1. Introduction
The endoplasmic reticulum (ER) is important in
maintaining intracellular calcium stores, steroid and lipid
biosynthesis, membrane regeneration, gluconeogenesis and the
folding and assembly of newly synthesized proteins (1,2).
Accumulation of misfolded or unfolded proteins in the ER induces ER
stress and results in the unfolded protein response (UPR).
Activation of the UPR is primarily a protective mechanism for cells
under stress, that results in the degradation of unfolded proteins
accumulated in the ER by ER-associated degradation. The UPR system
involves three major pathways dependent on inositol-requiring
enzyme-1, activating transcription factor 6 (ATF6) and
double-stranded RNA-dependent protein kinase-like endoplasmic
reticulum kinase to accelerate unfolded protein degradation,
increase the synthesis of chaperones and other proteins required
for processing and suppress the synthesis of new proteins,
respectively (3). UPR has been
observed to be activated in numerous liver diseases, such as
chronic viral hepatitis (4),
insulin resistance (5), alcoholic
liver disease (6),
ischemia-reperfusion injury (7,8) and
acute liver toxin insults (9).
However, when these responses are activated to a high level and/or
persistently, cells or organs undergo ER stress-induced injury
(10–12).
The cyclic adenosine monophosphate (cAMP)-responsive
element-binding protein H (CREBH), encoded by the CREB3L3 gene, is
a type of ER-residing transcription factor that has a region of
high sequence similarity with ATF6. It belongs to the CREB/ATF
family (13), which also includes
the following proteins: cAMP-responsive element-binding protein 3
(CREB3) also known as Luman or LZIP (14,15);
cAMP-responsive element binding protein 3-like 1 (CREB3L1) also
known as OASIS (16);
cAMP-responsive element binding protein 3-like 2 (CREB3L2) also
known as BBF2H7 (17); and
cAMP-responsive element binding protein 3-like 4 (CREB3L4) also
known as CREB4, AIBZIP or Tisp40 (18,19).
Although CREBH is robustly expressed in the liver, it is also
expressed at lower levels in the small intestine and stomach
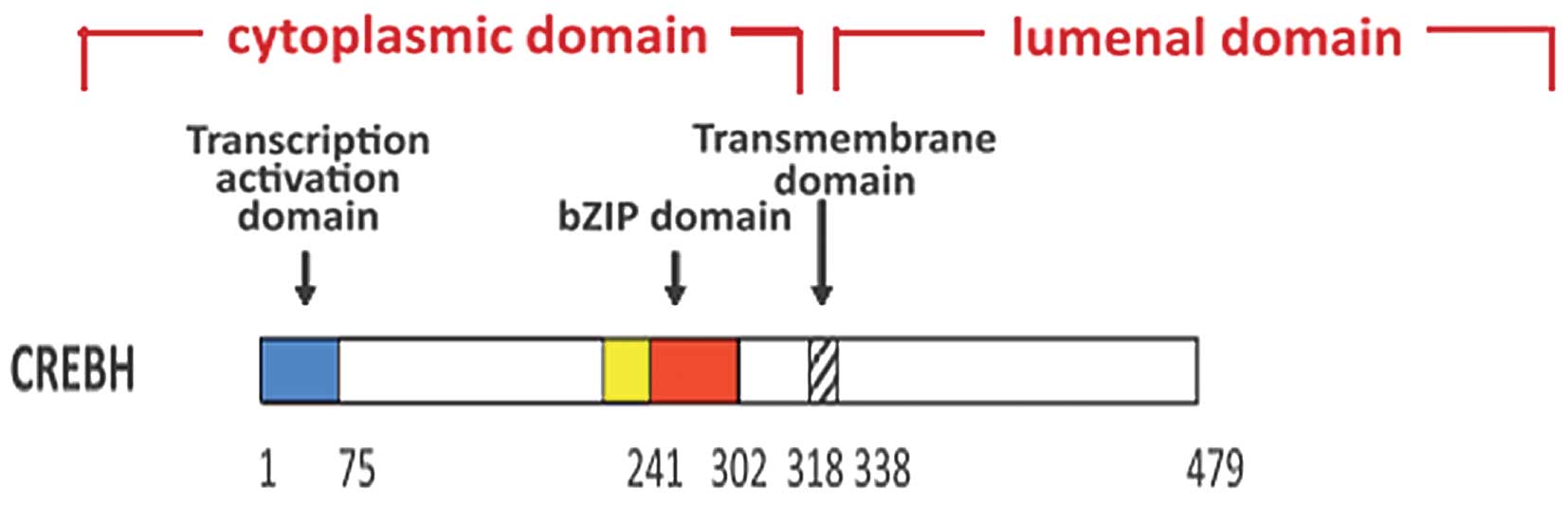
(13,20). CREBH contains an ER transmembrane
domain, a transcription-activation domain and a basic leucine
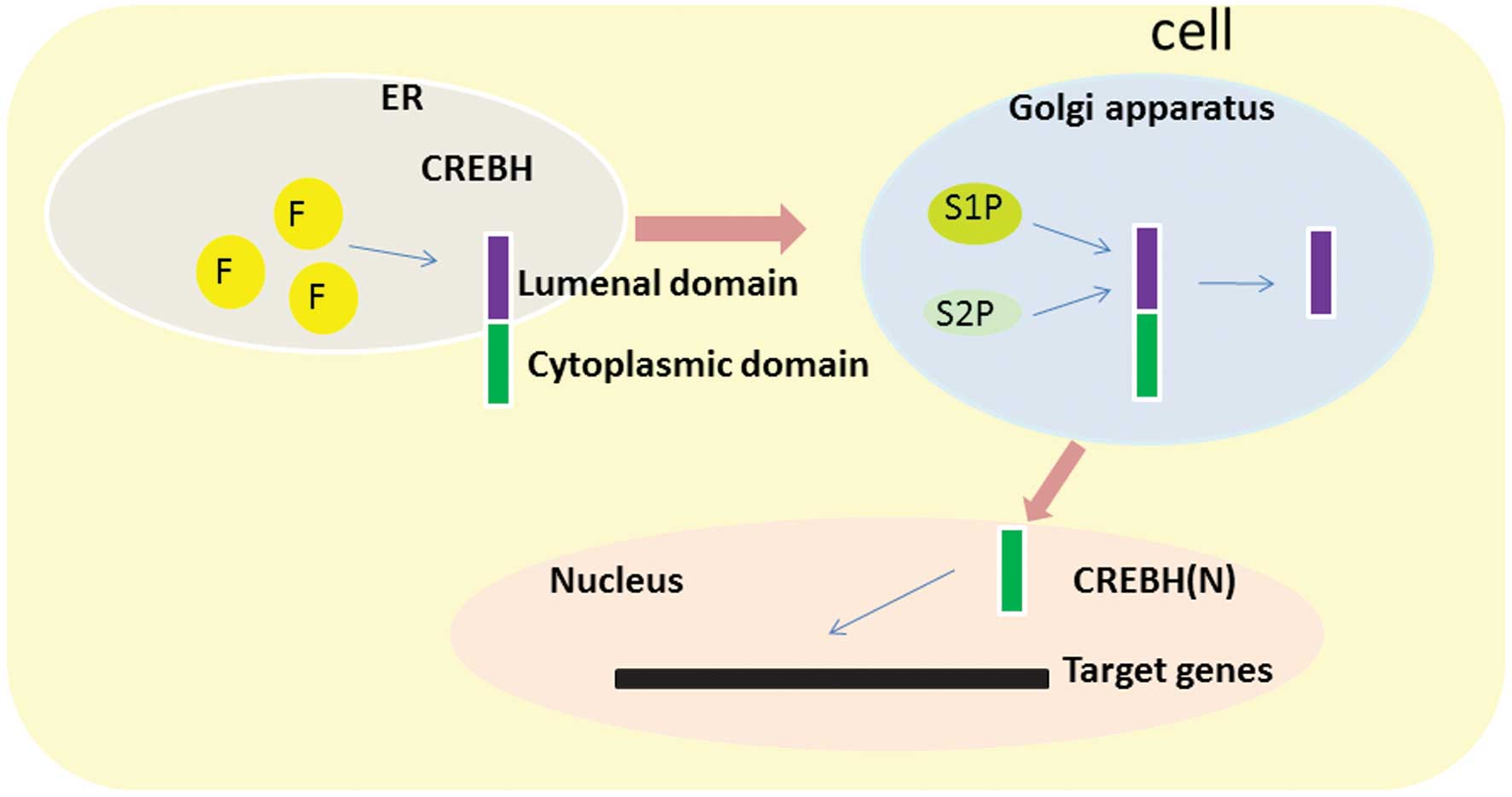
zipper (bZIP) domain. In response to ER stress, CREBH is activated
by regulated intramembrane proteolysis and translocates from the ER
to the Golgi apparatus, where it is cleaved by the site-specific
proteases, site-1 protease and site-2 protease, releasing bZIP
(13). This forms the
transcriptionally active form of CREBH, CREBH(N), which
translocates to the nucleus (21,22),
similar to sterol regulatory element-binding transcription factor 1
(SREBP) and ATF6 (23,24). Chan et al (25) observed that CREBH was modified at
three N-linked glycosylation sites in the luminal domain.
Disruption of all three sites by site-directed mutagenesis
abrogated N-linked glycosylation of CREBH, and it was observed that
N-linked glycosylation is necessary for the activation of CREBH via
intramembrane proteolysis (25).
Unglycosylated CREBH is largely uncleaved and retained in an
inactive form in the ER even if treated with an activator of ER
stress and the UPR, such as brefeldin A, which induces proteolytic
activation of CREBH (25).
Unglycosylated CREBH is less capable of activating the
transcription driven by the UPR element (25). Furthermore, Barbosa et al
(26) reported that the CREB3
class of proteins has a highly conserved region designated the ATB
domain, immediately adjacent to the N-terminal end of the bZIP
region and ~30 residues in length. It is observed only in the CREB3
class of proteins, however not in ATF6. The conserved ATB domain is
required for CREBH in the context of physiological activation of
target secretory pathway genes in human tissue culture cells and in
Drosophila embryos (Figs. 1
and 2) (26).
Previous studies have demonstrated that following
translocation to the nucleus, CREBH(N) binds to the
CREBH-responsive element (CRE), box B and ATF6-binding element
(13,27,28)
in the promoter of target genes involved in the regulation of the
hepatic iron metabolism (27,29),
gluconeogenesis (30–32), lipid metabolism (33,34)
and inflammation (22,35), and the pathophysiological procedure
of NAFLD (33) and associated
conditions. The present review summarizes physiological functions
of CREBH in NAFLD development and progression.
2. Regulation of CREBH expression
CREBH is activated by ER stress
An initial study demonstrated that CREBH expression
was upregulated and the levels of CREBH(N) increased when cells
were treated with ER stress inducers such as tunicamycin (TM),
thapsigargin and dithiothreitol (22). However, subsequent studies did not
observe the proteolytic activation of CREBH by ER stress inducer
(21,25,36).
Xu et al (36) demonstrated
that TM had no effect on CREBH mRNA expression levels and
suppressed the processing of CREBH to CREBH(N) in vitro and
in vivo, suggesting that CREBH processing is not increased
by chemical ER stress-inducers. As mentioned above, the activation
of CREBH via intramembrane proteolysis requires N-linked
glycosylation; TM treatment inhibits CREBH glycosylation, thus, it
is likely to result in the degradation of the unglycosylated CREBH
protein (21). However, CREBH has
been demonstrated to regulate the expression of hepcidin and
proinflammatory and acute phase response genes, thus, linking ER
stress to inflammation and iron metabolism (20,27,37)
and suggesting that the activation of CREBH in the context of ER
stress is dependent on the specific agent and experimental
conditions utilized. It remains to be determined whether CREBH
processing is regulated under any physiologically relevant
conditions in the liver.
CREBH is regulated by cannabinoid
receptor type 1 (CB1R) signaling
Previous studies investigating the underlying
mechanisms of CB1R signaling in the regulation of hepatic
gluconeogenesis demonstrated that CREBH is a downstream target gene
of CB1R signaling, and is key in regulating hepatic gluconeogenesis
(31), disrupting hepatic insulin
receptor signaling (32) and
mediating alcohol-induced regulation of bile acid enzyme gene
expression (38). Following
treatment with AM251 (a CB1R-specific antagonist), CB1R-mediated
induction, by 2-AG (a CB1R agonist) of CREBH was reversed in rat
hepatocytes (31). By pretreating
primary hepatocytes with multiple specific inhibitors of cell
signaling pathways prior to 2-AG treatment, Chanda et al
(31) observed that CB1R signaling
induces CREBH gene expression via the extracellular
signal-regulated kinase 1/2 and c-Jun N-terminal kinase (JNK)
pathways. Furthermore, it was also demonstrated that an activator
protein 1 binding site renders 2-AG responsive to the CREBH
promoter. Administration of 2-AG to cells with multiple serial
deletion constructs of the CREBH promoter demonstrated a
kinase-dead mutant of c-Jun (c-Jun KD; S63/73A) cotransfection
significantly inhibited 2-AG-mediated activation of the CREBH gene
promoter (31). Chanda et
al (38) additionally observed
that JNK phosphorylation and CREBH activation were significantly
reduced in CB1R knockout mice challenged with alcohol. Overall,
these data suggest that the CB1R is a critical component for
induction of CREBH via the JNK signaling transduction pathway.
CREBH is regulated by estrogen-related
receptor-γ (ERRγ)
The orphan nuclear receptor, ERRγ is a
constitutively active transcription factor that regulates genes
involved in the hepatic glucose metabolism, alcohol metabolism and
the ER stress response (39–41).
Misra et al (35) reported
that ERRγ directly regulated CREBH gene expression in response to
ER stress by binding to the ERRγ response element in the CREBH
promoter. Overexpression or knockdown of ERRγ significantly
increased or reduced, respectively, the expression of CREBH and
C-reactive protein (CRP) (35). It
also demonstrated the transcriptional coactivator, peroxisome
proliferator-activated receptor γ coactivator 1-α (PGC1α) was
required for ERRγ-mediated induction of the CREBH gene by binding
the CREBH promoter with ERRγ. The two proteins increased
template-associated H3 and histone H4 acetylation to facilitate
CREBH gene transcription (35).
Previous studies have demonstrated that SHP-interacting leucine
zipper protein (SMILE) inhibited the transactivation of ERRγ by
competition with transcriptional co-activator, PGC1α (42,43).
SMILE belongs to the bZIP family (44,45),
and the gene produces two isoforms, SMILE-L (long isoform, also
termed CREBZF) and SMILE-S (short isoform, previously termed
Zhangfei) (45). Misra et
al (46) reported that
curcumin and SMILE significantly inhibit the transcriptional
activity of CREBH, however did not repress ATF6 transactivity.
Following knockdown of endogenous SMILE, curcumin no longer
inhibited the transcriptional activity of CREBH on the reporter
gene, indicating that SMILE was a repressor of the transcription
factor, CREBH45. They also demonstrated that SMILE interacted with
CREBH via its bZIP domain, without being homodimerized, and
competed with PGC1α to inhibit CREBH transcriptional activity,
similar to ERRγ (46).
CREBH is regulated by nutrition
Hepatic CREBH is activated in a fasting state and
markedly suppressed following refeeding (36). Long-term intake of a high-fat diet
impairs the fasting/refeeding regulation (33) and these nutritional alterations in
CREBH expression levels were markedly associated with plasma levels
of free fatty acids (FFAs). Danno et al (47) demonstrated that CREBH expression
increased in primary hepatocytes with the administration of various
fatty acids (FAs). This is in agreement with the observations of
Gentile et al (48), who
also demonstrated that FAs upregulate CREBH via the activation of
gene transcription. This processing is blocked by inhibitors of
proteasome activity, suggesting that the upregulation of CREBH mRNA
expression levels by FAs requires proteasome activity. Notably,
insulin was indicated to prevent FFA-mediated upregulation of CREBH
and the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)
inhibitors affected the insulin-mediated suppression of the
upregulation of CREBH47 by FFAs. This suggests that insulin and
PI3K signaling are key determinants of the FFA-mediated regulation
of CREBH in liver cells. The present review hypothesized that the
suppression of CREBH mRNA expression levels in the fed state may be
due to postprandial hyperinsulinemia. Glucocorticoids promote
gluconeogenesis in an antagonistic effect to insulin by binding to
the glucocorticoid receptor (GR) (49). Notably, dexamethasone, a synthetic
corticosteroid, induces CREBH gene transcription by activating the
binding of GR to the glucocorticoid transcriptional response
element in the proximal promoter region (30). In addition, Danno et al
(47) observed that the CREBH gene
promoter contains a peroxisome proliferator responsive element for
peroxisome proliferator-activated receptor (PPAR)α transactivation
and indicated that administration of fenofibrate, a PPARα agonist,
increased CREBH expression. This induction was blocked by the
further addition of a PPARα inhibitor, MK886, suggesting that CREBH
was regulated by PPARα. As FAs are endogenous ligands for PPARα,
PPARα is key in the adaptive response to fasting and increased
concentrations of FAs, which may partially explain CREBH activation
by fasting and FAs. However, another study (48) observed that the regulation of CREBH
mRNA expression levels by FAs did not involve PPARα signaling.
Thus, it remains to be elucidated whether PPARα is essential for
FA- and fasting-induced upregulation of CREBH mRNA and protein
expression levels. Toll-like receptor 4 (TLR4), similarly to PPARα,
has emerged as an important mediator of the biological effects of
FAs (50,51). In H4IIE liver cells incubated with
lipopolysaccharide (LPS), which activates TLR4, CREBH mRNA
expression levels were observed to be significantly increased,
suggesting that TLR4 signaling is also involved in the regulation
of CREBH mRNA expression (48).
Via TLR4-dependent pathways, gut microbiota, which increase serum
LPS, have been suggested to be involved in the pathogenesis of
NAFLD, due to the induction of liver inflammation (52).
CREBH is regulated by hepatocyte nuclear
factor 4α (HNF4α)
PPARα has also been demonstrated to be regulated by
HNF4α (53), which is critical in
hepatocyte differentiation and liver function (54). Luebke-Wheeler et al
(20) demonstrated that HNF-4α
regulated CREBH directly via binding an HNF4α recognition element
site lying 3.7 kb upstream of CREBH exon 1, and that HNF4α was
important for the expression of CREBH in the liver, however not the
small intestine (20). A possible
explanation is that intestinal transcription factors, such as the
HNF4γ protein that is predominantly expressed in the gut, regulate
CREBH with no requirement for HNF4α. Further research to elucidate
this disparity in expression is required.
3. CREBH and hepatic lipid metabolism
CREBH is a transcription factor that is key in the
regulation of hepatic lipid accumulation. Following feeding with an
atherogenic high-fat (AHF) diet, greater accumulation of hepatic
lipid contents, such as hepatic triglycerides (TGs), plasma
cholesterol, high-density lipoproteins, and low-density
lipoproteins and an increase in plasma TG was observed in CREBH
knockout mice than in control wild-type (WT) mice. Notably, the
levels of plasma TG increased when feeding with a normal chow diet
(33). To further investigate the
involvement of CREBH in maintaining lipid homeostasis, CREBH null
and WT control mice were fasted for 16 h, and it was observed that
the levels of plasma TG in the CREBH null mice were markedly
increased compared with those of the control mice. Under nutrient
starvation, a condition that stimulates lipolysis, the levels of
ketone, 3-hydroxybutyric acid, a product of FA oxidation in the
plasma, were slightly reduced in the CREBH-null mice, compared with
the control mice, suggesting that CREBH deletion leads to a defect
in TG lipolysis resulting in higher levels of plasma TG (33). Zhang et al (33) and Lee et al (34) demonstrated that plasma TG levels
were significantly lower in CREBH(N) overexpression mediated by
adenoviral infection or transgenic mice than in WT littermates.
Zhang et al (33)
demonstrated adenoviral delivery of activated CREBH resulted in a
major increase in hepatic TGs.
CREBH regulates hepatic lipid accumulation
predominantly by impacting the genes encoding the key enzymes
involved in lipid metabolism. Gene microarray analysis and
quantitative polymerase chain reaction analysis indicated that the
deletion of CREBH in the liver reduced expression levels of five
groups of genes involved in lipid metabolism, including lipogenic
regulators, TG synthesis enzymes, enzymes or regulators in
lipolysis and lipid transport, FA elongation enzymes and FA
oxidation or cholesterol biosynthesis enzymes (33). Decreased expression levels of the
lipogenic regulators and TG synthesis enzymes resulted in reduced
de novo lipogenesis, and reduced expression levels of
enzymes or regulators involved in FA elongation. Oxidation or
cholesterol biosynthesis may be responsible for abnormal
accumulation of hepatic lipid metabolites, and defective expression
of enzymes required for lipolysis and lipid transport may account
for hypertriglyceridemia, reduced fat mass and body weight gain,
and massive steatosis in the CREBH null mice fed the AHF diet. The
genes involved in regulation of CREBH in lipid metabolism in the
liver include apolipoprotein (APO)C2, APOA4, APOA5, APOC3,
fibroblast growth factor 21 (FGF21), fat-specific protein 27
(FSP27) and lipin 132 (34). APOC3
is a lipoprotein lipase (LPL) inhibitor, while APOA5 and APOC2
activate LPL. The activity of LPL is further increased by APOA4,
facilitating the delivery of hydrolyzed FAs to peripheral cells,
thus, lowering plasma levels of TG (55,56).
Patients with genetic defects in APOC2, APOA5 or LPL have high
circulating TG levels due to impaired clearance (56–59).
FGF21 is predominantly produced by the liver (60). Studies indicate that its
concentration is correlated with hypertriglyceridemia, hepatic
steatosis and insulin resistance (61–63).
FSP27 is a lipid droplet (LD)-associated protein that promotes LD
growth and TG storage in white adipocytes; it is also highly
expressed in the steatotic liver and contributes to TG accumulation
(64–66). Lipin 1 is a cytosolic phosphatidic
acid phosphatase that generates diacylglycerol (DAG) in response to
an increased intracellular FFA level, and is also crucial in lipid
metabolism in the liver (67).
CREBH regulates the transcriptional activation of APOC2, APOA4,
APOA5, FGF21, FSP27 and lipin 1 directly by binding the CRE binding
motifs in their gene promoters (32,34,36,68–70).
It remains unclear whether the APOC3 promoter contains a CRE
binding site, however, the very low density lipoprotein
(VLDL)-associated APOC3 expression level is markedly higher in
CREBH−/− mice compared with WT mice, with no
significant alterations in mRNA expression level (34), suggesting post-transcriptional
control of APOC3 by CREBH. Therefore, CREBH has a crucial role in
the maintenance of hepatic TG homeostasis. Multiple nonsynonymous
mutations in CREBH, including W46X, 245fs, E240K, V180M, G105R and
P166L that produced nonfunctional or hypomorphic CREBH protein were
identified in patients with extreme hypertriglyceridemia (34), further demonstrating a critical
role for CREBH in the human TG metabolism. The identification of
CREBH as a stress-induced metabolic regulator has important
implications in the understanding and treatment of metabolic
diseases.
4. CREBH and hepatic glucose metabolism
Phosphoenolpyruvate carboxykinase (PEPCK-C) and
glucose-6-phosphatase (G6Pase) are important in the regulation of
gluconeogenesis (71,72). CREBH(N) has been demonstrated to
increase PEPCK-C and G6Pase transcription by binding CRE in the
promoters of these genes via a CREB/CREB-regulated transcriptional
coactivator 2 (CRTC2)-dependent manner (30). Notably, knockdown of hepatic CREBH
by small hairpin RNA results in reduced hepatic glucose production
by suppressing PEPCK-C and G6Pase expression (30). Chanda et al (31) observed that CB1R signaling mediated
by CREBH induced hepatic gluconeogenesis in primary hepatocytes;
and the knockdown of CREBH attenuated the CB1R signaling-mediated
upregulation of hepatic gluconeogenesis.
In addition, CREBH serves a potential role in the
regulation of insulin resistance (INR). Notably, iron excess has
been associated with reduced insulin sensitivity and with disease
progression, whereas iron removal has been demonstrated to be
beneficial (73–75). Iron metabolism is controlled by
hepcidin, which is a downstream target gene of CREBH (27). Thus, CREBH has a critical
association with insulin sensitivity. Hepatic CB1R and CREBH gene
expression levels are higher in various models of insulin
resistance (30,76). Chanda et al (32) demonstrated that CB1R-mediated
activation of CREBH increased DAG production and phosphorylation of
protein kinase C ε type, which induces INR effects via disrupting
the insulin receptor signaling pathway. CB1R activation under
CREBH-deficient conditions failed to induce DAG production,
ultimately leading to the recovery of insulin receptor signaling
component activity. Therefore, the suppression of CB1R/CREBH
signaling may reduce INR. However, the study by Chanda et al
(32) did not directly assess
insulin resistance in mice, such as with an insulin tolerance test.
As mentioned above, fenofibrate, a PPARα agonist, has been
demonstrated to reduce plasma glucose and insulin levels and to
increase insulin sensitivity mediated by CREBH (77). By contrast, Zhang et al
(33) observed that CREBH knockout
mice are less responsive to insulin than WT mice, which does not
support the hypothesis that targeted inhibition of CREBH would be
useful for improving insulin resistance. Therefore, the function of
CREBH in controlling insulin sensitivity remains to be determined,
however, these findings in rodent and human primary hepatocytes
indicate that CREBH is important in the hepatic glucose
metabolism.
5. CREBH and inflammation
CREBH is crucial in the activation of an acute
inflammatory response. The acute phase response (APR) is systemic
inflammatory component of innate immunity, which is an ancient
metazoan adaptation mechanism initiated by chemical structures
presented by invading microorganisms or exposed by damage to the
host (78–80). Zhang et al (22) demonstrated that the expression
levels of C-reactive protein (CRP) and serum amyloid P-component,
whose synthesis occurs predominantly in APR, were significantly
reduced in the fetal livers of CREBH knockdown mice compared with
the RNAi control mice. It was also observed that CREBH and ATF6
interact and bind to the same conserved element in the promoter of
CRP to synergistically activate expression upon ER stress,
suggesting that CREBH was required to activate the APR. In
addition, in line with other studies by Zhang et al
(22) and Misra et al
(35) it was demonstrated that
ERRγ increased CRP promoter activation via regulating CREBH. CREBH
expression was also observed to be induced by ER stress and
proinflammatory cytokines, including interleukin (IL)6, IL1β or
tumor necrosis factor α (TNFα), in hepatoma cells and in the liver.
Notably, in ER stress, cleavage of CREBH, and APR induced by
proinflammatory cytokines, occurred in the liver, but not in
hepatoma cell lines; suggesting this is impaired and not comparable
to that in the liver in vivo (22). Furthermore, hepcidin, another APR
protein, is important in the anemia of inflammation (81) and is also transcriptionally
regulated by CREBH (27,29). In addition, mRNA expression levels
of other major APR proteins, including serum amyloid A1, A2 and A3,
fibrinogen and α1-acid glycoprotein in CREBH knockdown fetal
livers, were similar to those in the control fetal livers,
suggesting that CREBH is not required for induction of all APR
genes (22).
6. Roles of CREBH on NAFLD
NAFLD is defined as a pathological accumulation of
fat in the form of TG in the liver, not as a result of alcohol
consumption (82). It is an
inclusive term that includes a spectrum of liver pathologies from
simple steatosis to non-alcoholic steatohepatitis (NASH). The liver
is important in the lipid metabolism, including importing and
manufacturing FFAs, storing TGs in LDs and exporting lipids as VLDL
to the serum. Alterations in any of these processes may result in
the development of NAFLD (83).
Donnelly et al (84)
observed that under pathophysiological conditions, ~60% of hepatic
TGs derive from FFAs from adipose tissues, 26% from de novo
lipogenesis and 15% from the diet. FFAs derived from adipose
tissues and de novo lipogenesis are termed non-esterified
fatty acids. The excess of FFAs stimulates TG synthesis. The TGs
are stored as LDs within hepatocytes, or lipidated by
apolipoprotein B100 within the lumen of ER and subsequently
secreted into the blood as VLDLs (85) via the Golgi apparatus (86). Therefore, an excess of FFAs and TG,
or impaired VLDL assembly or secretion, may result in excessive
lipid accumulation in the liver. Accumulation of lipids in the
liver further stimulates existing hepatic INR by generation of
lipid-derived secondary messengers, such as DAG and ceramides
(87). Furthermore, lipid
accumulation in the liver is also linked with the progression of ER
stress, mitochondria stress and impaired autophagy, resulting in
the condition termed lipotoxicity (88). This latter event may result in an
immune response by the Kupffer cells and hepatic stellate cells,
which leads to the progression of NASH, hepatic cirrhosis, and in
certain severe cases, hepatocellular carcinoma (89).
In vivo data indicates that in metabolic
syndrome disorders, such as NAFLD, levels of ER stress markers in
the liver and other tissues are increased, and liver damage occurs
(80–93). CREBH is an ER-bound transcription
factor activated by ER stress and it is a key metabolic regulator
required to activate expression of the genes involved in de
novo lipogenesis, TG and cholesterol biosynthesis, FA
elongation and oxidation, lipolysis and lipid transport in response
to ER stress (33). Previous
studies have demonstrated that fasting- and high fat diet
(HFD)-induced fatty liver was more pronounced in
CREBH−/− mice compared with the WT mice
(33,70), while adenoviral delivery of CREBH
inhibited HFD-induced steatosis in WT mice (77). In addition, NAFLD is associated
with inflammation and fibrosis, increased hepatocyte ballooning,
lobular and portal inflammation, Mallory bodies and collagen
deposition were observed in CREBH null mice following the AHF diet
(33). A histological scoring
system for NAFLD was used (94,95),
demonstrating that the CREBH null mice developed profound NASH
following the AHF diet. Increased levels of the key indicators of
hepatotoxicity were also observed, including alanine
aminotransferase and aspartate aminotransferase, and
NASH-associated pro-inflammatory cytokines (96), such as TNFα and IL6 (33). Notably, fasting induced hepatic
steatosis and an increase in CREBH expression levels (36), however, increased expression levels
of CREBH maintains lipid homeostasis by regulating expression of
the genes involved in the lipid metabolism (33). CREBH also inhibited HFD-induced
steatosis in WT mice (77),
suggesting there is a negative feedback control mechanism in the
involvement of CREBH in the development of NAFLD.
CREBH is suggested to be involved in the development
of NAFLD by regulating the lipid metabolism and maintaining insulin
sensitivity. Accumulation of lipids in the liver stimulates
existing hepatic insulin resistance, which further stimulates
hepatic SREBP-1c production, resulting in increased de novo
synthesis of fatty acids (97). As
mentioned above, CREBH regulates insulin sensitivity by regulating
hepcidin to control iron levels and mediating CB1R to disrupt
hepatic insulin receptor signaling. Fenofibrate has been
demonstrated to reduce plasma glucose and insulin levels and
increase the insulin sensitivity mediated by CREBH (77). In addition, CREBH prevents
insulin-induced SREBP-1c expression by markedly increasing the
promoter activity of insulin induced gene 2, whose downregulation
mediates insulin-stimulated transcriptional activity of SREBP-1c
(77). The previous studies
demonstrated that CREBH is key in the involvement of NAFLD by
regulating insulin sensitivity.
As mentioned above, CREBH is also associated with
inflammation in the liver (22,35).
The development of NAFLD involves insulin resistance and increased
inflammation. Furthermore, inflammation in the development of NASH
further impedes insulin signaling (98). Cytokine production of IL-6 and
TNF-α is increased in NASH and may be involved in its pathogenesis
(99). CREBH expression and
cleavage may be induced by IL-6 and TNF-α to activate the APR
(22), thus, the present review
hypothesized that CREBH is a protective factor in response to
inflammation by maintaining the balance of glucose and lipid
metabolism by mediating gene expression in the liver for the two
processes. In addition, TLR4 is activated by LPS or gut microbiota,
which is also associated with NAFLD. The activated TLR4 increases
hepatic expression levels of TNF-α and increases hepatic steatosis
and inflammation during the development of NAFLD (52). CREBH is regulated by TLR4 (48), suggesting that CREBH may be
activated by TLR4 and is, thus, involved in the development of
NAFLD. The biological relevance of this regulation remains to be
elucidated but CREBH may provide a potential therapeutic target in
NAFLD.
7. Conclusions
Various studies have elucidated the roles of CREBH.
Via the regulation by ER stress, CB1R signaling, ERRγ, SMILE,
nutrition, PPARα and HNF4α, CREBH promotes the genes encoding
lipogenic regulators, TG synthesis enzymes, enzymes or regulators
in lipolysis and lipid transport, FA elongation enzymes and FA
oxidation or cholesterol biosynthesis enzymes to regulate hepatic
lipid metabolism. By binding CRE in the promoter of PEPCK-C and
G6Pase, it is involved in hepatic glucose metabolism in a
CRTC2-dependent manner. In addition, CREBH regulates iron
metabolism and mediates CB1R signaling, thereby regulating insulin
sensitivity. Furthermore, CREBH is crucial in the activation of an
acute inflammatory response. As inflammation and disorders of
glucose and lipid metabolism are the predominant factors in NAFLD,
targeting CREBH appears to be a promising effective therapeutic
strategy.
References
|
1
|
Fujimoto M and Hayashi T: New insights
into the role of mitochondria-associated endoplasmic reticulum
membrane. Int Rev Cell Mol Biol. 292:73–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wagner M and Moore DD: Endoplasmic
reticulum stress and glucose homeostasis. Curr Opin Clin Nutr Metab
Care. 14:367–373. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esposito V, Grosjean F, Tan J, Huang L,
Zhu L, Chen J, Xiong H, Striker GE and Zheng F: CHOP deficiency
results in elevated lipopolysaccharide-induced inflammation and
kidney injury. Am J Physiol Renal Physiol. 304:F440–F450. 2013.
View Article : Google Scholar :
|
|
4
|
Benali-Furet NL, Chami M, Houel L, De
Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R,
Ichas F, Rizzuto R and Paterlini-Bréchot P: Hepatitis C virus core
triggers apoptosis in liver cells by inducing ER stress and ER
calcium depletion. Oncogene. 24:4921–4933. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ozcan U, Cao Y, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ji C and Kaplowitz NL: Betaine decreases
hyperhomocysteinemia, endoplasmic reticulum stress, and liver
injury in alcohol-fed mice. Gastroenterology. 124:1488–1499. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duvigneau JC, Kozlov AV, Zifko C, Postl A,
Hartl RT, Miller I, Gille L, Staniek K, Moldzio R, Gregor W, et al:
Reperfusion does not induce oxidative stress but sustained
endoplasmic reticulum stress in livers of rats subjected to
traumatic-hemorrhagic shock. Shock. 33:289–298. 2010. View Article : Google Scholar
|
|
8
|
Emadali A, Nguyên DT, Rochon C, Tzimas GN,
Metrakos PP and Chevet E: Distinct endoplasmic reticulum stress
responses are triggered during human liver transplantation. J
Pathol. 207:111–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim TH, Kim YW, Shin SM, Kim CW, Yu IJ and
Kim SG: Synergistic hepatotoxicity of N,N-dimethylformamide with
carbon tetrachloride in association with endoplasmic reticulum
stress. Chem Biol Interact. 184:492–501. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ron D: Translational control in the
endoplasmic reticulum stress response. J Clin Invest.
110:1383–1388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaufman RJ: Orchestrating the unfolded
protein response in health and disease. J Clin Invest.
110:1389–1398. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schröder M and Kaufman RJ: ER stress and
the unfolded protein response. Mutat Res. 569:29–63. 2005.
View Article : Google Scholar
|
|
13
|
Omori Y, Imai J, Watanabe M, Komatsu T,
Suzuki Y, Kataoka K, Watanabe S, Tanigami A and Sugano S: CREB-H: A
novel mammalian transcription factor belonging to the CREB/ATF
family and functioning via the box-B element with a liver-specific
expression. Nucleic Acids Res. 29:2154–2162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DenBoer LM, Hardy-Smith PW, Hogan MR,
Cockram GP, Audas TE and Lu R: Luman is capable of binding and
activating transcription from the unfolded protein response
element. Biochem Biophys Res Commun. 331:113–119. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang G, Audas TE, Li Y, Cockram GP, Dean
JD, Martyn AC, Kokame K and Lu R: Luman/CREB3 induces transcription
of the endoplasmic reticulum (ER) stress response protein Herp
through an ER stress response element. Mol Cell Biol. 26:7999–8010.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kondo S, Murakami T, Tatsumi K, Ogata M,
Kanemoto S, Otori K, Iseki K, Wanaka A and Imaizumi K: OASIS, a
CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat
Cell Biol. 7:186–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kondo S, Saito A, Hino S, Murakami T,
Ogata M, Kanemoto S, Nara S, Yamashita A, Yoshinaga K, Hara H and
Imaizumi K: BBF2H7, a novel transmembrane bZIP transcription
factor, is a new type of endoplasmic reticulum stress transducer.
Mol Cell Biol. 27:1716–1729. 2007. View Article : Google Scholar :
|
|
18
|
Nagamori I, Yabuta N, Fujii T, Tanaka H,
Yomogida K, Nishimune Y and Nojima H: Tisp40, a spermatid specific
bZip transcription factor, functions by binding to the unfolded
protein response element via the Rip pathway. Genes Cells.
10:575–594. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stirling J and O'Hare P: CREB4, a
transmembrane bZip transcription factor and potential new substrate
for regulation and cleavage by S1P. Mol Biol Cell. 17:413–426.
2006. View Article : Google Scholar :
|
|
20
|
Luebke-Wheeler J, Zhang K, Battle M,
Si-Tayeb K, Garrison W, Chhinder S, Li J, Kaufman RJ and Duncan SA:
Hepatocyte nuclear factor 4alpha is implicated in endoplasmic
reticulum stress-induced acute phase response by regulating
expression of cyclic adenosine monophosphate responsive element
binding protein H. Hepatology. 48:1242–1250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bailey D, Barreca C and O'Hare P:
Trafficking of the bZIP transmembrane transcription factor CREB-H
into alternate pathways of ERAD and stress-regulated intramembrane
proteolysis. Traffic. 8:1796–1814. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang K, Shen X, Wu J, Sakaki K, Saunders
T, Rutkowski DT, Back SH and Kaufman RJ: Endoplasmic reticulum
stress activates cleavage of CREBH to induce a systemic
inflammatory response. Cell. 124:587–599. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bailey D and O'Hare P: Transmembrane bZIP
transcription factors in ER stress signaling and the unfolded
protein response. Antioxid Redox Signal. 9:2305–2321. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Asada R, Kanemoto S, Kondo S, Saito A and
Imaizumi K: The signalling from endoplasmic reticulum-resident bZIP
transcription factors involved in diverse cellular physiology. J
Biochem. 149:507–518. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chan CP, Mak TY, Chin KT, Ng IO and Jin
DY: N-linked glycosylation is required for optimal proteolytic
activation of membrane-bound transcription factor CREB-H. J Cell
Sci. 123:1438–1448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barbosa S, Fasanella G, Carreira S,
Llarena M, Fox R, Barreca C, Andrew D and O'Hare P: An orchestrated
program regulating secretory pathway genes and cargos by the
transmembrane transcription factor CREB-H. Traffic. 14:382–398.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vecchi C, Montosi G, Zhang K, Lamberti I,
Duncan SA, Kaufman RJ and Pietrangelo A: ER stress controls iron
metabolism through induction of hepcidin. Science. 325:877–880.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Llarena M, Bailey D, Curtis H and O'Hare
P: Different mechanisms of recognition and ER retention by
transmembrane transcription factors CREB-H and ATF6. Traffic.
11:48–69. 2010. View Article : Google Scholar
|
|
29
|
Vecchi C, Montosi G, Garuti C, Corradini
E, Sabelli M, Canali S and Pietrangelo A: Gluconeogenic signals
regulate iron homeostasis via hepcidin in mice. Gastroenterology.
146:1060–1069. 2014. View Article : Google Scholar
|
|
30
|
Lee MW, Chanda D, Yang J, Oh H, Kim SS,
Yoon YS, Hong S, Park KG, Lee IK, Choi CS, et al: Regulation of
hepatic gluconeogenesis by an ER-bound transcription factor, CREBH.
Cell Metab. 11:331–339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chanda D, Kim DK, Li T, Kim YH, Koo SH,
Lee CH, Chiang JY and Choi HS: Cannabinoid receptor type 1 (CB1R)
signaling regulates hepatic gluconeogenesis via induction of
endoplasmic reticulum-bound transcription factor cAMP-responsive
element-binding protein H (CREBH) in primary hepatocytes. J Biol
Chem. 286:27971–27979. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chanda D, Kim YH, Kim DK, Lee MW, Lee SY,
Park TS, Koo SH, Lee CH and Choi HS: Activation of cannabinoid
receptor type 1 (Cb1r) disrupts hepatic insulin receptor signaling
via cyclic AMP-response element-binding protein H (Crebh)-mediated
induction of Lipin1 gene. J Biol Chem. 287:38041–38049. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang C, Wang G, Zheng Z, Maddipati KR,
Zhang X, Dyson G, Williams P, Duncan SA, Kaufman RJ and Zhang K:
Endoplasmic reticulum-tethered transcription factor cAMP responsive
element-binding protein, hepatocyte specific, regulates hepatic
lipogenesis, fatty acid oxidation, and lipolysis upon metabolic
stress in mice. Hepatology. 55:1070–1082. 2012. View Article : Google Scholar :
|
|
34
|
Lee JH, Giannikopoulos P, Duncan SA, Wang
J, Johansen CT, Brown JD, Plutzky J, Hegele RA, Glimcher LH and Lee
AH: The transcription factor cyclic AMP-responsive element-binding
protein H regulates triglyceride metabolism. Nat Med. 17:812–815.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Misra J, Chanda D, Kim DK, Cho SR, Koo SH,
Lee CH, Back SH and Choi HS: Orphan nuclear receptor Errγ induces
C-reactive protein gene expression through induction of ER-bound
Bzip transmembrane transcription factor CREBH. PLoS One.
9:e863422014. View Article : Google Scholar
|
|
36
|
Xu X, Park JG, So JS, Hur KY and Lee AH:
Transcriptional regulation of apolipoprotein A-IV by the
transcription factor CREBH. J Lipid Res. 55:850–859. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shin DY, Chung J, Joe Y, Pae HO, Chang KC,
Cho GJ, Ryter SW and Chung HT: Pretreatment with CO-releasing
molecules suppresses hepcidin expression during inflammation and
endoplasmic reticulum stress through inhibition of the STAT3 and
CREBH pathways. Blood. 119:2523–2532. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chanda D, Kim YH, Li T, Misra J, Kim DK,
Kim JR, Kwon J, Jeong WI, Ahn SH, Park TS, et al: Hepatic
cannabinoid receptor type 1 mediates alcohol-induced regulation of
bile acid enzyme genes expression via CREBH. PLoS One.
8:e688452013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim DK, Ryu D, Koh M, Lee MW, Lim D, Kim
MJ, Kim YH, Cho WJ, Lee CH, Park TS, et al: Orphan nuclear receptor
estrogen-related receptor γ (ERRγ) is key regulator of hepatic
gluconeogenesis. J Biol Chem. 287:21628–21639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim DK, Kim JR, Koh M, Kim YD, Lee JM,
Chanda D, Park SB, Min JJ, Lee CH, Park TS, et al: Estrogen-related
receptor γ (ERRγ) is a novel transcriptional regulator of
phosphatidic acid phosphatase, LIPIN1, and inhibits hepatic insulin
signaling. J Biol Chem. 286:38035–38042. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim DK, Kim YH, Jang HH, Park J, Kim JR,
Koh M, Jeong WI, Koo SH, Park TS, Yun CH, et al: Estrogen-related
receptor γ controls hepatic CB1 receptor-mediated CYP2E1 expression
and oxidative liver injury by alcohol. Gut. 62:1044–1054. 2013.
View Article : Google Scholar
|
|
42
|
Xie YB, Park JH, Kim DK, Hwang JH, Oh S,
Park SB, Shong M, Lee IK and Choi HS: Transcriptional corepressor
SMILE recruits SIRT1 to inhibit nuclear receptor estrogen
receptor-related receptor gamma transactivation. J Biol Chem.
284:28762–28774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xie YB, Nedumaran B and Choi HS: Molecular
characterization of SMILE as a novel corepressor of nuclear
receptors. Nucleic Acids Res. 37:4100–4115. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lu R and Misra V: Zhangfei: A second
cellular protein interacts with herpes simplex virus accessory
factor HCF in a manner similar to Luman and VP16. Nucleic Acids
Res. 28:2446–2454. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xie YB, Lee OH, Nedumaran B, Seong HA, Lee
KM, Ha H, Lee IK, Yun Y and Choi HS: SMILE, a new orphan nuclear
receptor SHP-interacting protein, regulates SHP-repressed estrogen
receptor transactivation. Biochem J. 416:463–473. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Misra J, Chanda D, Kim DK, Li T, Koo SH,
Back SH, Chiang JY and Choi HS: Curcumin differentially regulates
endoplasmic reticulum stress through transcriptional corepressor
SMILE (small heterodimer partner-interacting leucine zipper
protein)-mediated inhibition of CREBH (cAMP responsive
element-binding protein H). J Biol Chem. 286:41972–41984. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Danno H, Ishii KA, Nakagawa Y, Mikami M,
Yamamoto T, Yabe S, Furusawa M, Kumadaki S, Watanabe K, Shimizu H,
et al: The liver-enriched transcription factor CREBH is
nutritionally regulated and activated by fatty acids and PPARalpha.
Biochem Biophys Res Commun. 391:1222–1227. 2010. View Article : Google Scholar
|
|
48
|
Gentile CL, Wang D, Pfaffenbach KT, Cox R,
Wei Y and Pagliasotti MJ: Fatty acids regulate CREBh via
transcriptional mechanisms that are dependent on proteasome
activity and insulin. Mol Cell Biochem. 344:99–107. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vegiopoulos A and Herzig S:
Glucocorticoids, metabolism and metabolic diseases. Mol Cell
Endocrinol. 275:43–61. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wong SW, Kwon MJ, Choi AM, Kim HP,
Nakahira K and Hwang DH: Fatty acids modulate Toll-like receptor 4
activation through regulation of receptor dimerization and
recruitment into lipid rafts in a reactive oxygen species-dependent
manner. J Biol Chem. 284:27384–27392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schaeffler A, Gross P, Buettner R,
Bollheimer C, Buechler C, Neumeier M, Kopp A, Schoelmerich J and
Falk W: Fatty acid-induced induction of Toll-like
receptor-4/nuclear factor-kappaB pathway in adipocytes links
nutritional signalling with innate immunity. Immunology.
126:233–245. 2009. View Article : Google Scholar :
|
|
52
|
Miura K and Ohnishi H: Role of gut
microbiota and Toll-like receptors in nonalcoholic fatty liver
disease. World J Gastroenterol. 20:7381–7391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pineda Torra I, Jamshidi Y, Flavell DM,
Fruchart JC and Staels B: Characterization of the human PPARalpha
promoter: Identification of a functional nuclear receptor response
element. Mol Endocrinol. 16:1013–1028. 2002.PubMed/NCBI
|
|
54
|
Hwang-Verslues WW and Sladek FM:
HNF4α-role in drug metabolism and potential drug target? Curr Opin
Pharmacol. 10:698–705. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Goldberg IJ, Scheraldi CA, Yacoub LK,
Saxena U and Bisgaier CL: Lipoprotein ApoC-II activation of
lipoprotein lipase. Modulation by apolipoprotein A-IV. J Biol Chem.
265:4266–4272. 1990.PubMed/NCBI
|
|
56
|
Jong MC, Hofker MH and Havekes LM: Role of
ApoCs in lipoprotein metabolism: Functional differences between
ApoC1, ApoC2, and ApoC3. Arterioscler Thromb Vasc Biol. 19:472–484.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Marçais C, Verges B, Charrière S, Pruneta
V, Merlin M, Billon S, Perrot L, Drai J, Sassolas A, Pennacchio LA,
et al: Apoa5 Q139X truncation predisposes to late-onset
hyperchylomicronemia due to lipoprotein lipase impairment. J Clin
Invest. 115:2862–2869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Merkel M, Eckel RH and Goldberg IJ:
Lipoprotein lipase: Genetics, lipid uptake, and regulation. J Lipid
Res. 43:1997–2006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pennacchio LA, Olivier M, Hubacek JA,
Cohen JC, Cox DR, Fruchart JC, Krauss RM and Rubin EM: An
apolipoprotein influencing triglycerides in humans and mice
revealed by comparative sequencing. Science. 294:169–173. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nishimura T, Nakatake Y, Konishi M and
Itoh N: Identification of a novel FGF, FGF-21, preferentially
expressed in the liver. Biochim Biophys Acta. 1492:203–206. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dushay J, Chui PC, Gopalakrishnan GS,
Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML
and Maratos-Flier E: Increased fibroblast growth factor 21 in
obesity and nonalcoholic fatty liver disease. Gastroenterology.
139:456–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chavez AO, Molina-Carrion M, Abdul-Ghani
MA, Folli F, Defronzo RA and Tripathy D: Circulating fibroblast
growth factor-21 is elevated in impaired glucose tolerance and type
2 diabetes and correlates with muscle and hepatic insulin
resistance. Diabetes Care. 32:1542–1546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li H, Dong K, Fang Q, Hou X, Zhou M, Bao
Y, Xiang K, Xu A and Jia W: High serum level of fibroblast growth
factor 21 is an independent predictor of non-alcoholic fatty liver
disease: A 3-year prospective study in China. J Hepatol.
58:557–563. 2013. View Article : Google Scholar
|
|
64
|
Matsusue K, Kusakabe T, Noguchi T,
Takiguchi S, Suzuki T, Yamano S and Gonzalez FJ: Hepatic steatosis
in leptin-deficient mice is promoted by the PPARgamma target gene
Fsp27. Cell Metab. 7:302–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Puri V, Konda S, Ranjit S, Aouadi M,
Chawla A, Chouinard M, Chakladar A and Czech MP: Fat-specific
protein 27, a novel lipid droplet protein that enhances
triglyceride storage. J Biol Chem. 282:34213–34218. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jambunathan S, Yin J, Khan W, Tamori Y and
Puri V: FSP27 promotes lipid droplet clustering and then fusion to
regulate triglyceride accumulation. PLoS One. 6:e286142011.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Reue K: The lipin family: Mutations and
metabolism. Curr Opin Lipidol. 20:165–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Song KH, Park AY, Kim JE and Ma JY:
Identification and characterization of cyclic AMP response
element-binding protein H response element in the human
apolipoprotein A5 gene promoter. BioMed Res Int. 2013:8924912013.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kim H, Mendez R, Zheng Z, Chang L, Cai J,
Zhang R and Zhang K: Liver-enriched transcription factor CREBH
interacts with peroxisome proliferator-activated receptor α to
regulate metabolic hormone FGF21. Endocrinology. 155:769–782. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xu X, Park JG, So JS and Lee AH:
Transcriptional activation of Fsp27 by the liver-enriched
transcription factor CREBH promotes lipid droplet growth and
hepatic steatosis. Hepatology. 61:857–869. 2015. View Article : Google Scholar
|
|
71
|
Hall RK and Granner DK: Insulin regulates
expression of metabolic genes through divergent signaling pathways.
J Basic Clin Physiol Pharmacol. 10:119–133. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hanson RW and Reshef L: Regulation of
phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev
Biochem. 66:581–611. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Facchini FS, Hua NW and Stoohs RA: Effect
of iron depletion in carbohydrate-intolerant patients with clinical
evidence of nonalcoholic fatty liver disease. Gastroenterology.
122:931–939. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Fernández-Real JM, Peñarroja G, Castro A,
García-Bragado F, Hernández-Aguado I and Ricart W: Blood letting in
high-ferritin type 2 diabetes: Effects on insulin sensitivity and
beta-cell function. Diabetes. 51:1000–1004. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Valenti L, Moscatiello S, Vanni E,
Fracanzani AL, Bugianesi E, Fargion S and Marchesini G: Venesection
for non-alcoholic fatty liver disease unresponsive to lifestyle
counseling–a propensity score-adjusted observational study. QJM.
104:141–149. 2011. View Article : Google Scholar
|
|
76
|
Jeong WI, Osei-Hyiaman D, Park O, Liu J,
Bátkai S, Mukhopadhyay P, Horiguchi N, Harvey-White J, Marsicano G,
Lutz B, et al: Paracrine activation of hepatic CB1 receptors by
stellate cell-derived endocannabinoids mediates alcoholic fatty
liver. Cell Metab. 7:227–235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Min AK, Jeong JY, Go Y, Choi YK, Kim YD,
Lee IK and Park KG: cAMP response element binding protein H
mediates fenofibrate-induced suppression of hepatic lipogenesis.
Diabetologia. 56:412–422. 2013. View Article : Google Scholar
|
|
78
|
Gabay C and Kushner I: Acute-phase
proteins and other systemic responses to inflammation. N Engl J
Med. 340:448–454. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Medzhitov R and Janeway CR Jr: Decoding
the patterns of self and nonself by the innate immune system.
Science. 296:298–300. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yoo JY and Desiderio S: Innate and
acquired immunity intersect in a global view of the acute-phase
response. Proc Natl Acad Sci USA. 100:1157–1162. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kaplan J, Ward DM and De Domenico I: The
molecular basis of iron overload disorders and iron-linked anemias.
Int J Hematol. 93:14–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kawano Y and Cohen DE: Mechanisms of
hepatic triglyceride accumulation in non-alcoholic fatty liver
disease. J Gastroenterol. 48:434–441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Musso G, Gambino R and Cassader M: Recent
insights into hepatic lipid metabolism in non-alcoholic fatty liver
disease (NAFLD). Prog Lipid Res. 48:1–26. 2009. View Article : Google Scholar
|
|
84
|
Donnelly KL, Smith CI, Schwarzenberg SJ,
Jessurun J, Boldt MD and Parks EJ: Sources of fatty acids stored in
liver and secreted via lipoproteins in patients with nonalcoholic
fatty liver disease. J Clin Invest. 115:1343–1351. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Postic C and Girard J: Contribution of de
novo fatty acid synthesis to hepatic steatosis and insulin
resistance: Lessons from genetically engineered mice. J Clin
Invest. 118:829–838. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tiwari S and Siddiqi SA: Intracellular
trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol.
32:1079–1086. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jornayvaz FR and Shulman GI:
Diacylglycerol activation of protein kinase Cε and hepatic insulin
resistance. Cell Metab. 15:574–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Neuschwander-Tetri BA: Hepatic
lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis:
The central role of nontriglyceride fatty acid metabolites.
Hepatology. 52:774–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zámbó V, Simon-Szabó L, Szelényi P,
Kereszturi E, Bánhegyi G and Csala M: Lipotoxicity in the liver.
World J Hepatol. 5:550–557. 2013.PubMed/NCBI
|
|
90
|
Gregor MF, Yang L, Fabbrini E, Mohammed
BS, Eagon JC, Hotamisligil GS and Klein S: Endoplasmic reticulum
stress is reduced in tissues of obese subjects after weight loss.
Diabetes. 58:693–700. 2009. View Article : Google Scholar :
|
|
91
|
Puri P, Mirshahi F, Cheung O, Natarajan R,
Maher JW, Kellum JM and Sanyal AJ: Activation and dysregulation of
the unfolded protein response in nonalcoholic fatty liver disease.
Gastroenterology. 134:568–576. 2008. View Article : Google Scholar
|
|
92
|
Sharma NK, Das SK, Mondal AK, Hackney OG,
Chu WS, Kern PA, Rasouli N, Spencer HJ, Yao-Borengasser A and
Elbein SC: Endoplasmic reticulum stress markers are associated with
obesity in nondiabetic subjects. J Clin Endocrinol Metab.
93:4532–4541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang D, Wei Y and Pagliassotti MJ:
Saturated fatty acids promote endoplasmic reticulum stress and
liver injury in rats with hepatic steatosis. Endocrinology.
147:943–951. 2006. View Article : Google Scholar
|
|
94
|
Brunt EM, Janney CG, Di Bisceglie AM,
Neuschwander-Tetri BA and Bacon BR: Nonalcoholic steatohepatitis: A
proposal for grading and staging the histological lesions. Am J
Gastroenterol. 94:2467–2474. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kleiner DE, Brunt EM, Van Natta M, Behling
C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS,
Unalp-Arida A, et al Nonalcoholic Steatohepatitis Clinical Research
Network: Design and validation of a histological scoring system for
nonalcoholic fatty liver disease. Hepatology. 41:1313–1321. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sakaguchi S, Takahashi S, Sasaki T,
Kumagai T and Nagata K: Progression of alcoholic and non-alcoholic
steatohepatitis: Common metabolic aspects of innate immune system
and oxidative stress. Drug Metab Pharmacokinet. 26:30–46. 2011.
View Article : Google Scholar
|
|
97
|
Shimomura I, Bashmakov Y and Horton JD:
Increased levels of nuclear SREBP-1c associated with fatty livers
in two mouse models of diabetes mellitus. J Biol Chem.
274:30028–30032. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
de Luca C and Olefsky JM: Inflammation and
insulin resistance. FEBS Lett. 582:97–105. 2008. View Article : Google Scholar
|
|
99
|
Polyzos SA, Kountouras J and Zavos C:
Nonalcoholic fatty liver disease: The pathogenetic roles of insulin
resistance and adipocytokines. Curr Mol Med. 9:299–314. 2009.
View Article : Google Scholar : PubMed/NCBI
|