Introduction
Mevalonate kinase deficiency (MKD; 610377) is an
autosomal monogenic recessively-inherited disease, caused by
mutations in the MVK gene (12q24.11) coding for mevalonate
kinase (MK). MK is a key enzyme of the mevalonate pathway, which is
essential for the biosynthesis of isoprenoids and the decrease of
which is considered to lead to an overproduction of the specific
marker of disease, interleukin (IL)-1β (1–3).
These mutations lead to a shortage of intermediate compounds and
final products of the metabolic route of cholesterol (4–6).
The residual activity of MK defines different
degrees of MKD severity, ranging between an auto-inflammatory
phenotype (hyper IgD syndrome/HIDS; OMIM #260920) and severe
clinical presentation (mevalonic aciduria/MA; OMIM #610377)
(6). HIDS is characterized by
fever, which re-occurs every 2–8 weeks, variably accompanied by
malaise, headache, diarrhea, abdominal pain, vomiting, skin rash,
arthralgia, arthritis, tender lymphadenopathy, anemia normocytic,
hepatosplenomegaly, and as oral and genital ulcers. Urinary
mevalonic acid (UMA) is increased predominantly during inflammatory
episodes, whereas increased levels of IL-1β, IL-1α, IL-6, IL-10,
IL-18, tumor necrosis factor (TNF)-α and IFN-γ cytokines are also
observed between fever episodes (7,8).
Laboratory analyses show increased neutrophil counts and high acute
phase reactants, often associated with persistently high serum IgD
and IgA (9).
Patients with MA exhibit the above-mentioned
symptoms associated with dysmorphic features, cataracts, uveitis,
neurological impairments, and failure to thrive. UMA excretion is
also high between attacks (10).
There have been >70 different disease-causing mutations
identified in the MVK gene. The majority of these exhibit
poor genotype-phenotype correlation with respect to clinical
presentation and biochemical derangements (11). Notably, it has been found that
>95% of patients with HIDS are compound heterozygous for the
V377I MVK allele, whereas a second mutant allele, I268T, is
specific to patients with MA (12). Other mutations have been described
in patients with HIDS and MA without a reliable genotype/phenotype
correlation (13), although
patients carrying the same mutation often exhibit substantial
variability in symptoms and respond differently to treatment
(14).
Of note, previous studies have described a
correlation and/or an overlap between the MKD phenotype and/or the
MVK genotype with other diseases, including inflammatory
bowel diseases (15,16), possibly due to shared genetic
background (17), and retinitis
pigmentosa, adding novel features to the wide range of the already
known MKD phenotypes (18,19).
Whilst several reports suggest that the shortage of
isoprenoid intermediates, including geranylgeraniol, is involved in
the autoinflammatory phenotype of MKD, further data has led to a
focus of attention on the end products of this pathway, including
25-deoxycholesterol (CH25) (20–22).
Of note, 25-hydroxylcholesterol can act by modulating the
mevalonate pathway itself by suppressing sterol regulatory
element-binding proteins (SREBPs), thus highlighting a complex
feedback network, which may be relevant in the modulation of MKD
phenotypes (23,24). The present study hypothesized that
MKD may require consideration a polygenic, rather than a monogenic,
disease. The overlap and the wide range of clinical signs shared by
MKD and other auto-inflammatory diseases can cause delayed or
incorrect diagnosis. In order to verify this hypothesis, the
presents study performed whole exome sequencing (WES) analysis on
DNA samples obtained from patients with mild or severe MKD
phenotypes to determine any more or less pathogenetic variants,
which may be involved in determining the phenotype of the
patient.
Materials and methods
Study design
Initially, the present identified the small
nucleotide variants (SNVs)/small insertion/deletions (INDELs),
which had a minor allele frequency (MAF) <0.05, as reported in
the NHLBI Exome Sequencing Project (ESP) Exome Variant Server
(http://evs.gs.washington.edu/EVS/)
database and referred to the general population. In order to be
considered, the variants also had to be harbored by genes belonging
to the cholesterol biosynthetic pathway (Table I). The hypothesis underlying this
approach was that the variants harbored by these genes modulate
overall cholesterol biosynthetic activity, possibly affecting the
overall availability of cholesterol and/or its biosynthetic
intermediates, thus contributing to the modification of MKD
clinical phenotypes.
 | Table ICholesterol biosynthetic pathway
genes used for analysis. |
Table I
Cholesterol biosynthetic pathway
genes used for analysis.
| Abbreviation | Gene |
|---|
| HADHB | Hydroxyacyl-CoA
dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, β
subunit |
| ACAT2 | Acetyl-CoA
acetyltransferase 2 |
| ACAT1 | Acetyl-CoA
acetyltransferase 1 |
| HMGCS1 |
3-hydroxy-3-methylglutaryl-CoA synthase
1 |
| HMGCS2 |
3-hydroxy-3-methylglutaryl-CoA synthase
2 |
| HMGCR |
3-hydroxy-3-methylglutaryl-CoA
reductase |
| MVK | Mevalonate
kinase |
| PMVK | Phosphomevalonate
kinase |
| MVD | Mevalonate
(diphospho) decarboxylase |
| IDI2 |
Isopentenyl-diphosphate δ isomerase 2 |
| IDI1 |
Isopentenyl-diphosphate δ isomerase 1 |
| GGPS1 | Geranylgeranyl
diphosphate synthase 1 |
| FDPS | Farnesyl
diphosphate synthase |
| CYP51A1 | Cytochrome P450,
family 51, subfamily A, polypeptide 1 |
| EBP | Emopamil binding
protein (sterol isomerase) |
| SC5DL |
Sterol-C5-desaturase |
| DHCR7 |
7-dehydrocholesterol reductase |
| NSDHL | NAD(P) dependent
steroid dehydrogenase-like |
| HSD17B7 | Hydroxysteroid
(17-β) dehydrogenase 7 |
| SQLE | Squalene
epoxidase |
| LSS | Lanosterol
synthase |
| DHCR24 |
24-dehydrocholesterol reductase |
| LBR | Lamin B
receptor |
| TM7SF2 | Transmembrane 7
superfamily member 2 |
| SC4MOL | Methylsterol
monooxygenase 1 |
| FDFT1 |
Farnesyl-diphosphate farnesyltransferase
1 |
The present study also investigated an alternative
analytical strategy, selecting only the variants that are poorly
represented in the general population and, at the same time,
predicted as potentially pathogenic. For this purpose, the
SNVs/INDELs were selected, according the following inclusion
criteria: (a) variants with an MAF <0.03 in the general
population, as reported in the ESP database, (b) SNVs leading to a
non synonymous amino acid substitution, (c) SNVs/INDELs carried in
homozygous state, (d) SNVs/INDELs predicted as pathogenic by the
following in silico algorithms: Polyphen-2 (25), Mutation Taster (26) and likelihood relation test
(27), according to scores
recorded in the dbNSFP v2.0 database (http://sites.google.com/site/jpopgen/dbNSFP); (e)
SNVs/INDELs considered phylogenetically conserved, based on genomic
evolutionary rate profiling (GERP)++ scores reported in the dbNSFP
v2.0 database. The first analytical approach examined the
hypothesis that a number non-rare variants are involved in
modifying the MKD clinical phenotype, acting along the same
biochemical pathway, whereas the second was intended to identify
rare and pathogenic variants, carried in a homozygous state and
possibly associated to certain, more severe, MKD clinical
phenotypes.
The two strategies were performed taking into
consideration the phylogenetic nucleotide evolutionary
conservation, based on PhyloP (28) and GERP++ scores, as reported in
dbNSFP v2.0; and only substitutions of conserved nucleotides were
considered for their potential pathogenic role. This type of
alteration is a predictor of deleteriousness, being a variation
that reduces organism fitness, which is a property closely
associated with molecular pathogenicity (29).
DNA extraction
Genomic DNA (gDNA) was extracted from 1–2 ml
EDTA-anticoagulated blood from the proband and their parents using
an EZ1 DNA Blood kit (Qiagen, Milan, Italy), according to the
manufacturer's protocol.
Whole exome sequencing and bioinformatics
analysis
The technical and scientific review board of the
Institute for Maternal and Child Health-IRCCS 'Burlo Garofolo'
(Trieste, Italy; no.185/08; 19/08/2008) approved the present study.
For a child to be eligible, informed consent had to be obtained
from parents or caregivers. Furthermore, patients with MKD of any
age were excluded from the study if they had an acute or chronic
infectious disease, any clinically significant disorder, or if they
were currently on any medication with known effects on
immunological factors, including corticosteroids. Blood was
collected by venepuncture from five patients with mild or severe
MKD phenotypes, determined by a physician using a visual analogue
scale score (Table II). For each
patient, a specific control of the identical sex and the age was
used. Each control sample was collected, preserved and analyzed in
an identical method of the patient to precisely identify phenotypic
differences.
| Table IIMevalonate kinase deficiency
phenotypes based on the typical clinical signs and symptoms
observed in the patients. |
Table II
Mevalonate kinase deficiency
phenotypes based on the typical clinical signs and symptoms
observed in the patients.
| Patient | Gender | Abdominal pain | Frequency of
febrile attacks | Diarrhea | Rash | Associated
condition | Phenotype
(mild/severe) |
|---|
| P1 | M | 1 | 0 | 0 | 0 | None | Mild |
| P2 | M | 2 | 1 | 2 | 2 | Normocytic anemia
scheletric pain | Severe |
| P3 | F | 2 | 1 | 1 | 2 | Normocytic
anemia | Mild |
| P4 | F | 1 | 0 | 0 | 1 | None | Mild |
| P5 | M | 2 | 1 | 1 | 1 | Normocytic
anemia | Severe |
The samples obtained from the patients diagnosed
with MKD were analyZed using whole exome sequencing (WES). Starting
with 3 µg gDNA, collected from each subject, a TruSeq™ Exome
Enrichment 62 Mb kit (Illumina, Inc., San Diego, CA, USA) was used
to capture the whole exome; of which the target size was 62 Mb,
comprising the overall genes coding sequence, the 5′-untranslated
regions (UTRs) and 3′-UTRs. A fragment exome library was
constructed, according to the manufacturer's protocol, and a 100 bp
paired-end sequence was analyzed using the Illumina HiSeq 1000
platform (Illumina, Inc.). Raw sequencing data were collected as
unmapped reads in fast Q format. CLC Genomics Workbench ver. 6.5
software was used to assess the quality of reads, to map reads back
to the human reference genome, hg19, to calculate the overall
coverage, to perform local realignment and base quality
recalibration, and to identify SNVs and INDELs, all of which were
collected into standardized Variant Call Format version 4.1
(30). The SNVs/INDELs were
annotated using ANNOVAR software (31) referring to the following public
databases: refGene, NCBI dbSNP build137 (http://www.ncbi.nlm.nih.gov/SNP/), 1000 Genomes
Project (http://www.1000genomes.org/), NHLBI
ESP Exome Variant Server (http://evs.gs.washington.edu/EVS/), dbNSFP v2.0
(27) and NCBI ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Polymerase chain reaction (PCR) and
sanger analysis
PCR amplification was performed for the specific
gene coding sequence of the MVK and RAB40AL genes in
15 µl total volume. Each reaction contained 50 ng/µl
genomic DNA and KAPA 2 G Fast Hot Start Readymix (RESNOVA, Rome,
Italy). PCR amplification was performed using a common annealing
temperature in a touchdown thermocycler with a two-step cycle:
Initial denaturation at 96°C for 3 min, followed by a gradiant
(0.5°C reduction per cycle) of 10 cycles at 96°C for 15 sec, 63°C
for 10 sec and 72°C for 1 second. The second step was 28 cycles of
96°C for 15 sec, 53°C for 15 sec and 72°C for 1 sec. The PCR
products were purified with 2 µl ExoSAP (USB Corporation,
Cleveland, OH, USA) by incubation at 37°C for 20 min and 85°C for
10 min. All thermal cycling, PCR amplifications, ExoSAP
purifications and sequencing reactions were performed in a 2720
Thermal Cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The amplification products were directly
sequenced by the Sanger method using an ABI PRISM 3130XL automated
DNA sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The sequences were analyzed using Seqman II software (DNASar I
Lasergene, 7.0; DNAStar, Inc., Madison, WI, USA). The PCR
amplification and Sanger sequencing primers used to confirm the
variations identified in the PEX11 G gene were as follows:
PEX11GcDNA, forward 5′-TGA AAC TGA GAC AGA GGCTG-3′ and reverse
5′-AGT GTC AGG GGG TAG TGG-3; and Pex11gcDNA, forward CCT GTG GAC
AAT GCT GAAG and Pex11gcDNA, reverse TCA TCA AGG GCT GTC TGC.
The PCR amplification and Sanger sequencing primers
used to confirm the variations identified in the uracil-DNA
glycosylase (UNG) gene were as follows: UNG exon2,
forward 5′-CTG TCC GCT TTT GCT GGG-3′ and reverse 5′-CCG GCT ACA
CTA ACA AGAC-3′.
Urinary mevalonic acid measurement
Mevalonic acid levels were determined in the
patients with MKD using standard procedures, as described by
Shoemaker et al (32). The
urine samples were collected over 24 h and stored at 4°C until
analyzed.
Urinary mevalonic acid concentrations were
determined using gas chromatography/mass spectrometry. Spectra were
obtained using a Hewlett Packard gas chromatograph 6890 system
(Hewlett Packard, Palo Alto, CA, USA), equipped with a Hewlett
Packard 5973 quadrupole, operating in electron-impact mode at 70
eV, as described previously (32).
UMA is considered to be within a normal range between 34 and 323
µg/ml (internal laboratory reference).
B lymphocyte phenotype
The analysis of B lymphocyte phenotype was performed
using the whole peripheral blood samples from the patients and
controls. Briefly, 100 µl of blood was washed twice in
phosphate-buffered saline (PBS), and then stained with anti-IgD/IgM
fluorescein isothio-cyanate (2.5 µl/test; cat. no.
348206/314506; BioLegend, Inc., San Diego, CA, USA), anti-CD27
phycoerythrin (PE; 5 µl/test; cat. no. 130093185), anti-CD19
PE-Vio770 (5 µl/test; cat. no. 130096641); and anti-CD45 (5
µl/test; cat. no. 130092880), all from Miltenyi Biotec GmbH
(Bergisch Gladbach, Germany). Data were acquired using a Cyan ADP
cytometer (Beckman Coulter, Brea, CA, USA), and analysis was
performed using FlowJo software v7.6 (Tree Star, Inc., Ashland, OR,
USA). B-switched memory cells were identified as CD45/CD19/CD27
positive and IgM/IgM negative.
Results
A total of ~14 Gb of sequence data per sample were
produced using WES analysis, corresponding to 53X overall median
coverage, among all the samples. The overall target percentage
covered at least 20X, and was 80.7, 71, 73.6, 79, 75.3% of the
whole exome, in patients 1, 2, 3, 4 and 5, respectively. On
average, 59.404 variants per sample were identified.
As shown using Sanger Sequencing, the patients with
MKD in the present study carried at least one mutation in the
MVK gene. Data are present, together with the patients'
mevalonic acid levels, in Table
III.
| Table IIIMevalonate kinase deficiency genotype
and levels of mevalonic acid. |
Table III
Mevalonate kinase deficiency genotype
and levels of mevalonic acid.
| Patient | Mevalonic acid
(µg/ml) | Mutation | Genotype | dbSNP v137 |
|---|
| P1 | 5,000 | c.G394A;
p.V132I | Het | rs104895336 |
| | c.G1129A;
p.V377I | Hom | rs28934897 |
| P2 | 2,638 | c.T803C;
p.I268T | Het | rs104895304 |
| | c.G1129A;
p.V377I | Het | rs28934897 |
| P3 | >20,000 | c.16_34del;
p.6_12del | Het | rs104895334 |
| | c.G1129A;
p.V377I | Het | rs28934897 |
| P4 | 11,054 | c.G1129A;
p.V377I | Hom | rs28934897 |
| P5 | >20,000a | c.G1006A;
p.G336S | Hom | rs104895358 |
By selecting the SNVs/INDELs with an MAF <0.05,
the present study identified 10 exonic SNVs, listed in Table IV, six of which were harbored by
MVK, as already known, with the remaining four variants
identified in the SQLE, IDI1 and NSDHL genes,
even when none of them were in a homozygous or a compound
heterozygous state.
| Table IVVariants carried by genes belonging
to the cholesterol biosynthetic pathway. |
Table IV
Variants carried by genes belonging
to the cholesterol biosynthetic pathway.
| Refgene | Chr | AA change
(esp6500) | MAF | MAF (1,000 g) | dbSNP 137 | CVar | PolyPhen2 | LRT | Mutation
Taster | GERP++ | PhyloP | P1 | P2 | P3 | P4 | P5 |
|---|
| SQLE
NM_003129 | 8 |
exon6:c.A937T:p.N313Y | 0.0130 | 0.01 | rs118130263 | N | D | D | P | 0.08 (C) | 0.139 (N) | Het | WT | WT | WT | WT |
| |
exon6:c.A1042G:p.I348V | 0.0016 | 0.0005 | rs199608260 | N | B | P | P | −4.43 (N) | −1.574 (N) | WT | WT | Het | WT | WT |
| IDI1
NM_004508 | 10 |
exon1:c.G38A:p.C13Y | 0.0346 | 0.02 | rs7091756 | N | B | U | P | −3.54 (N) | −1.854 (N) | WT | WT | WT | Het | WT |
| MVK
NM_000431 | 12 |
exon2:c.16_34del:p.6_12del | NA | NA | rs104895334 | N | NA | NA | NA | NA | NA | WT | WT | Het | WT | WT |
| |
exon5:c.G394A:p.V132I | NA | NA | rs104895336 | N | B | P | P | −7.14 (N) | −1.322 (N) | Het | WT | WT | WT | WT |
| |
exon9:c.T803C:p.I268T | 0.0003 | NA | rs104895304 | Y | D | D | D | 4.60 (C) | 1.930 (C) | WT | Het | WT | WT | WT |
| |
exon10:c.G1006A:p.G336S | NA | NA | rs104895358 | N | D | D | D | 3.61 (C) | 1.095 (C) | WT | WT | WT | WT | Hom |
| |
exon11:c.G1129A:p.V377I | 0.0017 | NA | rs28934897 | Y | B | P | D | 3.17 (C) | 0.489 (N) | Hom | Het | Het | Hom | WT |
| NSDHL
NM_015922 | X |
exon4:c.G356A:p.R119K | 0.0003 | NA | rs200930841 | N | B | P | D | 3.97 (C) | 0.567 (N) | Het | WT | WT | WT | WT |
Subsequently, the SNVs/INDELs were selected,
according to the second filtering criteria, which can define SNPs
that are hard to confirm. The results of this filtering identified
four variants in patient 2 (P2) and two variants in patient 5 (P5),
as shown in Table V.
| Table VRare variants, predicted to be likely
pathogenic by three software packages. |
Table V
Rare variants, predicted to be likely
pathogenic by three software packages.
| RefGene | Chr | AA change | MAF (esp6500) | dbSNP 137 | PolyPhen2 | LRT | Mutation
Taster | GERP++ | PhyloP | P1 | P2 | P3 | P4 | P5 |
|---|
| FRA10AC1
NM_145246 | 10 | exon5:
c.G241A:p.V81I | 0.0008 | rs146931399 | P | D | D | 4.1 (C) | 1.095 (C) | WT | Hom | WT | WT | WT |
| GLT8D2
NM_031302 | 12 | exon10:
c.A817G:p.M273V | 0.0085 | rs145520946 | D | D | D | 4.26 (C) | 0.869 (N) | WT | Hom | WT | WT | WT |
| PEX11G
NM_080662 | 19 | exon5:
c.C646T:p.L216F | 0.0242 | rs11668511 | D | D | D | 5.2 (C) | 2.405 (C) | WT | Hom | WT | WT | WT |
| GLOD5
NM_001080489 | X | exon3:
c.G304A:p.D102N | 0.0014 | rs2022334 | D | D | D | 4.74 (C) | 2.077 (C) | WT | Hom | WT | WT | WT |
| UNG
NM_080911 | 12 | exon1:
c.C262T:p.R88C | 0.0012 | rs151095402 | P | D | D | 4.21 (C) | 2.182 (C) | WT | WT | WT | WT | Hom |
| MVK
NM_000431 | 12 | exon10:
c.G1006A:p.G336S | NA | rs104895358 | D | D | D | 3.61 (C) | 1.095 (C) | WT | WT | WT | WT | Hom |
Variants in P2
The analyses revealed an SNV in the PEX11γ
gene (19p13.2), carried in a homozygous state by patient P2. A
missense variation, rs11668511 (NM_080662 c.C646T; p.L216F), was
found in exon 5 of the PEX11γ gene. Several in silico
tools, including Polyphen-2, MutationTaster and LTR, predicted this
variant to be pathogenic or disease causing (Table V).
The present study also found the following variants
in different genes: rs146931399 (NM_145246.4 c.G241A; p.V81I) on
the FRA10AC1 gene, rs145520946 (NM_031302.3 c.A817G; p.M273
V) on the GLT8D2 gene and, finally, rs202215334
(NM_001080489 c.G304A:p.D102 N) on the GLOD5 gene. The
functions of the proteins encoded by these genes remain to be
elucidated and limited previous data exists. Therefore, it is
difficult to correlate these variants with the clinical phenotype
of P2.
Variants in P5
The present study detected a variant in the
UNG gene (12q23-q24.1), which was carried in a homozygous
state in patient P5. In this patient, the missense variation,
rs151095402 (NM_080911.2 c.262C>T; p.R88C), in exon 2, was found
in homozygosis for the first time, to the best of our knowledge..
Despite being rare (MAF, ~0,0012), this missense mutation had no
effect on the IgG, IgA or IgE concentrations. In addition, a normal
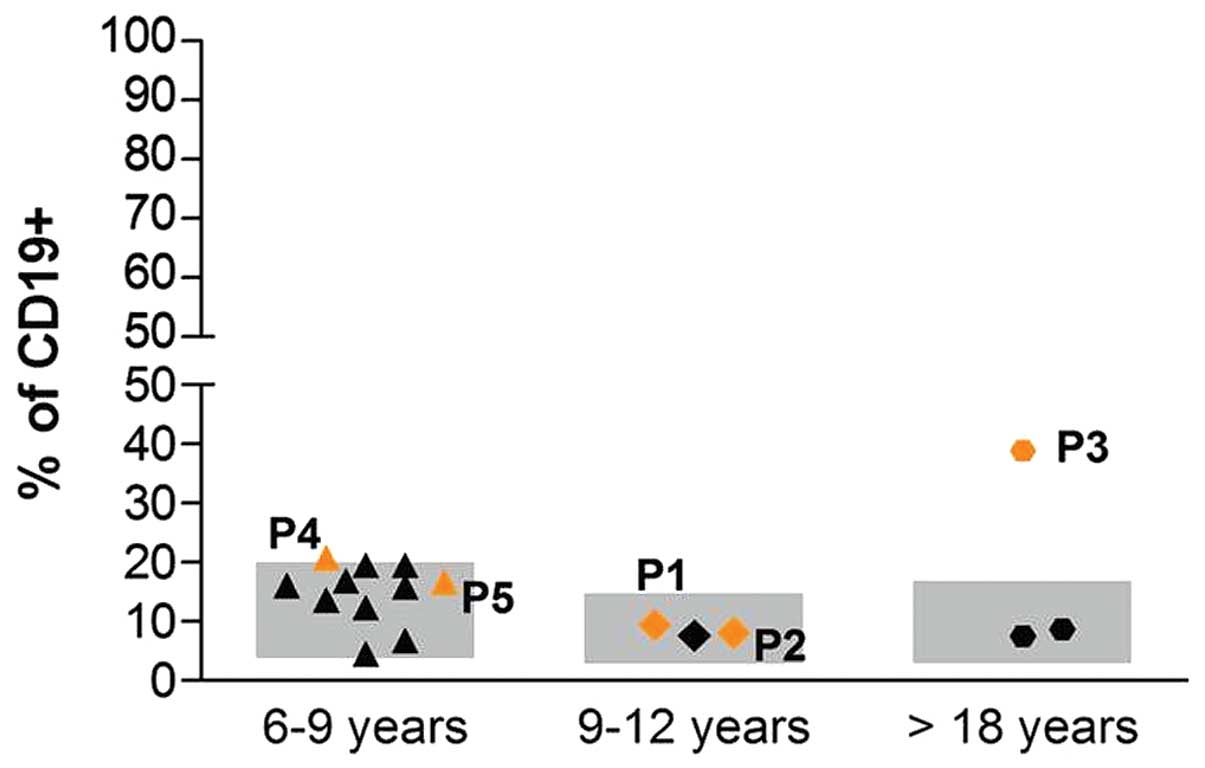
percentage of B-switched memory lymphocytes was observed (Fig. 1).
Discussion
Several studies have documented the phenotypic
heterogeneity of patients carrying mutations in the MVK gene
(15–19).
In the patients recruited in the present study, a
correlation was observed between MVK genotype and UMA urine
levels, and, of note, p.V132I has already been (33) described as being associated with
lower UMA levels. However, the present study found a poor
correlation between the' clinical phenotypes of the patients and
MVK mutations, even taking into account the mutations, which
change amino acids located close to each other in the protein
sequence, for example, p.V377I and p.G336S, and that were
considered to affect the same MK protein domain. Of considerable
clinical importance is the p.G336S mutation which, unlike other
mutations, determines inhibition of the metabolic pathway, causing
high UMA levels, which also occur outside of fever attacks
(5).
With the aim of identifying novel potential modifier
genes/variations associated with MKD phenotype variability, the
present study analyzed the entire exome of the five patients with
MKD, hypothesizing that MKD may be a polygenic disease. Considering
the heterogeneity of the clinical phenotypes, the present study
investigated the possible presence of modifier genes with potential
to affect the phenotype of the patients, in any way.
The variants obtained from a first analysis, which
filtered variants with an MAF <0.05 did not explain the wide
range of MKD phenotypes observed in the patients
When the analysis was performed with the second
filtering criteria, two putative genes were identified, which
potentially explained the clinical phenotype of two MKD patients
(P2 and P5).
Patient P2 was identified with a mutation in the
PEX11γ gene, which is a member of the PEX11 family, whose
predominant function is associated with the tabulation, enlargement
and clustering of peroxisomes, thus being important in human
metabolism (34). Peroxisomal gene
defects are also known to be associated with severe disorders
(35–38).
In mammals, there are three PEX11-associated genes,
PEX11-α, PEX11-β and PEX11-γ. Several previous
studies analyzing expression have been performed on PEX11-α,
and a few studies using animal models have been performed examining
the β proteins (35,39). The γ protein has been reported to
interact with α and β, and, by being present at the crossroads of
PEX11, activates the peroxisome proliferation pathways, possibly
being involved in homotypic interactions. In addition, PEX11-γ
overexpression has been suggested to have the ability to induce
early formation of juxtaposed elongated peroxisomes, suggesting
that the γ protein either acts upstream of PEX11-α and PEX11-β, or
that is their limiting factor. PEX11-γ appears to recruit the other
PEX11 proteins to facilitate aggregation, and is required for the
elongation of the peroxisome membrane (40).
Previously, a case of a patient carrying a
homozygous nonsense mutation in the PEX11-β gene has been
described. Using immunofluorescence microscopical analyses in
over-expression experiments at 37°C/40°C, it was suggested that the
occurrence of the enlarged catalase-containing peroxisomes is
associated with the PEX11-γ gene (38).
The PEX11γ gene and the c.C646T; p.L216F variant
have received limited attention in previous studies. The present
study hypothesized that this variant, rarely detected in the
general population (MAF, ~0.024), affected and modified the
phenotype of P2. Patient P2 presented with elevated UMA values due
to mutations in the MVK gene. However, in addition to
showing all the typical signs of a MKD phenotype, the patient
presented with visual blurring, not associated with cataracts
(37).
A mutation in the UNG gene encoding an
important DNA repair enzyme, was also found in P2.
The deletions in the UNG gene (c.391_393delC,
c.426_429delAT c.568_571delTA) and the missense mutations
(c.752T>C p.F251S) have been previously described to be
associated with hyper IgM immunodeficiency type 4 (40,41).
Hyper-IgM syndrome is characterized by normal or increased levels
of IgM, and is associated with low or absent serum levels of IgG,
IgA and IgE, indicating a defect in the class-switch recombination
process (42). In 2003, Imai et
al (43) reported on three
patients with HIGM5, in which deleterious mutations within the
catalytic domain of the UNG protein were identified. Functional
investigations of immunoprecipitation and immunoblot assays
confirmed the absence of expression in the three patients,
supporting the hypothesis of a correlation between the identified
mutation and protein instability.
In the present study, the p.R88C missense mutation
in the UNG gene identified in P5, was previously described
by Torseth et al (41), but
only in a heterozygotic state. Despite the fact that this mutation
is rare, its presence in homozygosis in the patient in the present
study had no effect on the levels of IgG, IgA and IgE (Fig. 1). In addition, the patient was
found to have a normal percentage of B-switched memory lymphocytes,
indicating normal immunity, with regards to the B-cell compartment.
Furthermore, patient four (P4) who was not a carrier of mutations
in the UNG gene, had high levels of IgA, as previously
reported described in several patients with MKD (44,45)
but with normal levels of IgG and IgM. Taken together, these
results suggested that investigation of the polymorphism found on
the UNG gene requires caution, as it does not correlate
directly with any specific disease phenotype.
The primary aim of the present study was to identify
novel potential modifier genes for MKD disease. A number of
preliminary results have shown that the clinical profile of
heterogeneous phenotypes of patients with MKD may be associated
with novel genes involved in modulating the MKD clinical picture
(15,18).
In the present study, following WES in five patients
with MKD, it was found that variations in genes encoding proteins
of the cholesterol pathway were not associated with the modulation
of MKD clinical phenotypes.
A missense variation, namely the c.C646T; p.L216F
(NM_080662) in exon 5 of the PEX11γ gene was observed in
homozygosis in P2, possibly correlating with visual blurring. A
rare UNG gene variant, namely (c.C262T; p.R88C), was
detected in homozygosis in P5, however this did not correlate with
a specific clinical phenotype. A number of other variants were
found in the five MKD patients analyzed, however, no correlations
with phenotype were observed. The lack of a direct correlation
between genetic variations and phenotype suggest a possible role of
post-transcriptional mechanisms, which may affect protein
expression or function.
In the patients examined in the present study, no
mutation was identified, but a a synonymous variation on
NM_003956:exon1:c.C657T:p.N219 N in the gene coding for the
CH25H gene was identified in the P2, P4 and P5 patients.
The patients in the present study were also analyzed
using direct sequencing to verify the presence of the intronic
polymorphism, identified by Moura et al (46). The intronic NM_001510.3:c.89-32007
A>G polymorphism (rs1450500) of the human glutamate receptor δ-2
(GRID-2) gene was not identified in any of the patient.
Therefore, it was concluded that, at least in these patients, and
in patients with more severe features, GRID2 does not appear
to be significantly involved.
Taking into account the primary limitation of the
present study lacking patients with MKD exhibiting different
clinical phenotypes, but sharing the same MVK mutations, the
use of WES, despite being an attractive approach, only provided
evidence of an association between one genetic variant, in the
PEX11γ gene, and one clinical characteristic, visual
blurring, in one of five patients. Therefore, further analysis
required, using NGS approaches on a larger sample size of patients
with MKD sharing the same MVK mutations and, ideally,
exhibiting extreme clinical phenotypes, in order to identify genes
and variants that correlate with clinical features. Considering the
fact that MKD is a rare, orphan disease, this approach may be
possible providing there are a sufficient number of patients with
the above-mentioned characteristics. The identification of modifier
genes specific for HIDS and MA may assist in the diagnoses of these
two forms of the same disease at an earlier stage.
Acknowledgments
This study was supported by a grant from the
Institute for Maternal and Child Health IRCCS 'Burlo Garofolo'
(grant no. RC 42/11) and the Associazione Azzurra Malattie Rare and
Beneficentia Stiftung (Vaduz, Liechtenstein).
Abbreviations:
|
MVK
|
mevalonate kinase gene
|
|
MK
|
mevalonate kinase
|
|
MKD
|
mevalonate kinase deficiency
|
|
HIDS
|
hyper IgD syndrome
|
|
MA
|
mevalonic aciduria
|
|
WES
|
whole exome sequencing
|
|
UTR
|
untranslated
|
|
SNVs
|
single nucleotide variants
|
|
INDELs
|
small insertion/deletions
|
|
VCF
|
variant call format
|
References
|
1
|
Mandey SH, Schneiders MS, Koster J and
Waterham HR: Mutational spectrum and genotype-phenotype
correlations in mevalonate kinase deficiency. Hum Mutat.
27:796–802. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Celec P and Behuliak M: The lack of
non-steroid isoprenoids causes oxidative stress in patients with
mevalonic aciduria. Med Hypotheses. 70:938–940. 2008. View Article : Google Scholar
|
|
3
|
Marcuzzi A, Decorti G, Pontillo A, Ventura
A and Tommasini A: Decreased cholesterol levels reflect a
consumption of anti-inflammatory isoprenoids associated with an
impaired control of inflammation in a mouse model of mevalonate
kinase deficiency. Inflamm Res. 59:335–338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Drenth JP, Cuisset L, Grateau G, Vasseur
C, van de Velde-Visser SD, de Jong JG, Beckmann JS, van der Meer JW
and Delpech M: Mutations in the gene encoding mevalonate kinase
cause hyper-IgD and periodic fever syndrome. International
Hyper-IgD Study Group. Nat Genet. 22:178–181. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Houten SM, Wanders RJ and Waterham HR:
Biochemical and genetic aspects of mevalonate kinase and its
deficiency. Biochim Biophys Acta. 1529:19–32. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haas D and Hoffmann GF: Mevalonate kinase
deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D
syndrome. Orphanet J Rare Dis. 1:132006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Caso F, Rigante D, Vitale A, Lucherini OM,
Costa L, Atteno M, Compagnone A, Caso P, Frediani B, Galeazzi M,
Punzi L and Cantarini L: Monogenic autoinflammatory syndromes:
state of the art on genetic, clinical, and therapeutic issues. Int
J Rheumatol. 2013:5137822013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Almeida de Jesus A and Goldbach-Mansky R:
Monogenic auto-inflammatory diseases: concept and clinical
manifestations. Clin Immunol. 147:155–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Frenkel J, Houten SM, Waterham HR, Wanders
RJ, Rijkers GT, Kimpen JL, Duran R, Poll-The BT and Kuis W:
Mevalonate kinase deficiency and Dutch type periodic fever. Clin
Exp Rheumatol. 18:525–532. 2000.PubMed/NCBI
|
|
10
|
Prietsch V, Mayatepek E, Krastel H, Haas
D, Zundel D, Waterham HR, Wanders RJ, Gibson KM and Hoffmann GF:
Mevalonate Kinase deficiency: enlarging the clinical and
biochemical spectrum. Pediatrics. 111:258–261. 2013. View Article : Google Scholar
|
|
11
|
Vuch J, Marcuzzi A, Bianco AM, Tommasini
A, Zanin V and Crovella S: Evolutionary hypothesis of the
Mevalonate Kinase Deficiency. Med Hypotheses. 80:67–69. 2013.
View Article : Google Scholar
|
|
12
|
Stabile A, Compagnone A, Napodano S,
Raffaele CG, Patti M and Rigante D: Mevalonate kinase genotype in
children with recurrent fevers and high serum IgD level. Rheumatol
Int. 33:3039–3042. 2013. View Article : Google Scholar
|
|
13
|
D'Osualdo A, Picco P, Caroli F, Gattorno
M, Giacchino R, Fortini P, Corona F, Tommasini A, Salvi G, Specchia
F, et al: MVK mutations and associated clinical features in Italian
patients affected with autoinflammatory disorders and recurrent
fever. Eur J Hum Genet. 13:314–320. 2005. View Article : Google Scholar
|
|
14
|
Shendi HM, Walsh D and Edgar JD:
Etanercept and anakinra can prolong febrile episodes in patients
with hyper-immunoglobulin D and periodic fever syndrome. Rheumatol
Int. 32:249–251. 2012. View Article : Google Scholar
|
|
15
|
Uhlig HHL: Monogenic diseases associated
with intestinal inflammation: implications for the understanding of
inflammatory bowel disease. Gut. 62:1795–1805. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levy M, Arion A, Berrebi D, Cuisset L,
Jeanne-Pasquier C, Bader-Meunier B and Jung C: Severe early-onset
colitis revealing mevalonate kinase deficiency. Pediatrics.
132:e779–e783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bianco AM, Girardelli M, Vozzi D, Crovella
S, Kleiner G and Marcuzzi A: Mevalonate kinase deficiency and IBD:
shared genetic background. Gut. 63:1367–1368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Siemiatkowska AM, Van den Born LI, Van
Hagen PM, Stoffels M, Neveling K, Henkes A, Kipping-Geertsema M,
Hoefsloot LH, Hoyng CB, Simon A, et al: Mutations in the mevalonate
kinase (MVK) gene cause nonsyndromic retinitis pigmentosa.
Ophthalmology. 120:2697–2705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Balgobind B, Wittebol-Post D and Frenkel
J: Retinitis pigmentosa in mevalonate kinase deficiency. J Inherit
Metab Dis. 28:1143–1145. 2005. View Article : Google Scholar
|
|
20
|
Tricarico PM, Kleiner G, Valencic E,
Campisciano G, Girardelli M, Crovella S, Knowles A and Marcuzzi A:
Block of the mevalonate pathway triggers oxidative and inflammatory
molecular mechanisms modulated by exogenous isoprenoid compounds.
Int J Mol Sci. 15:6843–6856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marcuzzi A, Zanin V, Piscianz E, Tricarico
PM, Vuch J, Girardelli M, Monasta L, Bianco AM and Crovella S:
Lovastatin-induced apoptosis is modulated by geranylgeraniol in a
neuroblastoma cell line. Int J Dev Neurosci. 30:451–456. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marcuzzi A, De Leo L, Decorti G, Crovella
S, Tommasini A and Pontillo A: The farnesyltransferase inhibitors
tipifarnib and lonafarnib inhibit cytokines secretion in a cellular
model of mevalonate kinase deficiency. Pediatr Res. 70:78–82. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simon A: Cholesterol metabolism and
immunity. N Engl J Med. 371:1933–1935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reboldi A, Dang EV, McDonald JG, Liang G,
Russell DW and Cyster JG: Inflammation. 25-Hydroxycholesterol
suppresses interleukin-1-driven inflammation downstream of type I
interferon. Science. 345:679–684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet Chapter. 7:Unit7.202013.
|
|
26
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: Mutation Taster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X, Jian X and Boerwinkle E: dbNSFP
v2.0: A database of human non-synonymous SNVs and their functional
predictions and annotations. Hum Mutat. 34:E2393–E2402. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pollard KS, Hubisz MJ, Rosenbloom KR and
Siepel A: Detection of nonneutral substitution rates on mammalian
phylogenies. Genome Res. 20:110–121. 2010. View Article : Google Scholar :
|
|
29
|
Kimura M: The neutral theory of molecular
evolution: a review of recent evidence. Jpn J Genet. 66:367–386.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Danecek P, Auton A, Abecasis G, Albers CA,
Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST,
et al: The variant call format and VCFtools. Bioinformatics.
27:2156–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shoemaker JD and Elliott WH: Automated
screening of urine samples for carbohydrates, organic and amino
acids after treatment with urease. J Chromatogr. 562:125–138. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simon A, Kremer HP, Wevers RA, Scheffer H,
De Jong JG, Van Der Meer JW and Drenth JP: Mevalonate kinase
deficiency: Evidence for a phenotypic continuum. Neurology.
62:994–997. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Opaliński Ł, Veenhuis M and van der Klei
IJ: Peroxisomes: membrane events accompanying peroxisome
proliferation. Int J Biochem Cell Biol. 43:847–851. 2011.
View Article : Google Scholar
|
|
35
|
Li X and Gould SJ: PEX11 promotes
peroxisome division independently of peroxisome metabolism. J Cell
Biol. 156:643–651. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wanders RJ and Waterham HR: Biochemistry
of mammalian peroxisomes revisited. Annu Rev Biochem. 75:295–332.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Steinberg SJ, Dodt G, Raymond GV,
Braverman NE, Moser AB and Moser HW: Peroxisome biogenesis
disorders. Biochim Biophys Acta. 1763:1733–1748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ebberink MS, Koster J, Visser G, Spronsen
Fv, Stolte-Dijkstra I, Smit GP, Fock JM, Kemp S, Wanders RJ and
Waterham HR: A novel defect of peroxisome division due to a
homozygous non-sense mutation in the PEX11β gene. J Med Genet.
49:307–313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wiemer EA, Wenzel T, Deerinck TJ, Ellisman
MH and Subramani S: Visualization of the peroxisomal compartment in
living mammalian cells: dynamic behavior and association with
microtubules. J Cell Biol. 136:71–80. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koch J, Pranjic K, Huber A, Ellinger A,
Hartig A, Kragler F and Brocard C: PEX11 family members are
membrane elongation factors that coordinate peroxisome
proliferation and maintenance. J Cell Sci. 123:3389–3400. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Torseth K, Doseth B, Hagen L, Olaisen C,
Liabakk NB, Græsmann H, Durandy A, Otterlei M, Krokan HE, Kavli B
and Slupphaug G: The UNG2 Arg88Cys variant abrogates RPA-mediated
recruitment of UNG2 to single-stranded DNA. DNA Repair (Amst).
11:559–569. 2012. View Article : Google Scholar
|
|
42
|
Al-Saud BK, Al-Sum Z, Alassiri H,
Al-Ghonaium A, Al-Muhsen S, Al-Dhekri H, Arnaout R, Alsmadi O,
Borrero E, Abu-Staiteh A, Rawas F, Al-Mousa H and Hawwari A:
Clinical, immunological, and molecular characterization of
hyper-IgM syndrome due to CD40 deficiency in eleven patients. J
Clin Immunol. 33:1325–1335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Imai K, Slupphaug G, Lee WI, Revy P,
Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, et
al: Human uracil-DNA glycosylase deficiency associated with
profoundly impaired immunoglobulin class-switch recombination. Nat
Immunol. 4:1023–1028. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
44
|
Drenth JP, Haagsma CJ and van der Meer JW:
Hyperimmunoglobulinemia D and periodic fever syndrome The clinical
spectrum in a series of 50 patients International Hyper-IgD Study
Group. Medicine (Baltimore). 73:133–144. 1994. View Article : Google Scholar
|
|
45
|
Klasen IS, Göertz JH, van de Wiel GA,
Weemaes CM, van der Meer JW and Drenth JP: Hyper-immunoglobulin A
in the hyper-immunoglobulinemia D syndrome. Clin Diagn Lab Immunol.
8:58–61. 2001.PubMed/NCBI
|
|
46
|
Moura R, Tricarico PM, Campos Coelho AV
and Crovella S: GRID2 a novel gene possibly associated with
mevalonate kinase deficiency. Rheumatol Int. 35:657–659. 2015.
View Article : Google Scholar
|