On average, every 34 sec someone in the United
States suffers from a coronary event, and every 40 sec someone has
a stroke (1). Cardiovascular
disease and stroke are pathologies associated with the development
of atherosclerosis, a chronic inflammatory process that affects the
walls of large- and medium-sized arteries. The systemic risk
factors associated with a higher prevalence of
atherosclerosis-related diseases include dyslipidemia,
hypertension, chronic kidney disease, metabolic syndrome and
diabetes (1). Unstable plaques may
rupture and block the bloodstream, ultimately leading to myocardial
infarction or stroke. Atherosclerotic plaques consist of fatty
materials, predominantly cholesterol; necrotic cores; calcified
regions; and various types of cells, including smooth muscle cells,
endothelial cells, immune cells, monocytes and foam cells. Among
these cells, lipid-laden macrophages, which are commonly known as
foam cells, are central components of the plaques, which have an
important role in the process of atherosclerotic plaque development
from the early to late stages.
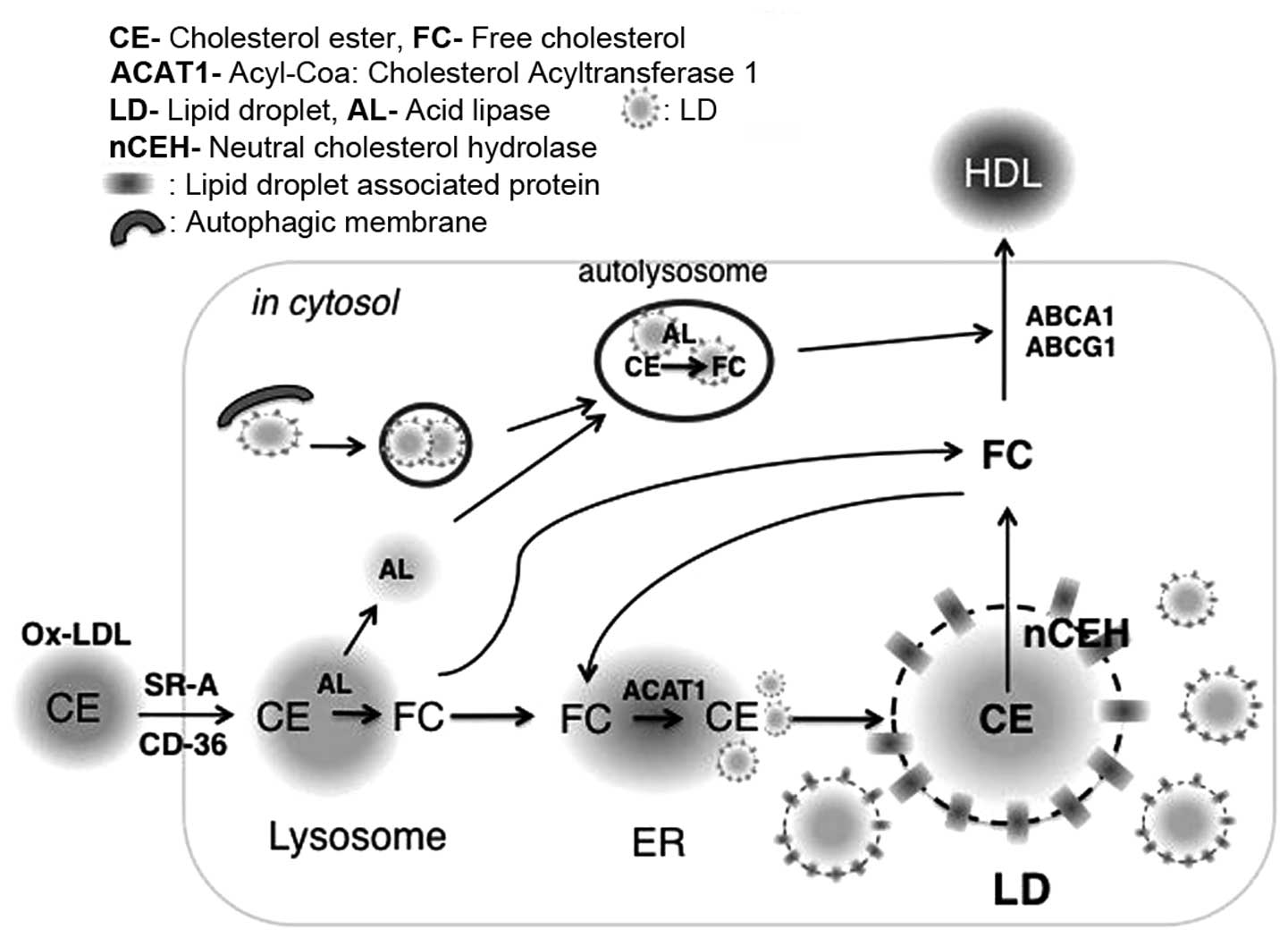
Following endocytosis of oxLDL by macrophages, the
cholesterol ester (CE) carried by these particles is hydrolyzed to
free cholesterol (FC) in the lysosomes, which is subsequently
released into the cytosol. Elevated FC levels in macrophages due to
uncontrolled LDL uptake can cause membrane damage and cytotoxicity
(5). However, toxicity can be
prevented by increasing FC efflux to high-density lipoprotein
(HDL), or by esterifying FC to CE, which is subsequently stored in
the core of cytoplasmic lipid droplets (LDs) (6) (Fig.
1). As a result of LD accumulation in the cytosol, macrophages
form ʻfoamyʼ looking shapes, hence their alternative name, foam
cells. While this mechanism may initially be protective, the
overwhelming accumulation of foam cells caused by unfettered LDL
uptake at the arterial wall results in inflammation and necrosis.
Therefore, foam cells have been the object of extensive research
efforts aiming to identify novel therapeutic strategies against
atherosclerosis. In this review, the general mechanisms of foam
cell formation are described, genes associated with LDs and their
roles in atherosclerotic development are investigated, and the
prospect of targeting foam cells to prevent and/or intervene
against atherosclerosis is discussed.
LDs consist of a phospholipid monolayer, lipid
droplet-associated proteins (LDAPs), and an inner core of neutral
lipids, including triacylglycerol (TAG), sterol esters, retinyl
esters, waxes and ether lipids (7,14). A
lipidomic study previously revealed that LDs are complexes that
contain >160 species of phospholipid. The most abundant
phospholipid is phosphatidylcholine, followed by
phosphatidylethanolamine, phosphatidylinositol and ether-linked
phosphatidlycholine (14). The
composition of neutral lipids in the core of LDs varies in
different cell types. For example, yeast cells contain almost an
equal proportion of TAG and CE, whereas adipocytes contain mostly
TAG; however, macrophages/foam cells contain mostly CE that
originates from LDL (14). In
eukaryotes, the prevalent theory for LD biogenesis is that LDs bud
off the endoplasmic reticulum (ER), where the majority of enzymes
for neutral lipid synthesis are located, including
acyl-CoA:cholesterol acyltransferase (ACAT) for sterol esters and
acyl-CoA:diacylglycerol acyltransferases for TAG (15,16).
Following synthesis of neutral lipids within the interspace of the
lipid bilayer of the ER membrane, lipids are enclosed by a
monolayer of phospholipids, which originates from the cytoplasmic
leaflet (17–20). The newly formed LDs increase in
size by incorporating lipids that are synthesized in situ by
enzymes localized at the LD surface, or by the fission of
pre-existing LDs (7,21).
LDAPs are usually located at the surface of LDs and
have an important role in the formation and degradation of LDs
(22). Proteomic analyses on LD
fractions of lipid-loaded cells have identified numerous LDAPs
(23,24). Relatively well-characterized LDAPs
include members of the perilipin, ADFP and Tip47 (PAT), and cell
death-inducing DNA fragmentation factor-like effector (CIDE)
families. In mammals, the PAT family comprises five members:
Perilipin 1 (PLIN1), Perilipin 2 (PLIN2/adipophilin/adipose
differentiation-related protein/ADFP), Perilipin 3 (PLIN3/Tip47),
Perilipin 4 (PLIN4/S3-12), and Perilipin 5 (PLIN5/lipid storage
droplet protein 5/myocardial lipid droplet protein/OXPAT/PAT1)
(25). The CIDE family comprises
three members: CIDEA, CIDEB and CIDEC (human)/fat-specific protein
of 27 kDa (mouse). While all cells have the ability to accumulate
LDs, the expression of LDAPs varies depending on cell and tissue
type. Therefore, different LDAPs are expected to replace the
function of others based on their expression pattern. PLIN1 was the
first LDAP to be identified (26),
and is highly expressed in white and brown adipose tissues, and
steroidogenic cells (26,27). In addition, PLIN1 is expressed in
detectable amounts in macrophages (28); however, its expression in mouse
macrophages remains controversial (29). Four splicing variants of PLIN1 have
been identified, namely perilipin A-D (26,30).
Perilipin A and B are expressed in adipocytes, whereas the C and D
isoforms are predominantly expressed in steroidogenic cells. Under
non-hydrolytic conditions, interaction of PLIN1 with comparative
gene identification-58 (CGI-58) blocks the access of hydrolases to
LDs, and protects TAG in LDs against hydrolysis. Under β-adrenergic
receptor activation-induced hydrolytic conditions, both PLIN1 and
cytoplasmic hormone sensitive lipase (HSL) are phosphorylated by
protein kinase A. Phosphorylated HSL gains access to LDs, whereas
phosphorylated PLIN1 dissociates from LDs and releases CGI-58 from
the LD surface. Interaction of CGI-58 with adipose triglyceride
lipase (ATGL) in the cytoplasm results in translocation of ATGL to
LDs, where it primarily hydrolyzes TAG, whereas HSL sequentially
breaks down the diacylglycerol generated by ATGL (31,32).
PLIN3 has been detected on the surface of LDs in
HeLa cells using a PLIN3 antibody to track its subcellular
localization (41). PLIN3 is
involved in the transport of mannose 6-phosphate receptors from
endosomes to the trans-Golgi network (42). Unlike PLIN1 and PLIN2, which are
fundamentally associated with LDs, PLIN3 is abundantly found in the
cytosol (43,44). Acetylated LDL (acLDL) loading of
PLIN2-depleted human THP-1 macrophages was shown to decrease CE
levels; however, PLIN3 knockdown reduced TAG levels in acLDL and
oleic acid-loaded cells (45).
Increased localization of PLIN3 to LDs was observed in
PLIN2-depleted THP-1 macrophages, without alterations to PLIN3
expression (29,45). Similarly, Chang et al
(46) detected increased PLIN3
localization to LDs in PLIN2-deficient hepatocytes. These results
suggest a differential and compensatory role of LDAPs in lipid
metabolism even within the same cells. PLIN4 is predominantly
detected in white adipose tissue, although lower amounts can be
detected in heart and skeletal muscle (41,47).
PLIN5 is expressed in heart, brown adipose tissue, liver and
skeletal muscle (34,47). A previous study, which used
microarrays to identify the expression of LDAPs in oxLDL-loaded
THP-1 macrophages, demonstrated that PLIN1 expression was not
altered, whereas PLIN2 was increased and PLIN3 was decreased.
Furthermore, PLIN4 and PLIN5 were upregulated by oxLDL in THP-1
cells (48).
With distinctive tissue distribution, the three
members of the CIDE family are involved in TAG metabolism and their
functions are highly associated with metabolic disorders. CIDEA and
CIDEB are abundantly expressed in brown adipose tissue and in the
liver, respectively (49,50). CIDEC is expressed in white and
brown adipose tissues, but not in normal liver tissue (49,51,52).
Deletion of each CIDE member in mice resulted in leaner mice due to
increased energy expenditure, and was associated with resistance to
diet-induced obesity and insulin resistance (53). All three members of the CIDE family
have recently been detected in THP-1 macrophages loaded with oxLDL
(48). However, the role of CIDEs
in cholesterol metabolism and atherosclerotic development has yet
to be elucidated.
The predominant form of lipid stored in the LDs of
macrophages/foam cells is CE. Whereas PAT and CIDE proteins have
central roles in LD metabolism, these proteins do not exert TAG or
CE hydrolyzing activities. HSL is the best-characterized CE
hydrolase in macrophages (54,55).
Despite its strong CE hydrolase activity, the absence of HSL in
human macrophages and plaques suggests the possible existence of
other CE hydrolase(s) to replace the role of HSL in human atheroma
(23,55). A recent proteomic analysis on the
LD fraction of Raw 264.7 macrophages identified a novel CE
hydrolase, lipid droplet-associated hydrolase (LDAH). Overexpressed
LDAH in RAW 264.7 macrophages decreased CE levels by increasing FC
efflux (23). LDAH was reported to
be ubiquitously expressed; however, it is significantly more
abundant in white and brown adipose tissues and in the liver.
Notably, LDAH is also highly expressed in both human and mouse
macrophages and atherosclerotic lesions (23).
In macrophages, oxLDL is notably taken-up via
endocytosis mediated by scavenger receptors (SRs), including SR-A
and cluster of differentiation 36 (CD36); however, additional
mechanisms may mediate oxLDL uptake (56,57).
CE derived from oxLDL is hydrolyzed to FC by lysosomal acid lipase
(LAL) and is then released into the cytosol. FC in the cytosol is
either effluxed by ATP-binding cassette (ABC) transporters,
including ABCA1 and ABCG1, or re-esterified by ACAT1 in the ER and
stored as CE in cytosolic LDs (Fig.
1). Two LD CE hydrolytic pathways have been reported. In the
first pathway, CE in LDs is hydrolyzed to FC by neutral cholesterol
ester hydrolases (nCEHs), which associate with LDs. The second
pathway involves autophagocytic engulfment of LDs, followed by
fusion with lysosomes and CE hydrolysis by LAL (58). FC generated from both pathways is
effluxed via ABC transporters and transferred to extracellular
acceptors for reverse transport to the liver and, ultimately, the
feces (Fig. 1).
Due to the central role of foam cells in
atherosclerotic development, foam cells have been the target of
interventions, in order to identify novel therapeutic strategies.
Some of the most significant approaches are described in this
review.
Native LDL is removed from circulation by the
process of endocytosis by LDL receptors (LDLRs); however, modified
LDL is recognized and taken-up by SRs (57,59).
Among these receptors, SR-A1 and CD36 are responsible for 75–90% of
the degradation of oxLDL in vitro (60). Therefore, blocking SRs may be a
promising strategy to ameliorate the development of
atherosclerosis. However, studies in SR-A1−/− or
CD36−/− mice with an apolipoprotein E knockout
(apoE−/−) or LDLR knockout (LDLR−/−)
background have exhibited contradictory results. Suzuki et
al (61) reported that
SR-A1−/−/apoE−/− mice exhibited decreased
atherosclerotic lesion development due to reduced cholesterol
uptake. However, these mice were more sensitive to infection, since
SRs bind a broad range of ligands expressed by bacterial pathogens
(61).
CD36−/−/apoE−/− mice also exhibited
significantly reduced lesion development; however, elevated plasma
LDL levels were detected due to loss of LDL uptake (62). In addition, SR-A or CD36 deficiency
in macrophages of LDLR−/− mice resulted in reduced
lesion development (63).
Conversely, Moore et al (56) detected increased lesion area in the
aortic sinus with abundant macrophages/foam cells in
apoE−/− mice lacking either SR-A1 or CD36, presumably
due to alternative LDL uptake mechanisms. Manning-Tobin et
al (64) also reported no
changes in lesion size and macrophage/foam cell content, but
observed reduced inflammatory gene expression and macrophage
apoptosis in
SR-A−/−/CD36−/−/apoE−/− mice.
Therefore, the benefit of targeting SRs remains controversial.
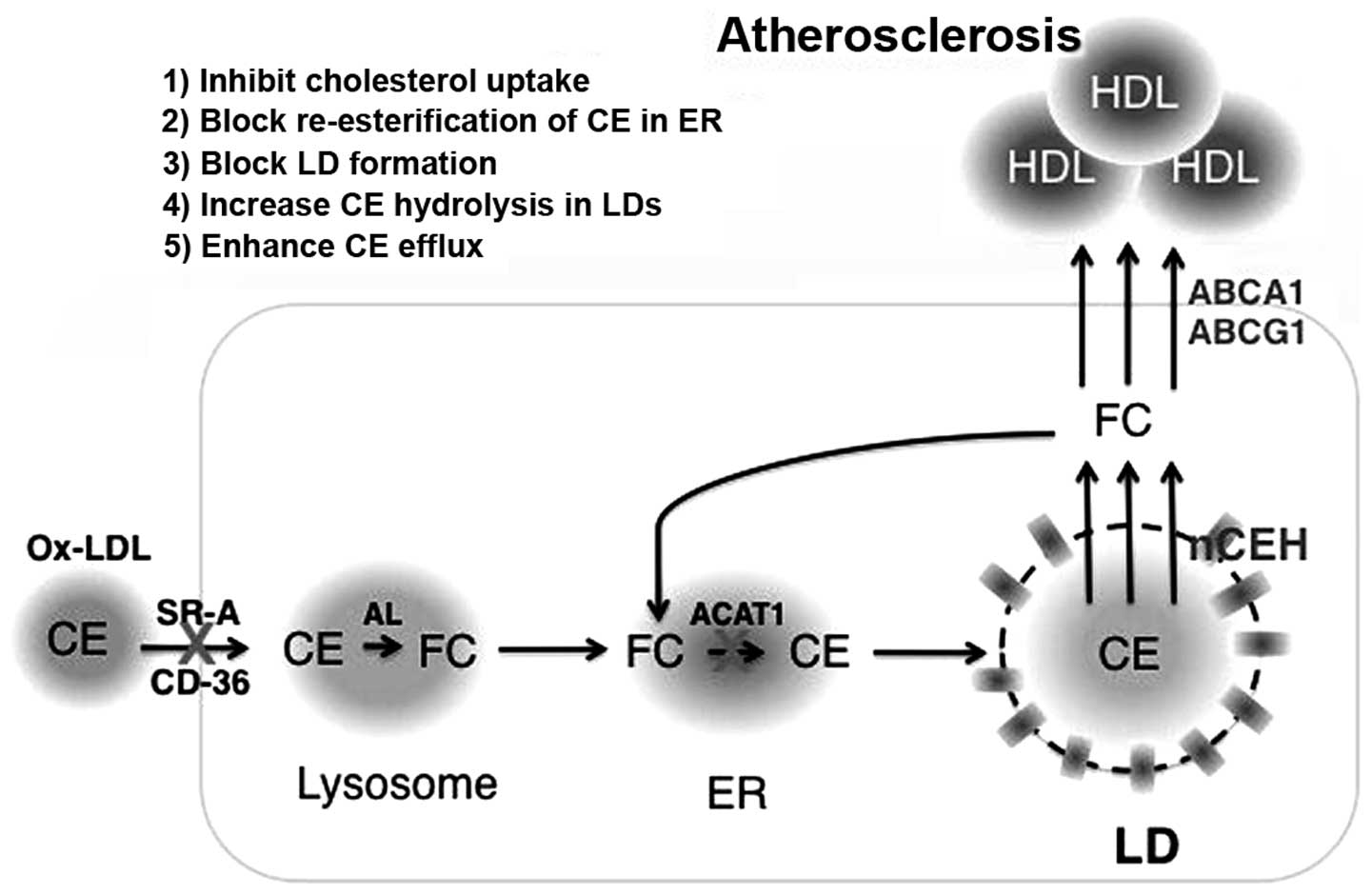
Excessive cytoplasmic FC can be re-esterified by
ACAT1 in the ER, and the generated CE can subsequently be stored in
LDs (Fig. 2). Since only FC, not
CE, can enter efflux pathways, blocking re-esterification of FC to
CE may be considered a promising strategy to inhibit foam cell
formation and facilitate cholesterol efflux. Unexpectedly,
ACTA1−/− mice, with either an LDLR−/− or
apoE−/− background, exhibited increased lesion size with
abundant necrotic cores due to macrophage apoptosis as a result of
toxicity from excessive FC accumulation in the ER (65–67).
Therefore, the therapeutic inhibition of ACAT1 does not appear to
be a desirable strategy for the treatment of atherosclerosis.
Since LDAPs are structurally or enzymatically
involved in LD homeostasis, their roles in pathologies associated
with dysfunctional lipid metabolism have been extensively studied.
As mentioned previously, abundant expression of certain LDAPs in
macrophages is closely associated with foam cell formation during
atherosclerotic development. Among known LDAPs, PLIN2 is highly
expressed in macrophages and its expression is increased by lipid
loading, whereas the expression levels of other members of the PAT
family remain very low or unchanged (29,48).
In line with the role of PLIN2 in TAG accumulation in non-monocytic
cells, PLIN2 increased CE accumulation in acLDL-loaded THP-1
macrophage by inhibiting cholesterol efflux (68). In addition, PLIN2 mRNA is highly
expressed in human and mouse atherosclerotic plaques compared with
healthy areas of the same arteries (68,69).
A global approach to identify cholesterol responsive genes in the
macrophages of LDLR−/− mice loaded with cholesterol
in vivo detected increased levels of PLIN2 (35). In agreement with these findings, a
significant role for PLIN2 in the development of atherosclerosis
was verified by Paul et al (29) using PLIN2 null mice with an
apoE−/− background.
PLIN2−/−/apoE−/− mice exhibited decreased
lesion development with reduced foam cell formation in lesions due
to increased FC efflux (29). In
addition, contrary to observations made under ACAT1 deficiency,
PLIN2 deficiency was well tolerated by the macrophages, thus
indicating that PLIN2 may be a safe target for the amelioration of
atherosclerotic development (70).
Macrophages/foam cells in the arterial wall generate
proinflammatory cytokines. The secretion of these cytokines is an
important predictor of atherosclerotic development. The expression
levels of proinflammatory cytokines, including tumor necrosis
factor-α, MCP-1 and interleukin-6, were increased by PLIN2
overexpression and decreased following knockdown of PLIN2 in THP-1
macrophages loaded with acLDL (71). Regarding the role of PLIN1 in the
development of atherosclerosis, contradictory results have been
reported. Langlois et al (72) detected increased atherosclerosis in
PLIN1−/− mice, whereas Zhao et al (28) reported that global and bone
marrow-specific PLIN1 deficiency reduced atherosclerosis. With
respect to CIDE proteins, CIDEB has been shown to control hepatic
cholesterol homeostasis, and CIDEB−/− mice exhibited
lower levels of plasma cholesterol and LDL, and increased hepatic
cholesterol levels, due to increased LDLR and ACAT expression
(73). These observations raise
the possibility that, in addition to a potential role in lipid
metabolism in macrophages, CIDE family proteins may have a role in
atherogenesis by regulating plasma cholesterol levels.
Reverse cholesterol transport (RCT) from arteries
involves transfer of cholesterol from macrophages/foam cells to HDL
(74). In order to be effluxed, CE
deposited in LDs must be hydrolyzed to FC; therefore, CE hydrolysis
may be considered the first step in RCT (75,76).
Since RCT from arteries is considered atheroprotective, enzymes
that hydrolyze CE stored in LDs, generally known as nCEHs, may have
high therapeutic potential. HSL is an intracellular neutral
hydrolase that is able to hydrolyze various esters, including CE in
macrophages (77,78). However, HSL knockdown in the bone
marrow macrophages of LDLR−/− mice did not induce
significant changes in lesion development, indicating the
possibility of compensatory mechanisms (79). Unexpectedly, rather than improving
atherosclerosis, macrophage-specific expression of transgenic rat
HSL in mice with an apoE−/− background accelerated
atherosclerosis. This paradoxical effect was not associated with
the excessive intracellular accumulation of FC, or with larger
necrotic core development within the lesions, but was attributed to
coupling of effective re-esterification of surplus FC to CE by
ACAT1 and to limited efflux by ABC transporters (77,80).
Notably, increasing cholesterol acceptors in HSL transgenic mice
reduced aortic lesion development (81). However, regardless of its role in
mice, HSL is not expressed in human atherosclerotic lesions
(23). Therefore, the identity of
the nCEH(s) in human atheroma remains unknown. A possible candidate
is LDAH, which is expressed in both human and mouse atherosclerotic
lesions, as well as in cultured and primary human and mouse
macrophages (23). LDAH
overexpression in macrophages has been reported to increase the
rate of CE hydrolysis and cholesterol efflux. However, to date, the
role of LDAH in genetically engineered mice has yet to be reported
(23).
The expression of ABCA1 and ABCG1 is regulated by
direct binding of liver X receptor (LXR) (92,93).
Administration of synthetic LXR agonists has been reported to
successfully attenuate atherosclerotic development (94). However, systemic administration of
LXR ligands causes unfavorable effects, including liver steatosis
and hypertriglyceridemia due to activation of enzymes associated
with fatty acid biosynthesis (95–98).
Therefore, several studies have attempted to discriminate the
mechanisms of LXR activation between liver and macrophages. Kim
et al (99) reported that
thyroid hormone receptor-associated protein 80 (TRAP80) selectively
activates LXR-mediated sterol regulatory element binding protein
1c, which causes liver steatosis, but not LXR-mediated ABCA1
expression. Combinatory treatments to concomitantly reduce TRAP80
activity and increase LXR activity could be of potential
therapeutic use against atherosclerosis (99). Furthermore, nanotechnology has
recently been employed for local delivery of LXR agonists to
macrophages/foam cells without systemic effects (100,101). The delivery of the LXR agonist
GW3965 encapsulated in poly(lactide-co-glycoli de)-b-poly(ethylene
glycol) copolymer nanoparticles to LDLR−/− mice markedly
reduced the number of macrophages and decreased the size of
atherosclerotic plaques by 50%, without increasing total
cholesterol and TAG levels in liver and plasma (100). Alternatively, Lim et al
(101) developed a novel
site-specific antibody-drug conjugate (ADC) to target and deliver
an aminooxy-modified LXR agonist conjugated to
anti-CD11-immunoglobulin G through a stable, cathepsin B cleavable
oxime linkage. The LXR agonist delivered by ADC was 3-fold more
powerful than the conventional LXR agonist T0901317 when tested in
THP-1 macrophages, but it did not induce LXR target genes in
hepatocytes (101). However, the
effect of this delivery system remains untested in vivo.
Since targeted delivery of LXR agonists effectively prevents
atherosclerosis while avoiding the unfavorable side effects of
conventional LXR agonists, additional research in this field is
strongly supported, and underscores the potential of nano-medicine
to treat atherosclerotic cardiovascular disease.
Atherosclerosis is a life threatening pathology,
which progresses as plaques grow in the arterial wall.
Macrophages/foam cells are found in the plaques from the early to
late stages of atherosclerotic development. Therefore, numerous
efforts to elucidate the mechanisms underlying foam cell formation,
and to target foam cells to prevent and/or reverse atherosclerosis
have been made. Presumably, given the complexity of advanced
plaques, effective interventions against atherosclerosis should
involve several pathways. Recent advances concerning the mechanisms
underlying foam cell formation have identified several LDAPs in
macrophages. Genetic modulation of some of these proteins in mice
has supported the hypothesis that LDAPs may represent plausible
novel targets for the amelioration of atherogenesis by preventing
foam cell formation, and promoting RCT with less side effects than
other interventions on non-LD foam cell proteins. The recent
identification of novel LDAPs in macrophages leaves much room for
research on the role of these proteins in lipid homeostasis and the
development of atherosclerosis. Unveiling the function of diverse
LDAPs and elucidating their molecular network may lead to novel
therapeutic strategies to overcome atherosclerosis.
The authors of the present study would like to
acknowledge the support from the National Institutes of Health R01
Grant (grant no. HL104251; to A. Paul) and the American Heart
Association Scientist Development Grant (grant no. 14SDG19690016;
to Y. Goo).
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al:
American Heart Association Statistics Committee and Stroke
Statisics: Executive summary: Heart disease and stroke statistics –
2013 update: A report from the American Heart Association.
Circulation. 127:143–152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ross R: The pathogenesis of
atherosclerosis: A perspective for the 1990s. Nature. 362:801–809.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ross R, Glomset J and Harker L: Response
to injury and atherogenesis. Am J Pathol. 86:675–684.
1977.PubMed/NCBI
|
|
4
|
Williams KJ and Tabas I: The
response-to-retention hypothesis of early atherogenesis.
Arterioscler Thromb Vasc Biol. 15:551–561. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maxfield FR and van Meer G: Cholesterol,
the central lipid of mammalian cells. Curr Opin Cell Biol.
22:422–429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maxfield FR and Tabas I: Role of
cholesterol and lipid organization in disease. Nature. 438:612–621.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Walther TC and Farese RV Jr: The life of
lipid droplets. Biochim Biophys Acta. 1791:459–466. 2009.
View Article : Google Scholar :
|
|
8
|
Thiam AR, Farese RV Jr and Walther TC: The
biophysics and cell biology of lipid droplets. Nat Rev Mol Cell
Biol. 14:775–786. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M and
Anderson RG: Chinese hamster ovary K2 cell lipid droplets appear to
be metabolic organelles involved in membrane traffic. J Biol Chem.
279:3787–3792. 2004. View Article : Google Scholar
|
|
10
|
Umlauf E, Csaszar E, Moertelmaier M,
Schuetz GJ, Parton RG and Prohaska R: Association of stomatin with
lipid bodies. J Biol Chem. 279:23699–23709. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kusminski CM, Shetty S, Orci L, Unger RH
and Scherer PE: Diabetes and apoptosis: Lipotoxicity. Apoptosis.
14:1484–1495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tabas I: Consequences of cellular
cholesterol accumulation: Basic concepts and physiological
implications. J Clin Invest. 110:905–911. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Herranz P, de Lucas R, Pérez-España L and
Mayor M: Lipodystrophy syndromes. Dermatol Clin. 26:569–578.
ix2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bartz R, Li WH, Venables B, Zehmer JK,
Roth MR, Welti R, Anderson RG, Liu P and Chapman KD: Lipidomics
reveals that adiposomes store ether lipids and mediate phospholipid
traffic. J Lipid Res. 48:837–847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Buhman KF, Accad M and Farese RV:
Mammalian acyl-CoA:Cholesterol acyltransferases. Biochim Biophys
Acta. 1529:142–154. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yen CL, Stone SJ, Koliwad S, Harris C and
Farese RV Jr: Thematic review series: Glycerolipids. DGAT enzymes
and triacylglycerol biosynthesis. J Lipid Res. 49:2283–2301. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Robenek H, Hofnagel O, Buers I, Robenek
MJ, Troyer D and Severs NJ: Adipophilin-enriched domains in the ER
membrane are sites of lipid droplet biogenesis. J Cell Sci.
119:4215–4224. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robenek MJ, Severs NJ, Schlattmann K,
Plenz G, Zimmer KP, Troyer D and Robenek H: Lipids partition
caveolin-1 from ER membranes into lipid droplets: Updating the
model of lipid droplet biogenesis. FASEB J. 18:866–868.
2004.PubMed/NCBI
|
|
19
|
Tauchi-Sato K, Ozeki S, Houjou T, Taguchi
R and Fujimoto T: The surface of lipid droplets is a phospholipid
monolayer with a unique fatty acid composition. J Biol Chem.
277:44507–44512. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ozeki S, Cheng J, Tauchi-Sato K, Hatano N,
Taniguchi H and Fujimoto T: Rab18 localizes to lipid droplets and
induces their close apposition to the endoplasmic reticulum-derived
membrane. J Cell Sci. 118:2601–2611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Long AP, Manneschmidt AK, VerBrugge B,
Dortch MR, Minkin SC, Prater KE, Biggerstaff JP, Dunlap JR and
Dalhaimer P: Lipid droplet de novo formation and fission are linked
to the cell cycle in fission yeast. Traffic. 13:705–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan Y, Li P and Ye J: Lipid homeostasis
and the formation of macrophage-derived foam cells in
atherosclerosis. Protein Cell. 3:173–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goo YH, Son SH, Kreienberg PB and Paul A:
Novel lipid droplet-associated serine hydrolase regulates
macrophage cholesterol mobilization. Arterioscler Thromb Vasc Biol.
34:386–396. 2014. View Article : Google Scholar :
|
|
24
|
Brasaemle DL, Dolios G, Shapiro L and Wang
R: Proteomic analysis of proteins associated with lipid droplets of
basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem.
279:46835–46842. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brasaemle DL: Thematic review series:
Adipocyte biology. The perilipin family of structural lipid droplet
proteins: Stabilization of lipid droplets and control of lipolysis.
J Lipid Res. 48:2547–2559. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Greenberg AS, Egan JJ, Wek SA, Garty NB,
Blanchette-Mackie EJ and Londos C: Perilipin, a major hormonally
regulated adipocyte-specific phosphoprotein associated with the
periphery of lipid storage droplets. J Biol Chem. 266:11341–11346.
1991.PubMed/NCBI
|
|
27
|
Servetnick DA, Brasaemle DL, Gruia-Gray J,
Kimmel AR, Wolff J and Londos C: Perilipins are associated with
cholesteryl ester droplets in steroidogenic adrenal cortical and
Leydig cells. J Biol Chem. 270:16970–16973. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao X, Gao M, He J, Zou L, Lyu Y, Zhang
L, Geng B, Liu G and Xu G: Perilipin1 deficiency in whole body or
bone marrow-derived cells attenuates lesions in
atherosclerosis-prone mice. PLoS One. 10:e01237382015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paul A, Chang BH, Li L, Yechoor VK and
Chan L: Deficiency of adipose differentiation-related protein
impairs foam cell formation and protects against atherosclerosis.
Circ Res. 102:1492–1501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu X, Gruia-Gray J, Copeland NG, Gilbert
DJ, Jenkins NA, Londos C and Kimmel AR: The murine perilipin gene:
The lipid droplet-associated perilipins derive from
tissue-specific, mRNA splice variants and define a gene family of
ancient origin. Mamm Genome. 12:741–749. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tansey JT, Huml AM, Vogt R, Davis KE,
Jones JM, Fraser KA, Brasaemle DL, Kimmel AR and Londos C:
Functional studies on native and mutated forms of perilipins. A
role in protein kinase A-mediated lipolysis of triacylglycerols. J
Biol Chem. 278:8401–8406. 2003. View Article : Google Scholar
|
|
32
|
Zimmermann R, Strauss JG, Haemmerle G,
Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger
G, Eisenhaber F, Hermetter A and Zechner R: Fat mobilization in
adipose tissue is promoted by adipose triglyceride lipase. Science.
306:1383–1386. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heid HW, Moll R, Schwetlick I, Rackwitz HR
and Keenan TW: Adipophilin is a specific marker of lipid
accumulation in diverse cell types and diseases. Cell Tissue Res.
294:309–321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dalen KT, Dahl T, Holter E, Arntsen B,
Londos C, Sztalryd C and Nebb HI: LSDP5 is a PAT protein
specifically expressed in fatty acid oxidizing tissues. Biochim
Biophys Acta. 1771:210–227. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Becker L, Gharib SA, Irwin AD, Wijsman E,
Vaisar T, Oram JF and Heinecke JW: A macrophage sterol-responsive
network linked to atherogenesis. Cell Metab. 11:125–135. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang HP and Serrero G: Isolation and
characterization of a full-length cDNA coding for an adipose
differentiation-related protein. Proc Natl Acad Sci USA.
89:7856–7860. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Masuda Y, Itabe H, Odaki M, Hama K,
Fujimoto Y, Mori M, Sasabe N, Aoki J, Arai H and Takano T:
ADRP/adipophilin is degraded through the proteasome-dependent
pathway during regression of lipid-storing cells. J Lipid Res.
47:87–98. 2006. View Article : Google Scholar
|
|
38
|
Xu G, Sztalryd C, Lu X, Tansey JT, Gan J,
Dorward H, Kimmel AR and Londos C: Post-translational regulation of
adipose differentiation-related protein by the ubiquitin/proteasome
pathway. J Biol Chem. 280:42841–42847. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Listenberger LL, Ostermeyer-Fay AG,
Goldberg EB, Brown WJ and Brown DA: Adipocyte
differentiation-related protein reduces the lipid droplet
association of adipose triglyceride lipase and slows
triacylglycerol turnover. J Lipid Res. 48:2751–2761. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Magne J, Aminoff A, Perman Sundelin J,
Mannila MN, Gustafsson P, Hultenby K, Wernerson A, Bauer G,
Listenberger L, Neville MJ, et al: The minor allele of the missense
polymorphism Ser251Pro in perilipin 2 (PLIN2) disrupts an α-helix,
affects lipolysis, and is associated with reduced plasma
triglyceride concentration in humans. FASEB J. 27:3090–3099. 2013.
View Article : Google Scholar
|
|
41
|
Wolins NE, Skinner JR, Schoenfish MJ,
Tzekov A, Bensch KG and Bickel PE: Adipocyte protein S3-12 coats
nascent lipid droplets. J Biol Chem. 278:37713–37721. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Diaz E and Pfeffer SR: TIP47: A cargo
selection device for mannose 6-phosphate receptor trafficking.
Cell. 93:433–443. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Barbero P, Buell E, Zulley S and Pfeffer
SR: TIP47 is not a component of lipid droplets. J Biol Chem.
276:24348–24351. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miura S, Gan JW, Brzostowski J, Parisi MJ,
Schultz CJ, Londos C, Oliver B and Kimmel AR: Functional
conservation for lipid storage droplet association among Perilipin,
ADRP and TIP47 (PAT)-related proteins in mammals, Drosophila and
Dictyostelium. J Biol Chem. 277:32253–32257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Buers I, Robenek H, Lorkowski S, Nitschke
Y, Severs NJ and Hofnagel O: TIP47, a lipid cargo protein involved
in macrophage triglyceride metabolism. Arterioscler Thromb Vasc
Biol. 29:767–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chang BH, Li L, Paul A, Taniguchi S,
Nannegari V, Heird WC and Chan L: Protection against fatty liver
but normal adipogenesis in mice lacking adipose
differentiation-related protein. Mol Cell Biol. 26:1063–1076. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wolins NE, Quaynor BK, Skinner JR, Tzekov
A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N,
Kern PA, Finck BN and Bickel PE: OXPAT/PAT-1 is a PPAR-induced
lipid droplet protein that promotes fatty acid utilization.
Diabetes. 55:3418–3428. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li H, Song Y, Li F, Zhang L, Gu Y, Zhang
L, Jiang L, Dong W, Ye J and Li Q: Identification of lipid
droplet-associated proteins in the formation of macrophage-derived
foam cells using micro-arrays. Int J Mol Med. 26:231–239. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou Z, Yon Toh S, Chen Z, Guo K, Ng CP,
Ponniah S, Lin SC, Hong W and Li P: Cidea-deficient mice have lean
phenotype and are resistant to obesity. Nat Genet. 35:49–56. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li JZ, Ye J, Xue B, Qi J, Zhang J, Zhou Z,
Li Q, Wen Z and Li P: Cideb regulates diet-induced obesity, liver
steatosis and insulin sensitivity by controlling lipogenesis and
fatty acid oxidation. Diabetes. 56:2523–2532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J,
Yao H, Zhang Y, Xue B, Li Q, et al: Up-regulation of mitochondrial
activity and acquirement of brown adipose tissue-like property in
the white adipose tissue of fsp27 deficient mice. PLoS One.
3:e28902008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nishino N, Tamori Y, Tateya S, Kawaguchi
T, Shibakusa T, Mizunoya W, Inoue K, Kitazawa R, Kitazawa S,
Matsuki Y, et al: FSP27 contributes to efficient energy storage in
murine white adipocytes by promoting the formation of unilocular
lipid droplets. J Clin Invest. 118:2808–2821. 2008.PubMed/NCBI
|
|
53
|
Gong J, Sun Z and Li P: CIDE proteins and
metabolic disorders. Curr Opin Lipidol. 20:121–126. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yeaman SJ: Hormone-sensitive lipase-a
multipurpose enzyme in lipid metabolism. Biochim Biophys Acta.
1052:128–132. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kraemer FB and Shen WJ: Hormone-sensitive
lipase: Control of intracellular tri-(di-)acylglycerol and
cholesteryl ester hydrolysis. J Lipid Res. 43:1585–1594. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Moore KJ, Kunjathoor VV, Koehn SL, Manning
JJ, Tseng AA, Silver JM, McKee M and Freeman MW: Loss of
receptor-mediated lipid uptake via scavenger receptor A or CD36
pathways does not ameliorate atherosclerosis in hyperlipidemic
mice. J Clin Invest. 115:2192–2201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ouimet M, Franklin V, Mak E, Liao X, Tabas
I and Marcel YL: Autophagy regulates cholesterol efflux from
macrophage foam cells via lysosomal acid lipase. Cell Metab.
13:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Goldstein JL, Ho YK, Basu SK and Brown MS:
Binding site on macrophages that mediates uptake and degradation of
acetylated low density lipoprotein, producing massive cholesterol
deposition. Proc Natl Acad Sci USA. 76:333–337. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kunjathoor VV, Febbraio M, Podrez EA,
Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF and
Freeman MW: Scavenger receptors class A-I/II and CD36 are the
principal receptors responsible for the uptake of modified low
density lipoprotein leading to lipid loading in macrophages. J Biol
Chem. 277:49982–49988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Suzuki H, Kurihara Y, Takeya M, Kamada N,
Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, et
al: A role for macrophage scavenger receptors in atherosclerosis
and susceptibility to infection. Nature. 386:292–296. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Febbraio M, Podrez EA, Smith JD, Hajjar
DP, Hazen SL, Hoff HF, Sharma K and Silverstein RL: Targeted
disruption of the class B scavenger receptor CD36 protects against
atherosclerotic lesion development in mice. J Clin Invest.
105:1049–1056. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mäkinen PI, Lappalainen JP, Heinonen SE,
Leppänen P, Lähteenvuo MT, Aarnio JV, Heikkilä J, Turunen MP and
Ylä-Herttuala S: Silencing of either SR-A or CD36 reduces
atherosclerosis in hyperlipidaemic mice and reveals reciprocal
upregulation of these receptors. Cardiovasc Res. 88:530–538. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Manning-Tobin JJ, Moore KJ, Seimon TA,
Bell SA, Sharuk M, Alvarez-Leite JI, de Winther MP, Tabas I and
Freeman MW: Loss of SR-A and CD36 activity reduces atherosclerotic
lesion complexity without abrogating foam cell formation in
hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 29:19–26. 2009.
View Article : Google Scholar :
|
|
65
|
Accad M, Smith SJ, Newland DL, Sanan DA,
King LE Jr, Linton MF, Fazio S and Farese RV Jr: Massive
xanthomatosis and altered composition of atherosclerotic lesions in
hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1.
J Clin Invest. 105:711–719. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fazio S, Major AS, Swift LL, Gleaves LA,
Accad M, Linton MF and Farese RV Jr: Increased atherosclerosis in
LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest.
107:163–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Warner GJ, Stoudt G, Bamberger M, Johnson
WJ and Rothblat GH: Cell toxicity induced by inhibition of acyl
coenzyme A:cholesterol acyltransferase and accumulation of
unesterified cholesterol. J Biol Chem. 270:5772–5778. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Larigauderie G, Furman C, Jaye M, Lasselin
C, Copin C, Fruchart JC, Castro G and Rouis M: Adipophilin enhances
lipid accumulation and prevents lipid efflux from THP-1
macrophages: Potential role in atherogenesis. Arterioscler Thromb
Vasc Biol. 24:504–510. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nuotio K, Isoviita PM, Saksi J, Ijäs P,
Pitkäniemi J, Sonninen R, Soinne L, Saimanen E, Salonen O, Kovanen
PT, et al: Adipophilin expression is increased in symptomatic
carotid atherosclerosis: Correlation with red blood cells and
cholesterol crystals. Stroke. 38:1791–1798. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Son SH, Goo YH, Chang BH and Paul A:
Perilipin 2 (PLIN2)-deficiency does not increase
cholesterol-induced toxicity in macrophages. PLoS One.
7:e330632012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chen FL, Yang ZH, Wang XC, Liu Y, Yang YH,
Li LX, Liang WC, Zhou WB and Hu RM: Adipophilin affects the
expression of TNF-alpha, MCP-1 and IL-6 in THP-1 macrophages. Mol
Cell Biochem. 337:193–199. 2010. View Article : Google Scholar
|
|
72
|
Langlois D, Forcheron F, Li JY, del
Carmine P, Neggazi S and Beylot M: Increased atherosclerosis in
mice deficient in perilipin1. Lipids Health Dis. 10:1692011.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li JZ, Lei Y, Wang Y, Zhang Y, Ye J, Xia
X, Pan X and Li P: Control of cholesterol biosynthesis, uptake and
storage in hepatocytes by Cideb. Biochim Biophys Acta.
1801:577–586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Brown AJ and Jessup W: Oxysterols and
atherosclerosis. Atherosclerosis. 142:1–28. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ishii I, Oka M, Katto N, Shirai K, Saito Y
and Hirose S: Beta-VLDL-induced cholesterol ester deposition in
macrophages may be regulated by neutral cholesterol esterase
activity. Arterioscler Thromb. 12:1139–1145. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kritharides L, Christian A, Stoudt G,
Morel D and Rothblat GH: Cholesterol metabolism and efflux in human
THP-1 macrophages. Arterioscler Thromb Vasc Biol. 18:1589–1599.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Escary JL, Choy HA, Reue K and Schotz MC:
Hormone-sensitive lipase overexpression increases cholesteryl ester
hydrolysis in macrophage foam cells. Arterioscler Thromb Vasc Biol.
18:991–998. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Buchebner M, Pfeifer T, Rathke N, Chandak
PG, Lass A, Schreiber R, Kratzer A, Zimmermann R, Sattler W,
Koefeler H, et al: Cholesteryl ester hydrolase activity is
abolished in HSL−/− macrophages but unchanged in macrophages
lacking KIAA1363. J Lipid Res. 51:2896–2908. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sekiya M, Osuga J, Nagashima S, Ohshiro T,
Igarashi M, Okazaki H, Takahashi M, Tazoe F, Wada T, Ohta K, et al:
Ablation of neutral cholesterol ester hydrolase 1 accelerates
atherosclerosis. Cell Metab. 10:219–228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Escary JL, Choy HA, Reue K, Wang XP,
Castellani LW, Glass CK, Lusis AJ and Schotz MC: Paradoxical effect
on atherosclerosis of hormone-sensitive lipase overexpression in
macrophages. J Lipid Res. 40:397–404. 1999.PubMed/NCBI
|
|
81
|
Choy HA, Wang XP and Schotz MC: Reduced
atherosclerosis in hormone-sensitive lipase transgenic mice
overexpressing cholesterol acceptors. Biochim Biophys Acta.
1634:76–85. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Rothblat GH, de la Llera-Moya M, Atger V,
Kellner-Weibel G, Williams DL and Phillips MC: Cell cholesterol
efflux: Integration of old and new observations provides new
insights. J Lipid Res. 40:781–796. 1999.PubMed/NCBI
|
|
83
|
Allahverdian S, Pannu PS and Francis GA:
Contribution of monocyte-derived macrophages and smooth muscle
cells to arterial foam cell formation. Cardiovasc Res. 95:165–172.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang N, Lan D, Chen W, Matsuura F and Tall
AR: ATP-binding cassette transporters G1 and G4 mediate cellular
cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci
USA. 101:9774–9779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
van Eck M, Bos IS, Kaminski WE, Orsó E,
Rothe G, Twisk J, Böttcher A, Van Amersfoort ES, Christiansen-Weber
TA, Fung-Leung WP, et al: Leukocyte ABCA1 controls susceptibility
to atherosclerosis and macrophage recruitment into tissues. Proc
Natl Acad Sci USA. 99:6298–6303. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Singaraja RR, Fievet C, Castro G, James
ER, Hennuyer N, Clee SM, Bissada N, Choy JC, Fruchart JC, McManus
BM, et al: Increased ABCA1 activity protects against
atherosclerosis. J Clin Invest. 110:35–42. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Burgess B, Naus K, Chan J,
Hirsch-Reinshagen V, Tansley G, Matzke L, Chan B, Wilkinson A, Fan
J, Donkin J, et al: Overexpression of human ABCG1 does not affect
atherosclerosis in fat-fed ApoE-deficient mice. Arterioscler Thromb
Vasc Biol. 28:1731–1737. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Basso F, Amar MJ, Wagner EM, Vaisman B,
Paigen B, Santamarina-Fojo S and Remaley AT: Enhanced ABCG1
expression increases atherosclerosis in LDLr-KO mice on a western
diet. Biochem Biophys Res Commun. 351:398–404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Baldán A, Pei L, Lee R, Tarr P, Tangirala
RK, Weinstein MM, Frank J, Li AC, Tontonoz P and Edwards PA:
Impaired development of atherosclerosis in hyperlipidemic Ldlr−/−
and ApoE−/− mice transplanted with Abcg1−/− bone marrow.
Arterioscler Thromb Vasc Biol. 26:2301–2307. 2006. View Article : Google Scholar
|
|
90
|
Out R, Hoekstra M, Hildebrand RB, Kruit
JK, Meurs I, Li Z, Kuipers F, Van Berkel TJ and Van Eck M:
Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar
macrophages and moderately influences atherosclerotic lesion
development in LDL receptor-deficient mice. Arterioscler Thromb
Vasc Biol. 26:2295–2300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yvan-Charvet L, Ranalletta M, Wang N, Han
S, Terasaka N, Li R, Welch C and Tall AR: Combined deficiency of
ABCA1 and ABCG1 promotes foam cell accumulation and accelerates
atherosclerosis in mice. J Clin Invest. 117:3900–3908.
2007.PubMed/NCBI
|
|
92
|
Sabol SL, Brewer HB Jr and
Santamarina-Fojo S: The human ABCG1 gene: Identification of LXR
response elements that modulate expression in macrophages and
liver. J Lipid Res. 46:2151–2167. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chawla A, Boisvert WA, Lee CH, Laffitte
BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, et
al: A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in
cholesterol efflux and atherogenesis. Mol Cell. 7:161–171. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Calkin AC and Tontonoz P: Liver x receptor
signaling pathways and atherosclerosis. Arterioscler Thromb Vasc
Biol. 30:1513–1518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Magana MM and Osborne TF: Two tandem
binding sites for sterol regulatory element binding proteins are
required for sterol regulation of fatty-acid synthase promoter. J
Biol Chem. 271:32689–32694. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Magana MM, Lin SS, Dooley KA and Osborne
TF: Sterol regulation of acetyl coenzyme A carboxylase promoter
requires two interdependent binding sites for sterol regulatory
element binding proteins. J Lipid Res. 38:1630–1638.
1997.PubMed/NCBI
|
|
97
|
Foretz M, Pacot C, Dugail I, Lemarchand P,
Guichard C, Le Lièpvre X, Berthelier-Lubrano C, Spiegelman B, Kim
JB, Ferré P and Foufelle F: ADD1/SREBP-1c is required in the
activation of hepatic lipogenic gene expression by glucose. Mol
Cell Biol. 19:3760–3768. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Cha JY and Repa JJ: The liver X receptor
(LXR) and hepatic lipogenesis. The carbohydrate-response
element-binding protein is a target gene of LXR. J Biol Chem.
282:743–751. 2007. View Article : Google Scholar
|
|
99
|
Kim GH, Oh GS, Yoon J, Lee GG, Lee KU and
Kim SW: Hepatic TRAP80 selectively regulates lipogenic activity of
liver X receptor. J Clin Invest. 125:183–193. 2015. View Article : Google Scholar :
|
|
100
|
Zhang XQ, Even-Or O, Xu X, van Rosmalen M,
Lim L, Gadde S, Farokhzad OC and Fisher EA: Nanoparticles
containing a liver X receptor agonist inhibit inflammation and
atherosclerosis. Adv Healthc Mater. 4:228–236. 2015. View Article : Google Scholar :
|
|
101
|
Lim RK, Yu S, Cheng B, Li S, Kim NJ, Cao
Y, Chi V, Kim JY, Chatterjee AK, Schultz PG, et al: Targeted
delivery of LXR agonist using a site-specific antibody-drug
conjugate. Bioconjug Chem. 26:2216–2222. 2015. View Article : Google Scholar : PubMed/NCBI
|