Introduction
Breast cancer is the second leading cause of
cancer-associated mortality, with high morbidity and mortality
rates in women worldwide. Chemotherapy remains an alternative
therapeutic option for breast cancer to surgery and radiation,
however, multidrug resistance and an unfavorable systemic toxicity
against normal cells limit their clinical effects, which highlights
the urgent requirement to identify novel therapeutic strategies and
agents (1).
Tumor necrosis factor-α (TNF-α) is one of the major
inflammatory cytokines, which can act as either a tumor promoter,
by linking inflammation with carcinogenesis, or as a tumor
inhibitor, through the induction of cancer cell death. TNF-α
signaling occurs via two cell-surface receptors, TNF-α factor
receptor 1 and 2 (TNFR1 and TNFR2). Complex I is formed when TNF-α
binds to TNFR-1, and this event leads to activation of nuclear
factor (NF)-κB, mitogen-activated protein kinases and Complex II,
ultimately leading to apoptosis (2,3). Of
note, Complex I and Complex II have disparate and opposing effects
on apoptosis. Complex I leads to activation of the NF-κB signaling
pathway, whereas Complex II leads to activation of caspase-3 and
caspase-9. Activated NF-κB can upregulate the expression levels of
downstream genes, which include anti-apoptotic genes, including
X-linked inhibitor of apoptosis (XIAP) and cellular inhibitors of
apoptosis 1 and 2 (cIAP1/2). Therefore, the balance between Complex
I and Complex II results in cell resistance to TNF-α-mediated
apoptosis. In the tumor microenvironment, sustained NF-κB
activation can have an anti-apoptotic effect on tumor cells, which
depends on Complex I signaling, whereas Complex II can attenuate
TNF-α-induced NF-κB activation.
Triptolide, a natural and biologically active
compound as a diterpenoid triepoxide, was originally purified from
the Chinese herb Tripterygium wilfordii Hook F (4). This natural product has been used in
traditional Chinese medicine for centuries, and has a myriad of
therapeutic applications against inflammation and autoimmune
diseases (5,6). Previous studies have reported that
triptolide has antitumor, anti-inflammatory and immunosuppressive
activities. In particular, triptolide has been investigated for
several different types of cancer cells in vitro and in
vivo (7–9). The inhibitory effects of triptolide
in the growth of cholangiocarcinoma cells in hamsters (10), and the growth of xenografts in
human breast cancer, bladder cancer, melanoma and gastric
carcinoma, have been shown in nude mice (9). It has also been demonstrated that the
antiproliferative properties of triptolide may be involved in
inhibiting NF-κB activity and inducing cell apoptosis (11). Taken together, these previous
studies have demonstrated that triptolide may be clinically
effective for tumor chemotherapy.
The TNF-α signaling pathway is important in tumor
development, and its antitumor properties may be exploited with
avoidance of its tumorigenic properties (2). Based on the essential requirement for
an inflammatory microenvironment in tumor formation, the present
study hypothesized that triptolide sensitizes human breast cancer
cells to TNF-α-induced apoptosis by inhibiting activation of the
NF-κB pathway. The results of the present study demonstrated that
TNF-α combined with triptolide effectively sensitized human breast
cancer cells to triptolide-mediated induction of apoptosis, by
targeting inhibitor of NF-κB (IκB), an effective signaling pathway
of TNF-α. Due to the inhibitory activities of IκB, triptolide
inhibited the NF-κB signaling pathway and further promoted the
activation of caspase-3. Finally, increased activation of caspase-3
resulted in apoptosis, which contributed to its anti-inflammatory
and anticancer activities. These observations suggested that this
may be a promising combination strategy for use in human breast
cancer therapeutics.
Materials and methods
Reagents
Triptolide was purchased from A.G. Scientific, Inc.
(San Diego, CA, USA) and dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich, St. Louis, MO, USA). Stock solutions (50 mM) were
prepared, and aliquots were stored at −20°C for further use in the
subsequent experiments. L15/1640, penicillin and streptomycin were
purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Fetal bovine serum (FBS) was obtained from Gibco (Thermo
Fisher Scientific, Inc).
3-[4,5-dimethyltiazol-2-yl]-2.5-diphenyl-tetrazolium bromide (MTT;
BTN111105), RNaseA and reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) kits were purchased from
Promega Corporation (San Luis Obispo, CA, USA).
Ac-Asp-Glu-Val-Asp-AMC and Z-Gly-Gly-Leu-AMC, the substrates for
caspase-3 and chymotrypsin (CT)-like activities, respectively, were
obtained from Calbiochem, Inc. (San Diego, CA, USA). Mouse
monoclonal anti-poly(ADP-ribose)polymerase (PARP) (46D11; dilution,
1:1,000), anti-caspase-3 (8G10; dilution, 1:1,000); anti-caspase-9
(9502; dilution, 1:1,000), anti-XIAP (2042; dilution, 1:1,000) and
anti-IAP1/2 (4952; dilution, 1:1,000) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Mouse monoclonal
anti-ubiquitin (P4D1; sc-8017; dilution, 1:1,000), rabbit
polyclonal anti-IκB-α (C-15; sc-203; dilution, 1:1,000), goat
polyclonal anti-β-actin (C-11; sc-1615; dilution, 1:5,000), and
secondary goat anti-rabbit IgG-horseradish peroxidase (dilution,
1:5,000; sc-2054) were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Velcade was purchased from A.G.
Scientific, Inc..
Cell culture and whole cell extract
preparation
Human breast cancer MDA-MB-231 and MCF-7 cells were
obtained from the American Type Culture Collection (Manassas, VA,
USA) and grown in L15/1640 supplemented with 10% FBS, 100 U/ml
penicillin and 100 µg/ml streptomycin
(5.0–8.0×105 cells/ml). All cells were maintained at
37°C with 5% CO2. Whole cell extracts were prepared, as
described previously (12,13). Briefly, cells were harvested,
washed with phosphate-buffered saline (PBS) and homogenized for 30
min at 4°C in lysis buffer, containing 50 mM Tris-HCl (pH 7.5), 150
mM NaCl, 0.5% NP-40, 0.5 mM phenylmethylsulfonyl fluoride and 0.5
mM dithiothreitol (Invitrogen; Thermo Fisher Scientific, Inc.). The
lysates were immediately centrifuged at 12,000 × g for 12 min at
4°C, and the supernatants were collected as whole cell extracts.
Protein concentration was determined using a bicinchoninic acid
protein assay (Pierce Biotechnology, Rockford, IL, USA). Bovine
serum albumin (BSA) was used as a standard (Invitrogen; Thermo
Fisher Scientific, Inc.).
MTT assay
MTT assays were performed in 96-well plates.
Briefly, 3.5×105 MDA-MB-231/MCF-7 cells were seeded per
well and incubated overnight at 37°C. The cells were treated with
triptolide (13, 25, 50, 100, 200 or 400 nm), alone or in
combination with TNF-α (10 ng/ml) for 48 h. DMSO (0.1%)-treated
cells were used as a control. Inhibition of cell proliferation was
determined using an MTT assay (10 µl MTT/100 µl
cells), as described previously (12). Absorbance was measured using a
Wallac Victor3™ multilabel counter (PerkinElmer, Walltham, MA, USA)
at 540 nm, and cell viability was determined relative to the
DMSO-treated control cells. In individual experiments, each
treatment condition was set up with four repeats, and each
experiment was repeated three times independently.
Caspase-3 activity determination, CT-like
activity assay and Western blot analysis
Caspase-3 activity was determined by measuring the
release of the AMC groups from the caspase-3-specific substrate,
Ac-Asp-Glu-Val-Asp-AMC. The MDA-MB-231/MCF-7 cells
(5.0–8.0×105/well) were plated and, following overnight
incubation at 37°C, were treated with indicated concentrations of
triptolide, either alone or in combination with TNF-α (10 ng/ml),
for 24 h, followed by preparation of whole cell extracts. The cell
extracts (25 mg) were then incubated in a 96-well plate in 100
µl assay buffer (50 mM Tris-HCl; pH 7.5) with 40 µM
Ac-Asp-Glu-Val-Asp-AMC. The reaction mixture was incubated at 37°C
for 2 h and the hydrolyzed fluorescent AMC groups were quantified,
as previously described (12,13).
The production of AMC groups was measured using a Victor3
Multilabel Counter (PerkinElmer) with an excitation filter of 380
nm and an emission filter of 460 nm.
Proteasomal CT-like activity was determined by
measuring the release of the AMC groups from the CT-like specific
substrate, Z-Gly-Gly-Leu-AMC. The MDA-MB-231 and MCF-7 cells
(5.0–8.0×105/well) were plated in 96-well plates and
treated with triptolide at the desired final concentrations, either
alone or in combination of TNF-α (10 ng/ml), for 8 h, or for the
indicated time periods, and incubated with Z-Gly-Gly-Leu-AMC (at 40
µM) for an additional 2 h at 37°C. The production of
hydrolyzed AMC groups was measured, as previously described
(12,13).
For Western blot analysis, the cells
(5.0–8.0×105/ml) were plated in 60 mm dishes and treated
with the desired final concentration of triptolide, either alone or
in combination with TNF-α, for different time periods, followed by
preparation of whole cell extracts. Protein was extracted from the
whole cell lysates using radioimmunoprecipitation assay buffer
(Beyotime Institute of Biotechnology, Haimen, China) supplemented
with 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 mM
NaF, 1 mM phenylmethylsulfonyl fluoride and 1X Roche Complete Mini
Protease Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN,
USA). After the determination of protein concentration, 5 µg
protein extracted from cultured cells was separated by sodium
dodecyl sulfate polyacrylamide gel electrophoresis and electrically
transferred onto polyvinylidene difluoride membranes. The blots
were blocked with 5% fat milk for 1 h at room temperature, and then
incubated with specific antibodies for 1 h at room temperature.
After washing, membranes were incubated for 1 h at room temperature
with goat anti-rabbit IgG-horseradish peroxidase and exposed to
X-ray film. Western blots were analyzed using enhanced
chemiluminescence reagent (Santa Cruz Biotechnology, Inc.), as
previously described (12,13).
RT-qPCR
Cells (5×106) were pretreated with TNF-α
(10 ng/ml) at the desired final concentrations, either alone or in
combination of triptolide for 12 h at 37°C. Total RNA was extracted
using TRIzol reagent (5×106 cells/ml; Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
The genomic DNA was digested using RNase-free DNaseI, and the
concentration of the RNA was detected using UV spectroscopy as
previously described (12).
Single-stranded cDNA was generated using random hexamer primers
with a Prime Script® RT Master mix first strand
synthesis system for RT-qPCR (A1250; Promega Corporation).
Following RT, qPCR was performed with SYBR® Premix Ex
Taq™ II (Tli RNaseH Plus). The 20.0 µl reaction mixture
contained 2.0 µl cDNA template, 10.0 µl
SYBR® Premix Ex Taq™ II (2×), 0.8 µl PCR forward
primer, 0.8 µl PCR reverse primer, 0.4 µl ROX
Reference Dye II and 6.0 µl dH2O. A GAPDH primer
set was used as an internal control. The products were detected by
agarose gel electrophoresis. The PCR primers used were as follows:
XIAP, forward ACATGGCTGTCAAGAAGG-AGAT and reverse
ACTGCAGCCTCGAACTTCTG (180 bp); cIAP1/2, forward
TTCCGTGGCTCTTATTCAAACT and reverse GCACAGTGGTAGGAACTTCTCAT (96 bp);
18sr, forward CCTGGATACCGCAGCTAGGA and reverse
GCGGCGCAATACGAATGCCCC (112 bp). Amplification cycles were as
follows: 95°C for 2 min, then 40 cycles at 95°C for 15 s, 60°C for
1 min, followed by 95°C for 10 min, and 60°C for 1 min. The
fluorescent signal of the PCR product was detected by ABI
PRISM® 7500 Real-Time PCR System (Thermo Fisher
Scientific, Inc.).
Statistical analysis
Statistical analysis was performed using SPSS
version 19.0 (IBM SPSS, Armonk, NY, USA). Data are expressed as the
mean ± standard deviation of the mean. Student's t-test was applied
to evaluate differences between the treated groups and controls.
Data from multiple groups were analyzed using one-way analysis of
variane, followed by the Tukey-Kramer multiple comparison test. For
all tests, P<0.05 was considered to indicate a statistically
significant difference between groups.
Results
TNF-α sensitizes breast cancer cells to
triptolide-induced cell death
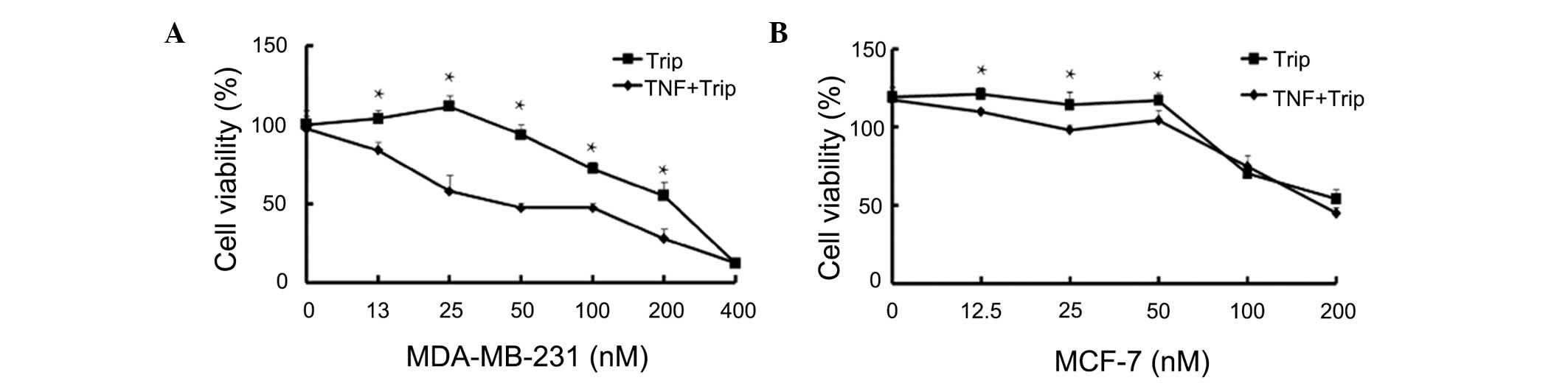
The antiproliferative effects of the combination of
TNF-α and triptolide against MDA-MB-231 and MCF-7 human breast
cancer cells were examined using an MTT assay. As shown in Fig. 1, TNF-α, at a low concentration of
10 ng/ml, had no significant effects on cell proliferation of
either human breast cancer cell lines. Exposure to different
concentrations of triptolide (25–200 nM) with or without TNF-α for
24 h induced a dose-dependent inhibition in cell proliferation. As
shown in Fig. 1A, triptolide
exerted inhibitory effects when combined with or without TNF-α, and
the inhibitory effects were more marked in the combination group.
The combination of TNF-α and triptolide exhibited increased
cytotoxic effects against the MDA-MB-231 human breast cells at
tripolide concentrations between 25 and 200 nM. The combined
effects were not marked when a higher concentration of triptolide
(400 nM) was used for treatment of the MDA-MB-231 cells. The
combination treatment had decreased cytotoxic effects against the
MCF-7 human breast cells (Fig.
1B), however, the combination of TNF-α and triptolide showed
potent antiproliferative effects against the two human breast
cancer cell lines.
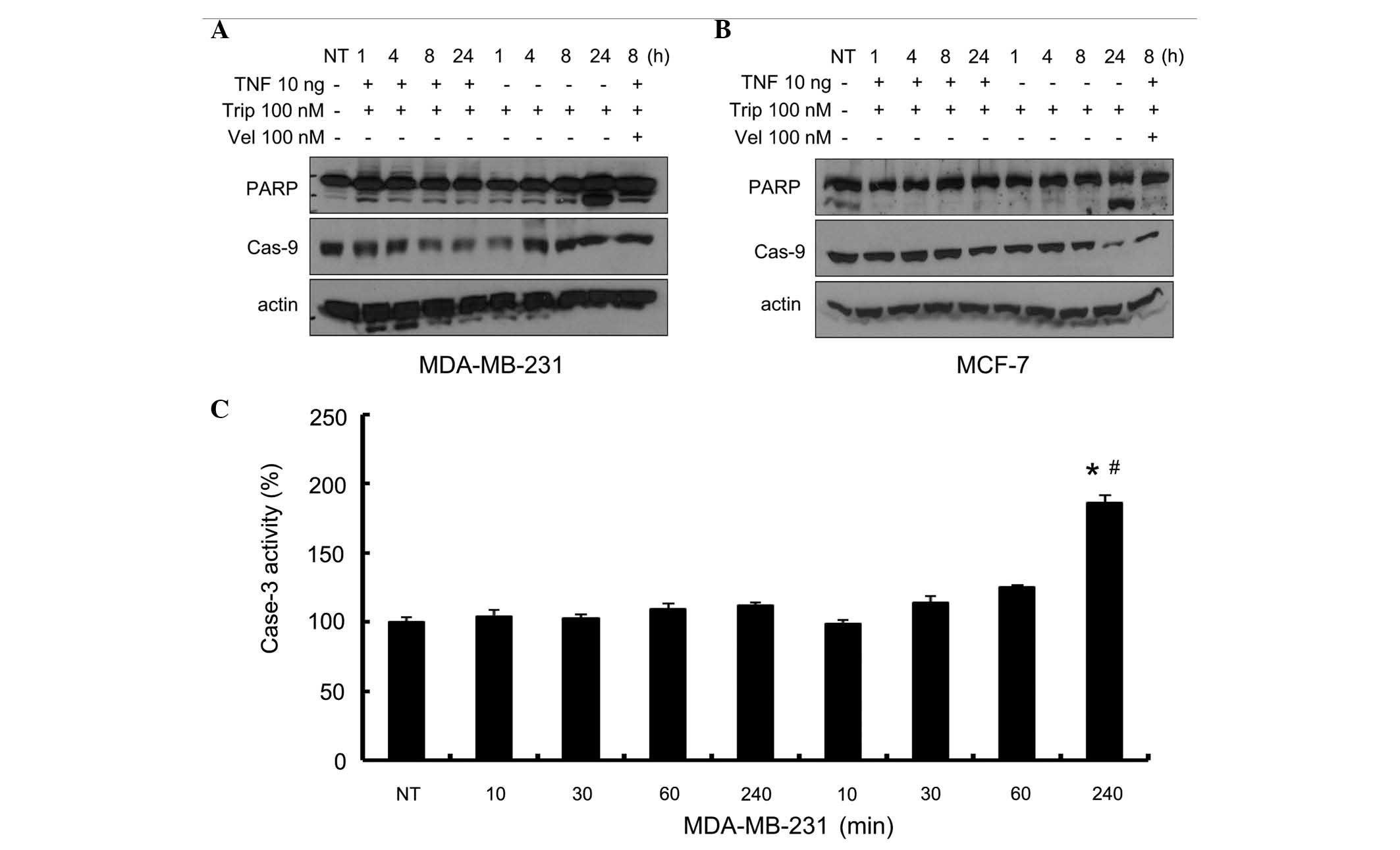
TNF-α sensitizes breast cancer cells to
triptolide-induced apoptosis
The present study then determined whether the cell
death induced by TNF-α+triptolide corresponded to the apoptotic
response. The MDA-MB-231 and MCF-7 cells were treated,
respectively, with triptolide (25, 50 and 100 nM), combined with or
without TNF-α (10 ng/ml) for 24 h. The nuclear enzyme, PARP, which
is one of the primary cleavage targets of caspase-3; and caspase-9,
which activates caspase-3, were detected. Velcade, the first
proteasome inhibitor to become a therapeutic modality for multiple
myeloma, was used as a positive control, as it has been reported to
induce the apoptosis of several types of cancer cell (14). As shown in Fig. 2A, TNF-α, at a low concentration of
10 ng, markedly increased the effect of velcade on PARP cleavage.
As shown in Fig. 2A and B, PARP
cleavage was enhanced in the triptolide+TNF-α-treated cells in a
dose-dependent manner and, to a lesser degree, in the
triptolide-treated cells. Triptolide also exhibited marked effects
on the levels of caspase-9 in a dose-dependent manner. Caspase-9
decreased markedly when the concentration of triptolide reached 100
nM. Following treatment with TNF-α combined with triptolide, the
degradation of caspase-9 was more marked, compared with
triptolide-only treatment in the two cell lines. As shown in
Fig. 2C, a marked increase in
caspase-3 activity was observed in the cells treated with
TNF-α+triptolide, compared with the cells treated with triptolide
alone, at triptolide concentrations up to 100 nM. Caspase-3
activity was not assessed in the MCF-7 cells, as MCF-7 cells do not
express caspase-3. These data demonstrated that the combination of
TNF-α and triptolide induced apoptosis in breast cancer cells,
associated with the activation of caspase-3.
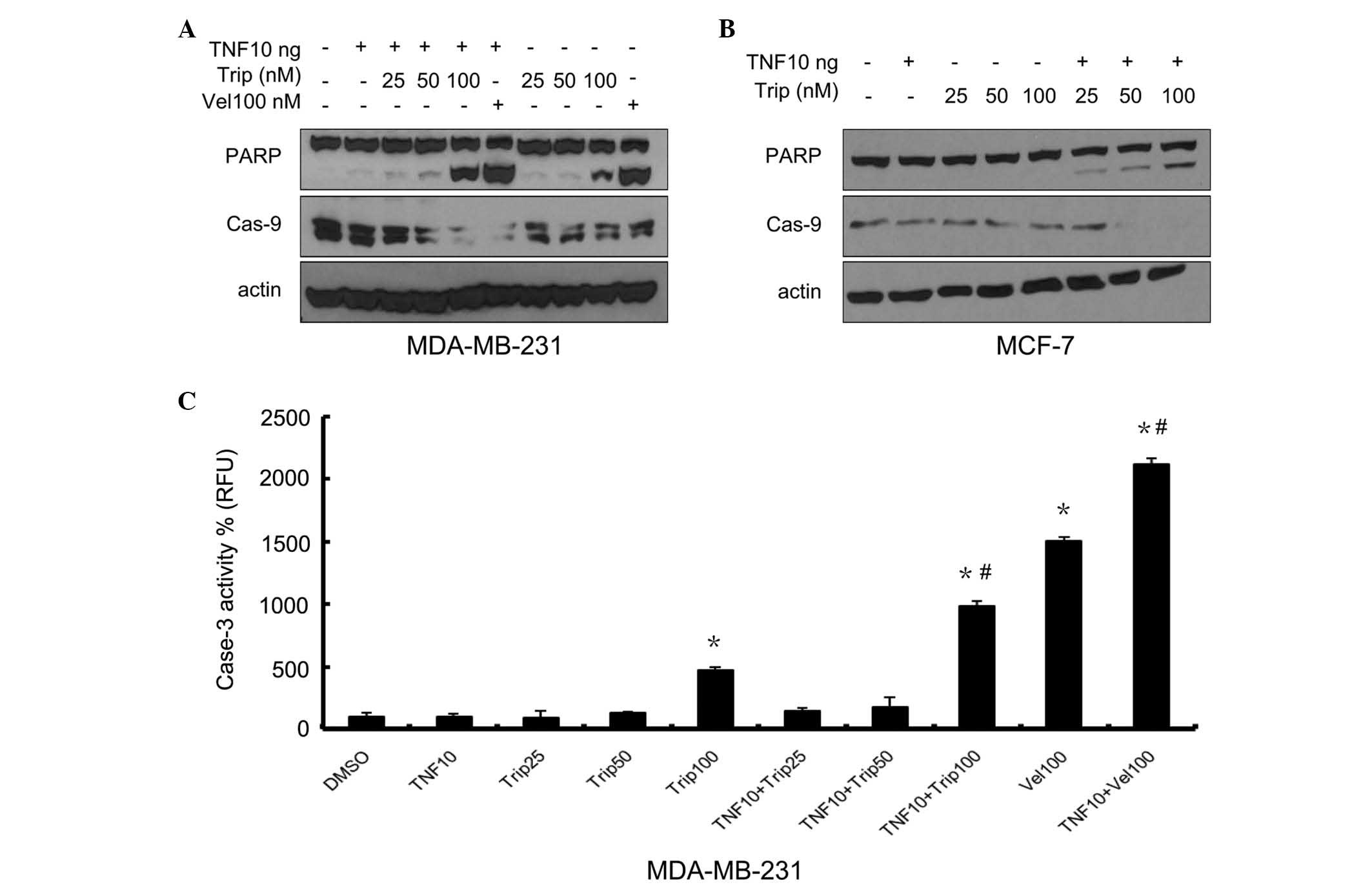 | Figure 2Effects of different concentrations of
triptolide in combination with TNF-α on PARP cleavage, and
caspase-3 and -9 activity. The MDA-MB-231 cells and MCF-7 cells
were treated either with different concentrations of triptolide, or
with TNF-α (10 ng/ml)+different concentrations of triptolide,
Velcade(100 nM) was used as a positive control. After 24 h, the
protein levels of PARP and caspase-9 were determined in the (A)
MDA-MB-231 cells and (B) MCF-7 cells by Western blot analysis using
specific antibodies. (C) Caspase-3 activity was determined in the
MDA-MB-231 cells. Data are expressed as the mean ± standard
deviation of three experiments. *P<0.05, vs.
DMSO-treated group; #P<0.05 Trip/Vel-treated group,
vs. Trip/Vel+TNF-α group. TNF-α, tumor necrosis factor-α; Trip,
triptolide; DMSO-dimethyl sulfoxide; Vel, Velcade; PARP, poly
(ADP-ribose) polymerase; Cas, caspase. |
TNF-α sensitizes breast cancer cells to
triptolide-induced apoptosis
The kinetic effects of the combination of TNF-α and
triptolide were further evaluated in the present study. As shown in
Fig. 3A and B, PARP fragments and
caspase-9 reduction were not detected up to 24 h when treated with
triptolide, with or without TNF-α, in either of the cell lines. At
24 h, the combination of TNF-α and triptolide markedly enhanced
quantity of PARP fragments, and reduced the quantity of caspase-9
in the two cell lines, which were consistent with the results of
Fig. 2. As shown in Fig. 3C, there were no changes in the
caspase-3 activity of the MDA-MB-231 cells prior to 240 mins when
treated with triptolide alone, however, levels of activity
increased at 240 mins when treated with TNF-α+triptolide. These
data indicated that the combination of TNF-α with triptolide
promoted the activation of caspase-3.
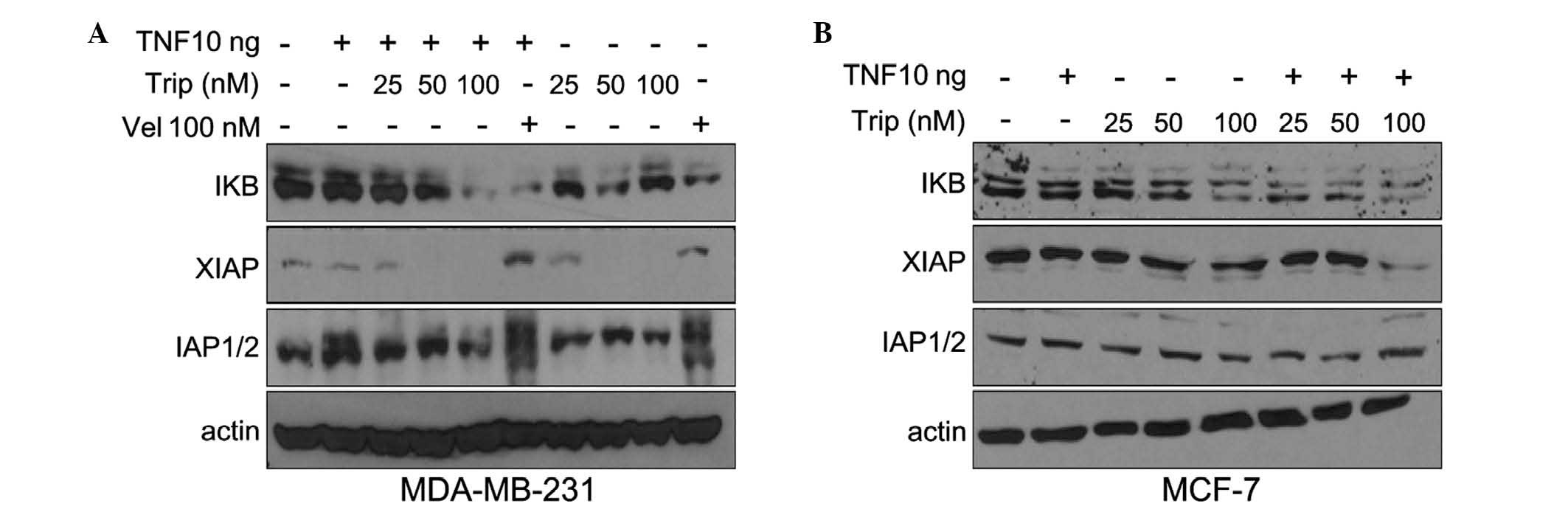
Effects of the combination of TNF-α with
triptolide on the NF-κB pathway
Previous reports have demonstrated that triptolide
is able to suppress the production of inflammatory mediators
induced by stimuli in various cell types (5,12,15).
As TNF-α and the NF-κB pathway are important in inflammation and
are involved in the anti-inflammatory effect of triptolide,
intracellular IκB, which is important in the NF-κB pathway was
further examined. MDA-MB-231 and MCF-7 cells were treated,
respectively, with triptolide (25, 50 and 100 nM), with or without
TNF-α (10 ng/ml), for 24 h. As shown in Fig. 4A and B, exposure of the cells to
triptolide, either alone or with TNF-α, inhibited the expression of
IκB in a dose-dependent manner. This inhibitory effect was more
marked when triptolide was used in combination with TNF-α. The
inhibitory effect of triptolide on IκB may be due to its inhibition
of inflammatory pathways, which is consistent with previous reports
(11,12).
According to previous reports, XIAP and cIAP1/2 are
target genes of NF-κB (16) and
exert effects on the caspase-9 and caspase-3 apoptotic pathways
(17,18). Therefore, the effects of TNF-α
combined with triptolide on the levels of XIAP and cIAP1/2 were
further examined in the present study. As shown in Fig. 4, in the MDA-MB-231 and MCF-7 cells,
exposure to TNF-α and triptolide had no significant effect on the
levels of cIAP1/2. However, triptolide caused a reduction in XIAP,
and TNF-α aggravated these effects.
Time-dependent effects of
TNF-α+triptolide on IκB
The present study also examined the kinetic effects
of TNF-α+triptolide treatment on IκB. The MDA-MB-231 and MCF-7
cells were treated, respectively, with triptolide (100 nM), with or
without TNF-α (10 ng/ml), and incubated for different durations. As
shown in Fig. 5A and B, the levels
of IκB and caspase-9 gradually decreased with increasing duration,
and the decrease in the levels of IκB in the
TNF-α+triptolide-treated cells were more marked, compared with
those in the triptolide only-treated cells. The results of the
kinetic effects confirmed that TNF-α combined with triptolide
inhibited the expression of IκB and promoted the degradation of
caspase-9.
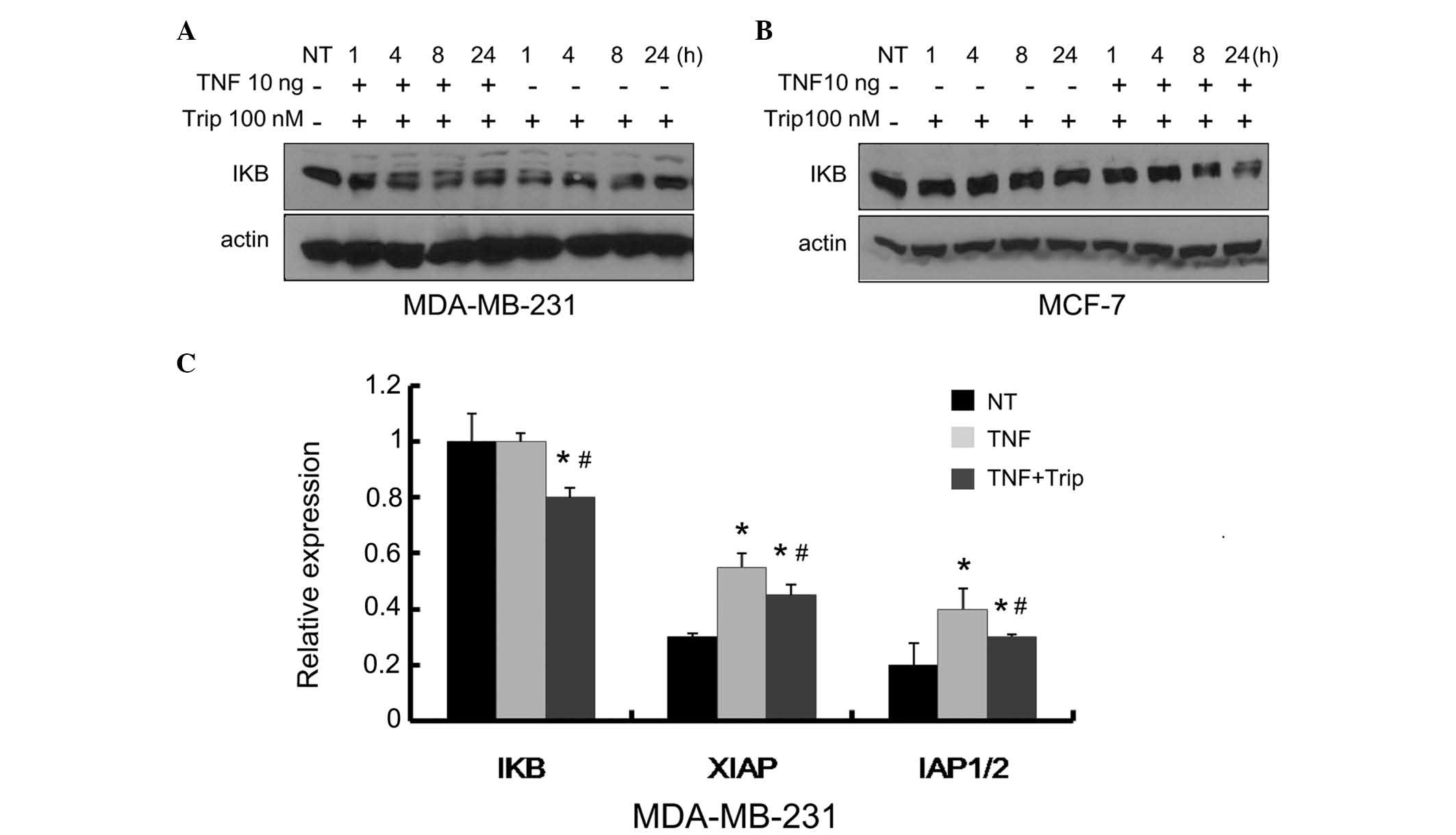 | Figure 5Kinetic effects of TNF-α+triptolide on
the levels of IκB, and the mRNA levels of NF-κB-targeting genes.
The (A) MDA-MB-231 cells and (B) MCF-7 cells were treated with 100
nM triptolide, or with TNF-α (10 ng/ml)+triptolidee (100 nM) for
the indicated durations. Velcade (20 nM) was used as a positive
control. The protein levels of IκB, XIAP and cIAP1/2 were
determined via Western blot analysis using specific antibodies. (C)
MDA-MB-231 cells were treated with TNF-α (10 ng/ml), or with TNF-α
(10 ng/ml)+triptolide (100 nM) for 12 h, followed by measurement of
the mRNA levels of IκB, XIAP and cIAP1/2 via reverse
transcription-quantitative polymerase chain reaction analysis. Data
are expressed as the mean ± standard deviation of three
experiments. *P<0.05, vs. NT group;
#P<0.05 Trip/Vel treated group, vs. Trip/Vel+TNF-α
group. TNF-α, tumor necrosis factor-α; Trip, triptolide; Vel,
velcade; NT, no treatment; IKB; inhibitor of NF-κB; XIAP; X-linked
inhibitor of apoptosis protein; IAP1/2, inhibitor of apoptosis
protein 1/2. |
Effects of TNF-α combined with triptolide
on the transcription of NF-κB target genes
In order to investigate whether the inhibition of
IκB, XIAP and IAP1/2 were caused by inhibiting gene expression, the
mRNA expression levels of IκB, XIAP and IAP1/2 were analyzed using
RT-qPCR. The MDA-MB-231 breast cancer cells were treated with TNF-α
(10 ng/ml) or TNF-α+triptolide (100 nM) for 6 h. As shown in
Fig. 5C, TNF-α resulted in a
marginal, but not significant increase in the mRNA level of IκB.
This increase may be a feedback effect of the cells to TNF-α.
TNF-α+triptolide caused a significant decrease in the mRNA level of
IκB, which may have been due to inhibition of the NF-κB pathway by
triptolide. The expression levels of XIAP and IAP1/2 increased when
the MDA-MB-231 cells were treated with TNF-α alone. Treatment with
TNF-α+triptolide reduced the expression levels of XIAP and IAP1/2,
with the effects on XIAP being more marked, compared with the
effects on IAP1/2. These results suggested that the inhibition of
IκB, XIAP and IAP1/2 may due to inactivation of the NF-κB
pathway.
Discussion
Resistance to conventional chemotherapy and systemic
toxicity remains a significant obstacle in improving the long-term
survival rates of patients with breast cancer. Therefore, it is
necessary to identify novel therapeutic strategies and agents to
improve treatment. In the present study, it was demonstrated that a
low dose of TNF-α sensitized MDA-MB-231 and MCF-7 cells to
triptolide, leading to apoptosis. The results showed that treatment
of cells with TNF-α (10 ng/ml) combined with triptolide (50–200 nM)
increased triptolide-mediated cell death (Fig. 1), suggesting that low doses of
TNF-α promoted triptolide to activate cell death. As shown in
Fig. 2, TNF-α+triptolide activated
caspase-3, and the activation of caspase-3 coincided with an
increase in the cleavage of PARP (Figs. 2 and 3). Caspase-3 led to activation of
downstream events, and promoted cell apoptosis. These observations
confirmed that combining TNF-α with triptolide may provide a novel
therapeutic combination for the treatment of patients with breast
cancer.
However, the mechanism underlying the combinational
effect of TNF-α with triptolide to induce apoptosis remains to be
fully elucidated. The response of cells to TNF-α, an important
modulator of IκB, is key in the resistance to caspase-3-mediated
apoptosis in several types of cancer cell (2,3). IκB
is the predominant inhibitory protein of NF-κB. Activation of IκB
kinases (IκKα, IκKβ and the NF-κB essential modulator, IκKγ)
results in phosphorylation of inhibitory IκBα, IκBβ and IκBε
proteins bound to NF-κB. NF-κB is consequently released, and
translocates to the nucleus where it interacts with other
transcription factors and transcriptional co-factors to regulate
the expression of an array of genes, several of which are involved
in inflammatory signaling, proliferation and apoptosis (19). The classical activation of the
NF-κB pathway can be initiated by a wide range of extracellular
stimuli. These agents can activate the cells and mediate the
phosphorylation of IκB, resulting in its degradation and rendering
the NF-κB dimer-free to translocate to the nucleus to regulate
several target genes (20),
including XIAP/cIAP1/2 (21).
NF-κB is activated by a variety of pro-inflammatory agents,
including TNF-α, phorbol esters and several growth factors
(22,23). Of these agents, the tumor
microenvironment has been implicated in TNF-α-mediated NF-κB
activation through the phosphorylation of IκB. In the present
study, it was found that triptolide significantly inhibited the
level of IκB induced by TNF-α. Following the use of a low dose of
TNF-α to imitate the tumor microenvironment, it was found that the
combination of TNF-α and triptolide was more effective, compared
with triptolide alone, at inhibiting the expression of IκB
(Fig. 4).
XIAP and cIAP-1/2 can inhibit death
receptor-mediated apoptosis (24).
These polypeptides belong to the IAP family, a group of
intracellular proteins containing one or more zinc-binding
baculovirus IAP repeat domains. Several IAPs, including XIAP,
cIAP-1 and cIAP-2, also contain a carboxy-terminal RING domain with
ubiquitin E3 ligase properties (25). Although all IAPs can potentially
bind to caspases, only XIAP is a direct inhibitor of caspases-9, -3
and -7 (17,18), whereas cIAP-1 and cIAP-2 are
considered to regulate receptor-mediated signaling pathways
upstream of mitochondria, through their interactions with TNF
receptor-associated factor (TRAF)1 and TRAF2 (26). The present study further examined
the association between TNF-α with triptolide, and the levels of
IκB, caspase-9, XIAP, and cIAP-1/2. The results demonstrated that
IκB, caspase-9, XIAP and cIAP1/2 were downregulated by the
combination of TNF-α and triptolide in the breast cancer cells
(Figs. 4 and 5). As XIAP is a direct inhibitor of
caspases-9, -3 and -7 (17,18),
the lower levels of XIAP and cIAP1/2, may contribute to the
increased degradation of caspase-9. Therefore, the present study
evaluated the effect of TNF-α combined with triptolide on the mRNA
expression levels of IκB, XIAP and cIAP1/2. The results revealed
that TNF-α+triptolide markedly downregulates the mRNA levels of
IκB, XIAP and cIAP1/2 in the breast cancer cells (Fig. 5A). This downregulation of IκB
eliminated the activation of the NF-κB-pathway that was induced by
TNF-α. In the presence of a low dose of TNF-α, the suppressive
effects of triptolide on XIAP and cIAP1/2 became necessary for the
apoptotic pathway to facilitate caspase-3 activation.
In conclusion, the present study found that the
cytotoxic effects of triptolide on breast cancer cells were
enhanced in the presence of TNF-α. The investigation of TNF-α in
the tumor microenvironment revealed that the levels of TNF-α are
generally higher in the tumor microenvironment, and that TNF-α is
important in the development of tumors. The results of the present
study provided evidence for the effect of the natural
anti-inflammatory drug, triptolide, in the treatment of
inflammatory tumors, which holds important implications for the
therapeutic strategies used in the treatment of breast cancer.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81101747 to Dr Li Lu
and grant no. 81372855 to Dr Gui Hi Wang).
References
|
1
|
Labovsky V, Martinez LM, Davies KM,
García-Rivello H, Calcagno Mde L, Matas A, Fernández Vallone VB,
Wernicke A, Choi H and Chasseing NA: Association between ligands
and receptors related to the progression of early breast cancer in
tumor epithelial and stromal cells. Clin Breast Cancer. 15:e13–e21.
2015. View Article : Google Scholar
|
|
2
|
Sung B, Park B, Yadav VR and Aggarwal BB:
Celastrol, a triterpene, enhances TRAIL-induced apoptosis through
the down-regulation of cell survival proteins and up-regulation of
death receptors. J Biol Chem. 285:11498–11507. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mandal C, Dutta A, Mallick A, Chandra S,
Misra L, Sangwan RS and Mandal C: Withaferin A induces apoptosis by
activating p38 mitogen-activated protein kinase signaling cascade
in leukemic cells of lymphoid and myeloid origin through
mitochondrial death cascade. Apoptosis. 13:1450–1464. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang DW, Lee JY, Oh DH, Park SY, Woo TM,
Kim MK, Park MH, Jang YH and Min do S: Triptolide-induced
suppression of phospholipase D expression inhibits proliferation of
MDA-MB-231 breast cancer cells. Exp Mol Med. 41:678–685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen BJ: Triptolide, a novel
immunosuppressive and anti-inflammatory agent purified from a
Chinese herb Tripterygium wilfordii Hook F. Leuk Lymphoma.
42:253–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu WY and Wang YP: Pharmacological actions
and therapeutic applications of Salvia miltiorrhiza depside salt
and its active components. Acta Pharmacol Sin. 33:1119–1130. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kiviharju TM, Lecane PS, Sellers RG and
Peehl DM: Antiproliferative and proapoptotic activities of
triptolide (PG490), a natural product entering clinical trials, on
primary cultures of human prostatic epithelial cells. Clin Cancer
Res. 8:2666–2674. 2002.PubMed/NCBI
|
|
8
|
Fidler JM, Li K, Chung C, Wei K, Ross JA,
Gao M and Rosen GD: PG490-88, a derivative of triptolide, causes
tumor regression and sensitizes tumors to chemotherapy. Mol Cancer
Ther. 2:855–862. 2003.PubMed/NCBI
|
|
9
|
Yang S, Chen J, Guo Z, Xu XM, Wang L, Pei
XF, Yang J, Underhill CB and Zhang L: Triptolide inhibits the
growth and metastasis of solid tumors. Mol Cancer Ther. 2:65–72.
2003.PubMed/NCBI
|
|
10
|
Tengchaisri T, Chawengkirttikul R,
Rachaphaew N, Reutrakul V, Sangsuwan R and Sirisinha S: Antitumor
activity of triptolide against cholangiocarcinoma growth in vitro
and in hamsters. Cancer Lett. 133:169–175. 1998. View Article : Google Scholar
|
|
11
|
Liu H, Liu ZH, Chen ZH, Yang JW and Li LS:
Triptolide: A potent inhibitor of NF-kappa B in T-lymphocytes. Acta
Pharmacol Sin. 21:782–786. 2000.
|
|
12
|
Lu L, Kanwar J, Schmitt S, Cui QC, Zhang
C, Zhao C and Dou QP: Inhibition of tumor cellular proteasome
activity by triptolide extracted from the Chinese medicinal plant
'thunder god vineʼ. Anticancer Res. 31:1–10. 2011.PubMed/NCBI
|
|
13
|
Yang H, Shi G and Dou QP: The tumor
proteasome is a primary target for the natural anticancer compound
Withaferin A isolated from 'Indian winter cherryʼ. Mol Pharmacol.
71:426–437. 2007. View Article : Google Scholar
|
|
14
|
Perrone G, Hideshima T, Ikeda H, Okawa Y,
Calabrese E, Gorgun G, Santo L, Cirstea D, Raje N, Chauhan D, et
al: Ascorbic acid inhibits antitumor activity of bortezomib in
vivo. Leukemia. 23:1679–1686. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao G, Vaszar LT, Qiu D, Shi L and Kao
PN: Anti-inflammatory effects of triptolide in human bronchial
epithelial cells. Am J Physiol Lung Cell Mol Physiol.
279:L958–L966. 2000.PubMed/NCBI
|
|
16
|
Sethi G, Ahn KS and Aggarwal BB: Targeting
nuclear factor-kappa B activation pathway by thymoquinone: Role in
suppression of antiapoptotic gene products and enhancement of
apoptosis. Mol Cancer Res. 6:1059–1070. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eckelman BP, Salvesen GS and Scott FL:
Human inhibitor of apoptosis proteins: Why XIAP is the black sheep
of the family. Embo Rep. 7:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Srinivasula SM, Hegde R, Saleh A, Datta P,
Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y
and Alnemri E: A conserved XIAP-interaction motif in caspase-9 and
Smac/DIABLO regulates caspase activity and apoptosis. Nature.
410:112–116. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baeuerle PA and Baltimore D: NF-kappa B:
Ten years after. Cell. 87:13–20. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao Y, Fu Y, Hu J, Liu Y and Yin X: The
effect of tissue factor pathway inhibitor on the expression of
monocyte chemotactic protein-3 and IκB-α stimulated by tumour
necrosis factor-α in cultured vascular smooth muscle cells. Arch
Cardiovasc Dis. 106:4–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gyrd-Hansen M and Meier P: IAPs: From
caspase inhibitors to modulators of NF-kappaB, inflammation and
cancer. Nat Rev Cancer. 10:561–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takada Y, Singh S and Aggarwal BB:
Identification of a p65 peptide that selectively inhibits NF-kappa
B activation induced by various inflammatory stimuli and its role
in down-regulation of NF-kappaB-mediated gene expression and
up-regulation of apoptosis. J Biol Chem. 279:15096–15104. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vile GF, Tanew-Ilitschew A and Tyrrell RM:
Activation of NF-kappa B in human skin fibroblasts by the oxidative
stress generated by UVA radiation. Photochem Photobiol. 62:463–468.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Varfolomeev E, Goncharov T, Fedorova AV,
Dynek JN, Zobel K, Deshayes K, Fairbrother WJ and Vucic D: c-IAP1
and c-IAP2 are critical mediators of tumor necrosis factor alpha
(TNFalpha)-induced NF-kappaB activation. J Biol Chem.
283:24295–24299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Salvesen GS and Duckett CS: IAP proteins:
Blocking the road to death's door. Nat Rev Mol Cell Biol.
3:401–410. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rothe M, Pan MG, Henzel WJ, Ayres TM and
Goeddel DV: The TNFR2-TRAF signaling complex contains two novel
proteins related to baculoviral inhibitor of apoptosis proteins.
Cell. 83:1243–1252. 1995. View Article : Google Scholar : PubMed/NCBI
|