosis. Of note,
Mcl-1 was confirmed as a functional target of miR-101 in HCC,
demonstrating that miR-101 may enhance the sensitivity of cancer
cells by downregulating Mcl-1 expression. These results suggested
the potential application of miR-101 overexpression as an adjuvant
in DOX-based therapy.
Acknowledgments
The present study was supported by the Key
Laboratory of Cancer Prevention and Intervention, China National
Ministry of Education.
References
|
1
|
El-Serag Hb: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lyra-González I, Flores-Fong LE,
González-García I, Medina-Preciado D and Armendáriz-Borunda J:
MicroRNAs dysregulation in hepatocellular carcinoma: Insights in
genomic medicine. World J Hepatol. 7:1530–1540. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Z, Zhang X, Wang G and Zheng H: Role
of MicroRNAs in hepatocellular carcinoma. Hepat Mon. 14:e186722014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chung V: Systemic therapy for
hepatocellular carcinoma and cholangiocarcinoma. Surg Oncol Clin N
Am. 24:187–198. 2015. View Article : Google Scholar
|
|
5
|
Yang F, Teves SS, Kemp CJ and Henikoff S:
Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys
Acta. 1845:84–89. 2014.
|
|
6
|
Marczak A, Denel-Bobrowska M, Rogalska A,
Łukawska M and Oszczapowicz I: Cytotoxicity and induction of
apoptosis by formamidinodoxorubicins in comparison to doxorubicin
in human ovarian adenocarcinoma cells. Environ Toxicol Pharmacol.
39:369–383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu F, Wang F, Yang T, Sheng Y, Zhong T and
Chen Y: Differential drug resistance acquisition to doxorubicin and
paclitaxel in breast cancer cells. Cancer Cell Int. 14:5382014.
View Article : Google Scholar
|
|
8
|
Teply BA and Kim JJ: Systemic therapy for
bladder cancer-a medical oncologist's perspective. J Solid Tumors.
4:25–35. 2014. View Article : Google Scholar :
|
|
9
|
Zhao X, Chen Q, Liu W, Li Y, Tang H, Liu X
and Yang X: Codelivery of doxorubicin and curcumin with lipid
nanoparticles results in improved efficacy of chemotherapy in liver
cancer. Int J Nanomedicine. 10:257–270. 2014.
|
|
10
|
Shuhendler AJ, Prasad P, Zhang RX, Amini
MA, Sun M, Liu PP, Bristow RG, Rauth AM and Wu XY: Synergistic
nanoparticulate drug combination overcomes multidrug resistance,
increases efficacy and reduces cardiotoxicity in a
nonimmunocompromised breast tumor model. Mol Pharm. 11:2659–2674.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohtsuka M, Ling H, Doki Y, Mori M and
Calin GA: MicroRNA processing and human cancer. J Clin Med.
4:1651–1667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Donzelli S, Mori F, Biagioni F, Bellissimo
T, Pulito C, Muti P, Strano S and Blandino G: MicroRNAs: Short
non-coding players in cancer chemoresistance. Mol Cell Ther.
2:162014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: MicroRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tian H, Hou L, Xiong YM, Huang JX, She YJ,
Bi XB and Song XR: MiR-218 suppresses tumor growth and enhances the
chemosensitivity of esophageal squamous cell carcinoma to
cisplatin. Oncol Rep. 33:981–989. 2015.
|
|
15
|
Zeng Q, Jin C, Chen W, Xia F, Wang Q, Fan
F, Du J, Guo Y, Lin C, Yang K, et al: Downregulation of serum
miR-17 and miR-106b levels in gastric cancer and benign gastric
diseases. Chin J Cancer Res. 26:711–716. 2014.
|
|
16
|
Shen Q, Bae HJ, Eun JW, et al: MiR-101
functions as a tumor suppressor by directly targeting nemo-like
kinase in liver cancer. Cancer Lett. 344:204–211. 2014. View Article : Google Scholar
|
|
17
|
Lv P, Zhang P, Li X and Chen Y: Micro
ribonucleic acid (RNA)-101 inhibits cell proliferation and invasion
of lung cancer by regulating cyclooxygenase-2. Thorac Cancer.
6:778–784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li JT, Jia LT, Liu NN, Zhu XS, Liu QQ,
Wang XL, Yu F, Liu YL, Yang AG and Gao CF: MiRNA-101 inhibits
breast cancer growth and metastasis by targeting CX chemokine
receptor 7. Oncotarget. 6:30818–30830. 2015.PubMed/NCBI
|
|
19
|
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee
DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Tong SJ, Liu J, Wang X and Qu LX:
MicroRNA-181 promotes prostate cancer cell proliferation by
regulating DAX-1 expression. Exp Ther Med. 8:1296–1300.
2014.PubMed/NCBI
|
|
22
|
Misra R, Das M, Sahoo BS and Sahoo SK:
Reversal of multidrug resistance in vitro by co-delivery of MDR1
targeting siRNA and doxorubicin using a novel cationic poly
(lactide-co-glycolide) nanoformulation. Int J Pharm. 475:372–384.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takasaki S: Roles of microRNAs in cancers
and development. Methods Mol Biol. 1218:375–413. 2015. View Article : Google Scholar
|
|
24
|
Hu Z, Lin Y, Chen H, Mao Y, Wu J, Zhu Y,
Xu X, Xu X, Li S, Zheng X and Xie L: MicroRNA-101 suppresses
motility of bladder cancer cells by targeting c-Met. Biochem
Biophys Res Commun. 435:82–87. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chiang CW, Huang Y, Leong KW, Chen LC,
Chen HC, Chen SJ and Chou CK: PKCalpha mediated induction of
miR-101 in human hepatoma HepG2 cells. J Biomed Sci. 17:352010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Guo X, Xiong L, Kong X, Xu Y, Liu
C, Zou L, Li Z, Zhao J and Lin N: MicroRNA-101 suppresses
SOX9-dependent tumorigenicity and promotes favorable prognosis of
human hepatocellular carcinoma. FEBS Lett. 586:4362–4370. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang HJ, Ruan HJ, He XJ, et al:
MicroRNA-101 is downregulated in gastric cancer and involved in
cell migration and invasion. Eur J Cancer. 46:2295–2303. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Quinn BA, Dash R, Azab B, et al: Targeting
Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs.
20:1397–1411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun JG, Xiang J, Zeng XL, Li X, Wu P, Fung
KP and Liu FY: Clitocine induces apoptosis and enhances the
lethality of ABT-737 in human colon cancer cells by disrupting the
interaction of Mcl-1 and Bak. Cancer Lett. 355:253–263. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo L, Zhang T, Liu H, Lv T, Yuan D, Yao
Y, Lv Y and Song Y: MiR-101 and Mcl-1 in nonsmall-cell lung cancer:
Expression profile and clinical significance. Med Oncol.
29:1681–1686. 2012. View Article : Google Scholar
|
|
31
|
Zhang T, Zhao C, Luo L, Zhao H, Cheng J
and Xu F: The expression of Mcl-1 in human cervical cancer and its
clinical significance. Med Oncol. 29:1985–1991. 2012. View Article : Google Scholar
|
|
32
|
Doi K, Gowda K, Liu Q, Lin JM, Sung SS,
Dower C, Claxton D, Loughran TP Jr, Amin S and Wang HG: Pyoluteorin
derivatives induce Mcl-1 degradation and apoptosis in hematological
cancer cells. Cancer Biol Ther. 15:1688–1699. 2014. View Article : Google Scholar : PubMed/NCBI
|