Introduction
Improvements in Neonatal Intensive Care Units, and
advancements made in neonatal resuscitation and respiratory support
and monitoring systems, have increased the survival rates of
very-low-birth-weight (<1,500 g) and premature (<28 weeks)
infants. However, this increase in survival rates has not resulted
in improved neurodevelopmental outcomes (1,2).
Previous studies have shown that ~40% of the surviving infants
suffered from neurological deficits, including periventricular
leukomalacia (PVL), a severe type of brain injury in premature
infants characterized by extensive oligodendrocyte precursor cells
(OPCs) migration and maturation (3–5). One
of the reasons for the occurrence of neurological deficits may be
oxygen administration. It is known that oxygen is widely
administered to premature infants. Previous studies have shown that
hyperoxia is important in the development of bronchial pulmonary
hypoplasia and retinopathy of prematurity (3–5).
Certain previous studies reported that hyperoxia-induced nerve cell
death was observed during the developmental phase (6,7). To
the best of our knowledge, the influence of hyperoxia on the human
brain has not been reported.
PVL can reduce the levels of neurotrophic factor and
nerve growth factor (NGF), resulting in chronic disability of
cerebral white matter (8,9). Certain previous studies have shown
that the reduced NGF may upregulate c-Jun NH2-terminal kinases
(JNK)-p53-Bcl-2-associated X protein (Bax) cell death and the Fas
cell surface death receptor (Fas) apoptosis pathway, which comprise
the predominant parts of the extrinsic apoptotic signal
transduction system (10). A
reduction in the tyrosine kinase receptor cell survival pathway, as
well as the increased JNK-p53-Bax and Fas apoptotic pathways may
therefore promote brain cell death (11–13).
Paired immunoglobin-like receptor B (PirB) is a
receptor expressed on myeloid cells. The inhibition of PirB in
animal models of spinal cord injury was revealed to promote the
regeneration of damaged nerve cells (14). Our previous study (15) also suggested that PirB, which is
induced by hypoxic-ischemic brain injury, inhibited nerve cell
regeneration.
17β-Estradiol (E2) is a neuroprotective agent
(16,17), which is important in the
development and function of the nervous system (18), particularly at the beginning of the
neural precursor cell differentiation around the ventricle
(19). Several hypoxia-ischemia
and excitatory toxin models have demonstrated the neuroprotective
effects of E2 on mature cells in vivo (20,21).
In addition, E2 can also affect the apoptotic process at several
different stages (21). During
pregnancy, the E2 levels in the placenta exhibited an ~100-fold
increase; however, they dropped rapidly within 24 h following the
removal of the infant's umbilical cord. Premature babies
experienced hormone withdrawal earlier compared with the full-term
infants, which further increases the brain tissue damage by oxygen
stimulation (22,23).
Despite previous reports suggesting that
hyperoxia-induced apoptosis is crucial in brain damage (24), that hyperoxia induces immature or
OPC apoptosis (25), that PirB
inhibits nerve cell regeneration (26) and that E2 has neuroprotective
effects (27), the association
between hyperoxia, PirB and E2 remains to be elucidated. Therefore
the aim of the present study was to investigate the effects of
hyperoxia on OPCs, as well as the effects of E2 on OPC
apoptosis.
Materials and methods
Animals
A total of 20 Sprague-Dawley rats, aged 2-days-old
were purchased from the Laboratory Animal Center of Sichuan
University (Sichuan, China) and used in the present study. Animal
care, maintenance and surgery were performed in accordance with the
regulations dictated by the Institutional Animal Care and Use
Committee of Sichuan University. Rats were immediately sacrificed
by decapitation under diethylether (Sigma-Aldrich, St. Louis, MO,
USA) anesthesia.
Primary cultures of OPCs
For the in vitro experiments, primary OPCs
were isolated, as previously described (28). Following sacrifice, the rat's
scalps and meninges were subsequently removed and the cortices were
dissected, rinsed twice in ice-cold Hank's buffered salt solution
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
incubated at 37°C for 15 min with 0.01% trypsin and DNase (both
Beyotime Institute of Biotechnology, Haimen, China). The tissue was
subsequently triturated and filtered through a 40 µm sterile
cell strainer to remove insoluble debris. The cells were plated
into poly-D-lysine coated T75 culture flasks, containing Dulbecco's
modified Eagle's medium (DMEM) with 20% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), until the cells reached confluence
(~10 days), by which time a bed of astrocytes had grown with a
layer of OPCs on top. The flasks were subsequently agitated at 200
rpm for 1 h to dislodge dead cells and microglia. The media was
changed and the flasks were agitated overnight at 200 rpm to
dislodge the OPCs. The OPCs were collected and plated into poly-D,
L-ornithine coated culture dishes with serum-free DMEM,
supplemented with hormones and growth factors (10 nM of each
platelet-derived growth factor-α and basic fibroblast growth
factor). To induce differentiation, the growth factors were
withdrawn from the medium and ciliary neurotrophic factor was
added. To avoid spontaneous differentiation, the cells were not
used beyond one passage. Cell viability was determined using a
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
(MTT) assay, and the apoptosis rate was analyzed by flow
cytometry.
Drug treatment
Stock solutions of E2 (10−6,
10−7, 10−8, 10−9 M) were prepared
in dimethyl sulfoxide (DMSO). E2 was added to the culture medium
for 0, 8, 16, 24 or 48 h, and subsequently the cells were collected
and the mRNA expression of PirB was detected using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). The
concentration of E2 at which the PirB mRNA level dropped the most
was selected as the appropriate concentration.
Small interfering (si)RNA and
transfection
PriB siRNA (5′-GTGTTCAGTTGTTCCCTTGACATGA-3′) and
negative control (NC) siRNA were purchased from Shanghai Jima
Biotechnology Co., Ltd. (Shanghai, China). The cells were plated at
50% confluence and transfected with 100 nM siRNA using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol.
OPC grouping
The OPCs were randomly divided into five groups: i)
Normal culture condition plus DMSO treatment group (normal), where
cells were cultured under normal conditions and treated with 1%
DMSO for 48 h; ii) the hyperoxia plus DMSO treatment group
(hyperoxia), where cells were first treated with 1% DMSO for 24 h
under normal conditions and were subsequently cultured under
hyperoxia conditions for 24 h; iii) the hyperoxia plus NC siRNA
group (hyperoxia + NC), where the cells were first transfected with
NC siRNA for 24 h under normal conditions and were subsequently
cultured under hyperoxic conditions for 24 h; iv) the hyperoxia
plus PirB siRNA group (hyperoxia + siRNA), where cells were first
transfected with PirB siRNA for 24 h under normal conditions and
were subsequently cultured under hyperoxic conditions for 24 h; v)
the hyperoxia plus E2 group (hyperoxia + E2), where cells were
first treated with E2 for 24 h under normal conditions and were
subsequently cultured under hyperoxic conditions for 24 h. The
hyperoxic groups were maintained at 37°C in a humidified air
incubator containing 80% O2, 5% CO2 and 15%
N2. For the normal culture condition group, the cells
were placed in a humidified incubator containing <21%
O2 and 5% CO2 at 37°C. For the E2 treatment
groups, the cells were cultured in culture medium supplemented with
the appropriate concentration of E2.
RNA extraction and RT-qPCR
Cell pellets of 6×105 cells from each
group were lysed using ice-cold TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. The RNA was reverse transcribed into cDNA
using PrimeScript RT reagent kit with cDNA Eraser (Takara Bio,
Inc., Dalian, China) in a 20 µl reaction, according to the
manufacturer's protocol. Equal quantities of cDNA were used as
templates for RT-qPCR to detect the expression level of PirB
expression relative to that of actin (endogenous control) using a
Mx3000P Real-Time PCR system (Stratagene, La Jolla, CA, USA) and a
SYBR Premix Ex TaqII PCR kit (Takara Bio, Inc.). Experiments were
performed in duplicate and repeated three times. The fold induction
of gene expression was calculated using the 2−ΔΔCt
method.
OPC cell viability measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The MTT assay is a laboratory test that measures
changes in color for measuring the activity of an enzyme that
reduces MTT (yellow color) to formazan (purple color). Following 48
h of incubation, 20 µl (5 mg/ml) MTT reagent was added to
each well and incubated for an additional 4 h. DMSO solution (200
µl) was subsequently added to each well to solubilize the
formazan crystals. The plates were read for optical density at 570
nm using a Multiskan MK3 plate reader (Thermo Fisher Scientific,
Inc.). The cell survival rate was calculated based on the optical
density of the cells.
Apoptosis analysis of OPCs using flow
cytometry
Each OPC group was seeded into a 6-well plate, at a
density of 104cells/well. The treated cells were washed
twice with cold phosphate-buffered saline and the cell pellet
(1–5×105) resuspended in binding buffer (Keygen Biotech
Co., Ltd., Nanjing, China) at a concentration of 106
cells/ml. The cells were mixed with 10 µl fluorescein
isothiocyanate-conjugated annexin-V reagent and 10 µl of 3
mM propidium iodide. Following incubation for 15 min at room
temperature in the dark, flow cytometry was performed using a BD
Accuri C6 FACScan analyzer (BD Pharmingen, San Diego, CA, USA).
Western blotting
The cells were lysed in radioimmunoprecipitation
assay buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% (v/v)
Nonidet P-40, 0.5% (v/v) sodium deoxycholate and 0.1% sodium
dodecyl sulfate (SDS)], supplemented with a mixture of protease and
phosphatase inhibitors. The lysates were sonicated for 5 sec,
centrifuged for 20 min at 12,000 × g at 4°C and stored at −80°C.
Equal concentrations of protein (50–60 µg total protein)
were electrophoretically separated by 10% SDS-polyacrylamide gel
(Beyotime Institute of Biotechnology) electrophoresis and
transferred onto nitrocellulose membranes (Pall Life Sciences, Port
Washington, NY, USA). The membranes were blocked with 5% non-fat
dry milk in Tris-buffered saline (TBS), containing 0.01% Tween-20
(TBS-T) for 1 h at 37°C. The membranes were subsequently incubated
with the following primary monoclonal antibodies overnight at 4°C:
Rabbit anti-PriB (1:1,000; ta323298; OriGene Technologies, Inc.,
Beijing, China), mouse anti-Fas (1:2,000; 05-351), rabbit
anti-caspase-3 (1:1,500; 04-439; both EMD Millipore, Billerica, MA,
USA), rabbit anti-caspase-8 (1:1,000; ab119892; Abcam, Cambridge,
MA, USA), mouse anti-phosphorylated (p)-Akt (1:2,500; MAB887) and
mouse anti-Akt (1:2,000; MAB2055; both R&D Systems,
Minneapolis, MN, USA). The membranes were subsequently washed three
times with TBS-T, followed by a 2–3 h incubation with horseradish
peroxidase-conjugated goat anti-rabbit (1:20,000; BA1055) or
anti-mouse (1:25,000; BA1050; both BosterBio, Wuhan, China)
immunoglobulin G secondary antibodies at 37°C. Following washing
three times with TBS-T, the protein bands were visualized using
Super Signal West Pico substrate (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The film was scanned and densitometric analysis
was performed using Image Pro-Plus 6.0 software (Media Cybernetics,
Inc., Rockville, MD, USA). The results of densitometric analysis
were expressed as a relative ratio of the target protein to
reference protein. The relative ratio of the target protein of the
control group was arbitrarily presented as 1.
Statistical analysis
Statistical analysis was performed using SPSS 18.0
software (SPSS, Inc., Chicago, IL, USA). All results are presented
as the mean ± standard deviation. Student's t-test was used to
determine the differences among the groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
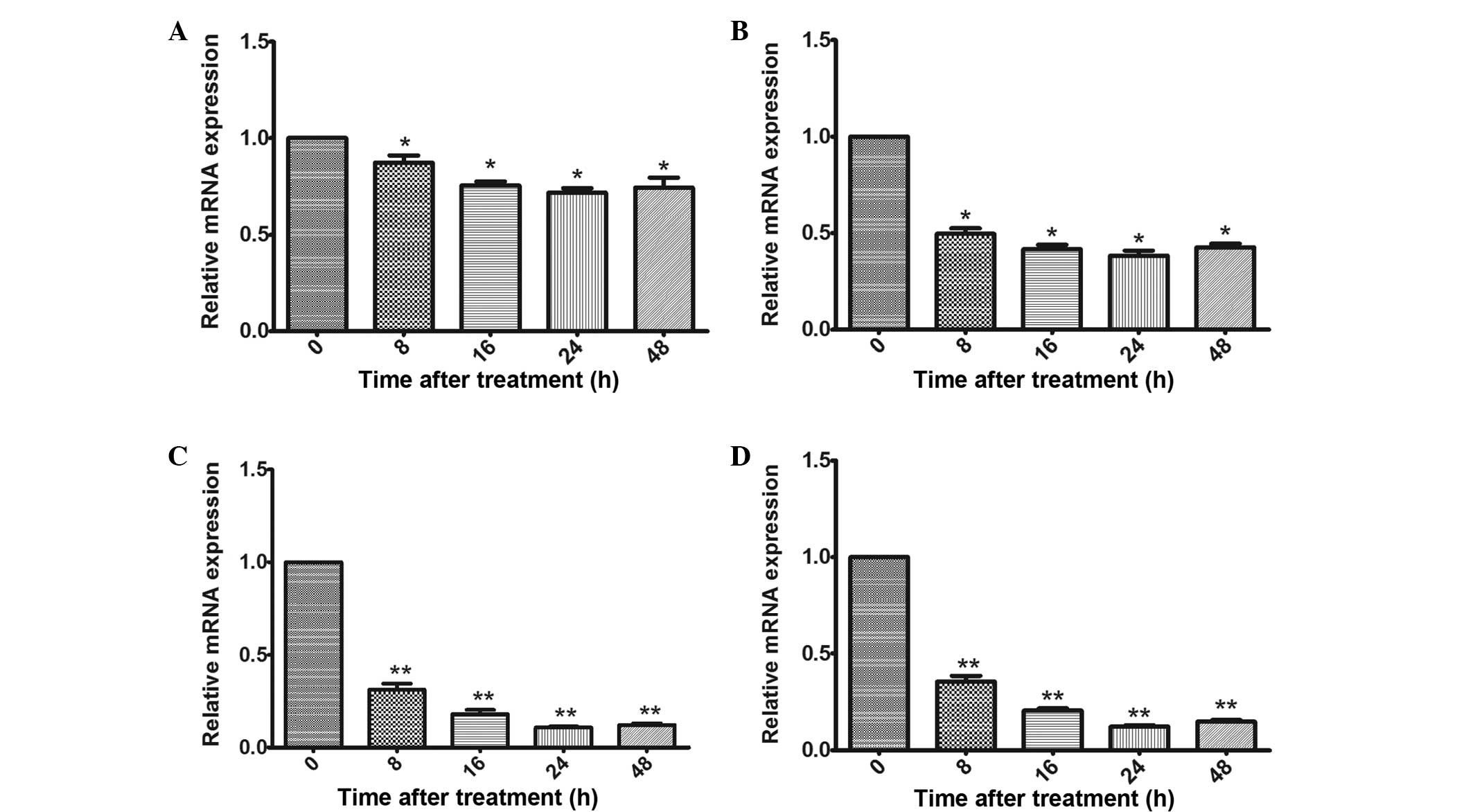
E2 treatment suppresses the mRNA
expression of PirB
To investigate the effect of E2 on OPCs, the mRNA
level of PriB following treatment with different concentrations of
E2 was first examined at different time-points. As shown in
Fig. 1A, the lowest concentration
of E2 (10−9 M) reduced the expression of PriB after 8 h,
and the PriB expression began to gradually decrease at 16 h
following treatment with E2. Compared with the 10−9 M
treatment, the mRNA expression of PriB exhibited a clear decrease
following treatment with 10−8, 10−7,
10−6 M E2 (Fig. 1B and
C). The 10−7 and 10−6 M concentrations of
E2 exhibited similar effects on the expression of PriB. Based on
these results, the treatment with 10−7 M E2 for 24 h was
selected for subsequent experiments.
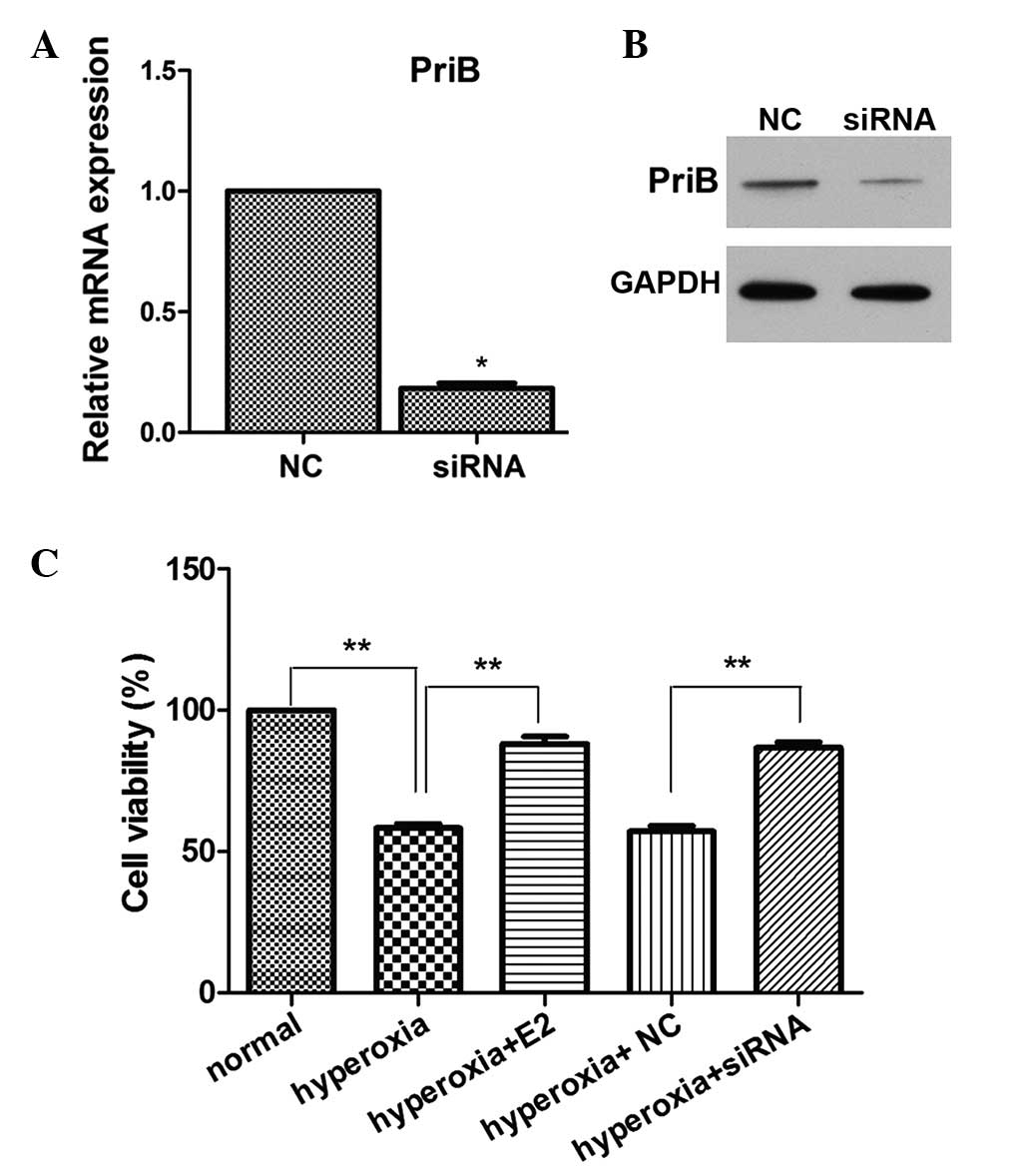
E2 treatment and silencing of PriB
increases OPC viability under hyperoxic stimulation
To further investigate the function of PriB on OPCs
under hyperoxia stimulation, PriB siRNA was used to transfect OPCs,
and the mRNA and expression levels of PriB were subsequently
measured. As shown in Fig. 2A and
B, PriB expression levels were successfully reduced. To
investigate the role of E2 on OPCs under hyperoxia stimulation, the
effect of E2 on cell viability was detected. As shown in Fig. 2C, hyperoxic stimulation
significantly decreased cell viability, as compared with the normal
culture controls. E2 pretreatment markedly increased cell
viability, as compared with the hyperoxia group. PriB siRNA
pretreatment also markedly increased cell viability, as compared
with the negative control siRNA pretreatment group under hyperoxic
stimulation (Fig. 2C).
E2 treatment and silencing of PriB
decreases cell apoptosis under hyperoxic stimulation
To further investigate the role of E2 on OPCs under
hyperoxia stimulation, the effect of E2 on cell apoptosis was
examined. As shown in Fig. 3A and
B, hyperoxic stimulation markedly increased both early and late
apoptosis, as compared with the normal culture condition controls.
E2 pretreatment notably decreased both early and late apoptosis
compared with the hyperoxia group. Furthermore, PriB siRNA
pretreatment also markedly decreased both early and late apoptosis
compared with the negative control siRNA pretreatment group under
hyperoxic stimulation (Fig. 3A and
B).
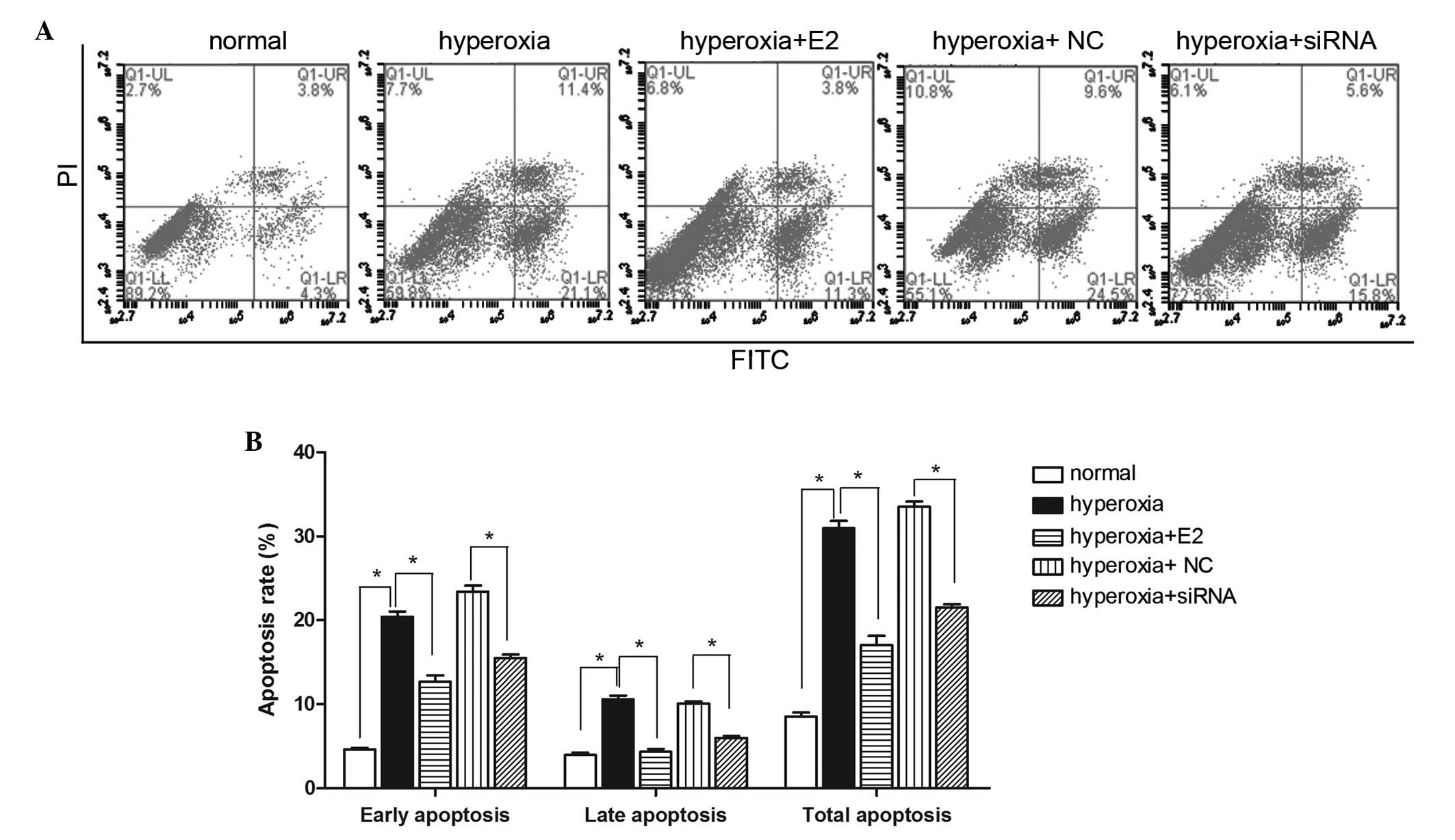 | Figure 3Effect of E2 treatment and PriB siRNA
pre-treatment on oligodendrocyte precursor cell apoptosis under
hyperoxia stimulation. (A) Flow cytometric analysis using annexin
V-FITC/PI staining following the different treatments. (B) Graph
showing the percentage of early, late and total apoptotic cells
(*P<0.05). Data are presented as the mean ± standard
deviation of three independent measurements. Q1-LL, healthy cells;
Q1-LR, early apoptotic cell; Q1-UL, necrotic cell; Q1-UR, late
apoptotic cell; E2, 17β-Estradiol; si, small interfering; FITC,
fluorescein isothiocyanate; PI, propidium iodide; NC, negative
control. |
E2 treatment and silencing of PirB
significantly reduces the expression of caspases-3 and -8, Fas and
Akt, however induces p-Akt
To further investigate the role of E2 and PirB on
OPC apoptosis, western blot analysis was performed to measure the
protein expression of caspases-3 and -8, Fas, Akt and p-Akt. As
shown in Fig. 4, hyperoxia
stimulation significantly induced the expression of caspases-3 and
-8, and Fas; however, hyperoxia stimulation significantly reduced
p-Akt. E2 pretreatment significantly decreased the
hyperoxia-induced expression of caspases-3 and -8, and Fas, and
increased the expression of p-Akt. Furthermore, PriB siRNA
pretreatment also significantly decreased the hyperoxia-induced
expression of caspases-3 and -8, and Fas and hyperoxia-induced
p-Akt reduction compared with the NC siRNA pretreatment group under
hyperoxic stimulation. No significant difference was identified in
the expression levels of Akt among all groups.
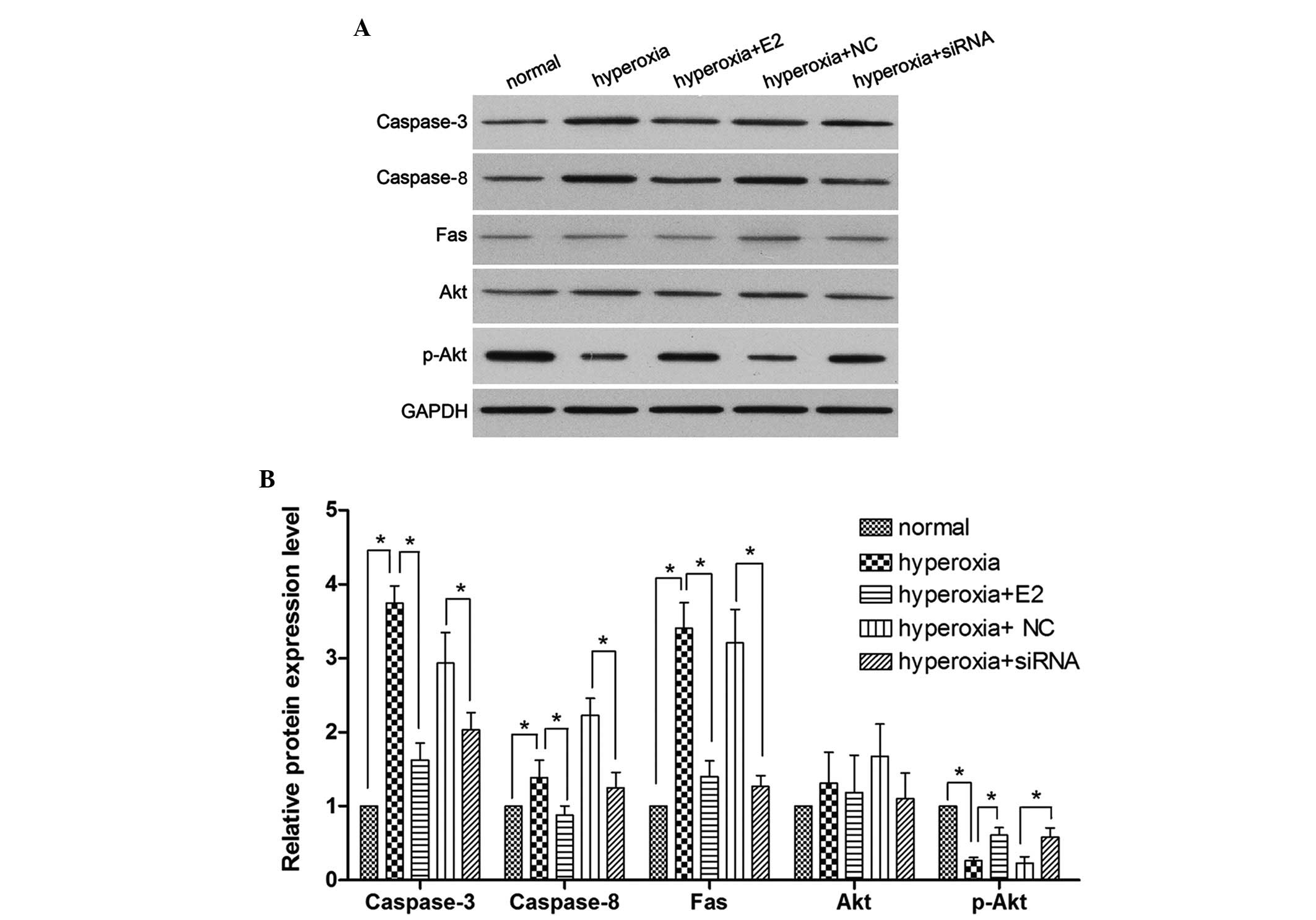 | Figure 4Effect of E2 treatment and PriB siRNA
pre-treatment on the expression of caspases-3 and -8, Fas, p-Akt
and Akt under hyperoxia stimulation. (A) Western blot analysis
following the different treatments. (B) The protein expression
levels of caspases-3 and -8, Fas, p-Akt, and Akt were quantified
using a densitometer (*P<0.05). Data are presented as
the mean ± standard deviation of three independent measurements.
E2, 17β-Estradiol; si, small interfering; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; NC, negative control; p-,
phosphorylated. |
Discussion
It has been shown that extremely low birth weight
remains an important risk factor for neurodevelopmental impairment
at 18 months of age (29,30). Oxygen is one of the most widely
used treatments in Neonatal Intensive Care Units, and previous
studies have shown that neonatal exposure to chronic hyperoxia
leads to reduced hippocampal size in adult mice (3). Although recent findings suggest that
neonatal rats, which have been exposed to supraphysiological
concentrations of oxygen are at high risk for neurodevelopmental
impairment in adults (6,7), they do not rule out the possibility
that hyperoxia may promote apoptosis in OPCs. Certain previous
studies have demonstrated that neuroactive steroids, including E2,
can protect neurons from various harmful compounds (31,32).
PirB is a receptor expressed on myeloid cells and is known to have
the ability to inhibit nerve cell regeneration (14,15).
The aim of the present study was to investigate the association
between hyperoxia, PirB and E2 treatment.
In the present study, OPCs were first treated with
various concentrations of E2 at different exposure durations, and
the ones during which the pirB mRNA expression reached its lowest
level were selected as the dosage and exposure durations of E2 for
the following assays. The results demonstrated that the optimum
concentration and exposure duration were 10−7 M and 24
h, respectively. Subsequently, the effect of E2 treatment on OPC
cell viability was analyzed using an MTT assay. Hyperoxia was
revealed to significantly decrease the cell viability of OPCs,
which was reversed by E2 treatment. Based on the knowledge that E2
treatment decreases the expression of PirB, the role of PirB in the
hyperoxia-induced decrease in OPC cell viability was investigated
next. PirB was successfully silenced by siRNA and this silencing
was revealed to abolish the hyperoxia-induced decrease in OPC cell
viability, indicating that E2 treatment and PirB silencing can
protect OPCs against hyperoxia-induced cell damage.
The effects of E2 treatment and PirB silencing on OL
apoptosis was also analyzed using flow cytometry. Hyperoxia was
found to significantly increase the apoptosis of OPCs, while E2
treatment was shown to partially reverse it. Furthermore, the
silencing of PirB was also found to abolish the hyperoxia-induced
apoptosis of OPCs, indicating that E2 treatment or silencing of
PirB can protect OPCs against hyperoxia-induced apoptosis, which
was consistent with previous studies (8,23,33,34).
In order to further investigate how hyperoxia
induces OPC apoptosis, western blot analysis was performed to
measure the protein expression levels of Akt, caspases-3 and -8,
Fas and p-Akt. GAPDH was used as the loading control. As shown in
Fig. 4, the expression levels of
caspases-3 and -8, and Fas, increased significantly following
oxygen treatment, whereas the expression of p-Akt significantly
decreased. E2 treatment partially reversed the hyperoxia-induced
upregulation of caspases-3 and -8, and Fas, as well as the
downregulation of p-Akt. Furthermore, the silencing of PirB was
also found to reverse the hyperoxia-induced upregulation of
caspases-3 and -8, and Fas, as well as the downregulation of p-Akt.
Based on the present and previous results (11–14),
it was concluded that hyperoxia resulted in the upregulation of
caspases-3 and -8, and Fas and the downregulation of p-Akt, leading
to cell damage and apoptosis.
Hyperoxia was found to increase the apoptosis and
decrease the survival rate of OPCs. OPC apoptosis was induced by
hyperoxia via the upregulation of caspases-3 and -8, and Fas. E2
treatment significantly downregulated the expression of PirB. The
downregulation of PirB, either by E2-treatment or PirB silencing,
markedly decreased hyperoxia-induced apoptosis, increased cell
viability, and decreased the expression of caspases-3 and -8, and
Fas in OPCs, indicating that E2 can protect against
hyperoxia-induced apoptosis, predominantly via the downregulation
of PirB in OPCs.
Acknowledgments
The present study was supported by funds from the
Natural Science Foundation of China (grant nos. 81401239 and
81401233).
References
|
1
|
Broitman E, Ambalavanan N, Higgins RD,
Vohr BR, Das A, Bhaskar B, Murray K, Hintz SR and Carlo WA;
National Institute of Child Health and Human Development Neonatal
Research Network: Clinical data predict neurodevelopmental outcome
better than head ultrasound in extremely low birth weight infants.
J Pediatr. 151:500–505. 505.e1–2. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neubauer AP, Voss W and Kattner E: Outcome
of extremely low birth weight survivors at school age: The
influence of perinatal parameters on neurodevelopment. Eur J
Pediatr. 167:87–95. 2008. View Article : Google Scholar
|
|
3
|
Auten RL, Mason SN, Auten KM and
Brahmajothi M: Hyperoxia impairs postnatal alveolar epithelial
development via NADPH oxidase in newborn mice. Am J Physiol Lung
Cell Mol Physiol. 297:L134–L142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dorfman A, Dembinska O, Chemtob S and
Lachapelle P: Early manifestations of postnatal hyperoxia on the
retinal structure and function of the neonatal rat. Invest
Ophthalmol Vis Sci. 49:458–466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Londhe VA, Sundar IK, Lopez B, Maisonet
TM, Yu Y, Aghai ZH and Rahman I: Hyperoxia impairs alveolar
formation and induces senescence through decreased histone
deacetylase activity and upregulation of p21 in neonatal mouse
lung. Pediatr Res. 69:371–377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramani M, Van Groen T, Kadish I, Bulger A
and Ambalavanan N: Neurodevelopmental impairment following neonatal
hyperoxia in the mouse. Neurobiol Dis. 50:69–75. 2013. View Article : Google Scholar :
|
|
7
|
Vottier G, Pham H, Pansioot J, Biran V,
Gressens P, Charriaut-Marlangue C and Baud O: Deleterious effect of
hyperoxia at birth on white matter damage in the newborn rat. Dev
Neurosci. 33:261–269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sosa MA, De Gasperi R, Paulino AJ, Pricop
PE, Shaughness MC, Maudlin-Jeronimo E, Hall AA, Janssen WG, Yuk FJ,
Dorr NP, et al: Blast overpressure induces shear-related injuries
in the brain of rats exposed to a mild traumatic brain injury. Acta
Neuropathol Commun. 1:512013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Østerholt HC, Dannevig I, Wyckoff MH, Liao
J, Akgul Y, Ramgopal M, Mija DS, Cheong N, Longoria C, Mahendroo M,
et al: Antioxidant protects against increases in low molecular
weight hyaluronan and inflammation in asphyxiated newborn pigs
resuscitated with 100% oxygen. Plos One. 7:e388392012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gerstner B, Bührer C, Rheinländer C,
Polley O, Schüller A, Ber ns M, Obladen M and Felderhoff-Mueser U:
Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia.
J Neurosci Res. 84:306–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanamoto T, Mota M, Takeda K, Rubin LL,
Miyazono K, Ichijo H and Bazenet CE: Role of apoptosis
signal-regulating kinase in regulation of the c-Jun N-terminal
kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol.
20:196–204. 2000. View Article : Google Scholar
|
|
12
|
DekkerS MP, Mikoletopoulou V and Barde YA:
Cell biology in neuroscience: Death of developing neurons: New
insights and implications for connectivity. J Cell Biol.
203:385–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akhter R, Sanphui P and Biswas SC: The
essential role of P53 up-regulated modulator of apoptosis (Puma)
and its regulation by FoxO3a transcription factor in β-amyloid
induced neuron death. J Biol Chem. 289:10812–10822. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Atwal JK, Pinkston-Gosse J, Syken J,
Stawicki S, Wu Y, Shatz C and Tessier-Lavigne M: PirB is a
functional receptor for myelin inhibitors of axonal regeneration.
Science. 322:967–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Xiong Y and Mu D: PirB restricts
neuronal regeneration in developing rat brain following
hypoxia-ischemia. Mol Med Rep. 6:339–344. 2012.PubMed/NCBI
|
|
16
|
Tyson JE, Parikh NA, Langer J, Green C and
Higgins RD; National Institute of Child Health and Human
Development Neonatal Research Network: Intensive care for extreme
prematurity-moving beyond gestational age. N Engl J Med.
358:1672–1681. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karki P, Smith K, Johnson J Jr and Lee E:
Astrocyte-derived growth factors and estrogen neuroprotection: Role
of transforming growth factor-α in estrogen-induced upregulation of
glutamate transporters in astrocytes. Mol Cell Endocrinol.
389:58–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang L, Andersson S, Warner M and
Gustafsson JA: Estrogen receptor (ER)beta knockout mice reveal a
role for ERbeta in migration of cortical neurons in the developing
brain. Proc Natl Acad Sci USA. 100:703–708. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mitra SW, Hoskin E, Yudkovitz J, Pear L,
Wilkinson HA, Hayashi S, Pfaff DW, Ogawa S, Rohrer SP, Schaeffer
JM, et al: Immunolocalization of estrogen receptor beta in the
mouse brain: Comparison with estrogen receptor alpha.
Endocrinology. 144:2055–2067. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Behl C, Widmann M, Trapp T and Holsboer F:
17-β estradiol protects neurons from oxidative stress-induced cell
death in vitro. Biochem Biophys Res Commun. 216:473–482. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Merchenthaler I, Dellovade TL and Shughrue
PJ: Neuroprotection by estrogen in animal models of global and
focal ischemia. Ann N Y Acad Sci. 1007:89–100. 2003. View Article : Google Scholar
|
|
22
|
Brännvall K, Korhonen L and L indholm D:
Estrogen-receptor-dependent regulation of neural stem cell
proliferation and differentiation. Mol Cell Neurosci. 21:512–520.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishihara Y, Fujitani N, Kawami T, Adachi
C, Ishida A and Yamazaki T: Suppressive effects of 17β-estradiol on
tributyltin-induced neuronal injury via Akt activation and
subsequent attenuation of oxidative stress. Life Sci. 99:24–30.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sifringer M, Bendix I, Börner C,
Endesfelder S, von Haefen C, Kalb A, Holifanjaniaina S, Prager S,
Schlager GW, Keller M, et al: Prevention of neonatal oxygen-induced
brain damage by reduction of intrinsic apoptosis. Cell Death Dis.
3:e2502012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stark S, Schuller A, Sifringer M, Gerstner
B, Brehmer F, Weber S, Altmann R, Obladen M, Buhrer C and
Felderhoff-Mueser U: Suramin induces and enhances apoptosis in a
model of hyperoxia-induced oligodendrocyte injury. Neurotox Res.
13:197–207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Atwal JK, Pinkston-Gosse J, Syken J,
Stawicki S, Wu Y, Shatz C and Tessier-Lavigne M: PirB is a
functional receptor for myelin inhibitors of axonal regeneration.
Science. 322:967–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim EJ, Kwon KJ, Park JY, Lee SH, Moon CH
and Baik EJ: Neuroprotective effects of prostaglandin E2 or cAMP
against microglial and neuronal free radical mediated toxicity
associated with inflammation. J Neurosci Res. 70:97–107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Y, Balasubramaniyan V, Peng J,
Hurlock EC, Tallquist M, Li J and Lu QR: Isolation and culture of
rat and mouse oligodendrocyte precursor cells. Nat Protoc.
2:1044–1051. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lorenz JM, Wooliever DE, Jetton JR and
Paneth N: A quantitative review of mortality and development
disability in extremely premature newborns. Arch Pediatr Adolesc
Med. 152:425–435. 1998.PubMed/NCBI
|
|
30
|
Vohr BR, Wright LL, Dusick AM, Mele L,
Verter J, Steichen JJ, Simon NP, Wilson DC, Broyles S, Bauer CR, et
al: Neurodevelopment and functional outcomes of extremely low birth
weight infants in the national institute of child health and human
development neonatal research network, 1993–1994. Pediatrics.
105:1216–1226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Acconcia F, Totta P, Ogawa S, Cardillo I,
Inoue S, Leone S, Trentalance A, Muramatsu M and Marino M: Survival
versus apoptotic 17beta-estradiol effect: Role of ER alpha and ER
beta activated non-genomic signaling. J Cell Physiol. 203:193–201.
2005. View Article : Google Scholar
|
|
32
|
Aguirre CC and Baudry M: Progesterone
reverses 17beta-estra diol-mediated neuroprotection and BDNF
induction in cultured hippocampal slices. Eur J Neurosci.
29:447–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Prins SA, Von Lindern JS, Van Dijk S and
Versteeqh FG: Motor development of premature infants born between
32 and 34 weeks. Int J Pediatr. 2010:pii: 462048. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miller SL, Yawno T, Alers NO,
Castillo-Melendez M, Supramaniam VG, VanZyl N, Sabaretnam T, Loose
JM, Drummond GR, Walker DW, et al: Antenatal antioxidant treatment
with melatonia to decrease newborn neurodevelopmental deficits and
brain injury caused by fetal growth restriction. J Pineal Res.
56:283–294. 2014. View Article : Google Scholar : PubMed/NCBI
|