Introduction
Intrahepatic cholangiocarcinoma (iCCA) is the second
most common type of aggressive malignancy of the liver, resulting
in a poor prognosis (1). Over the
past two decades, the incidence of iCCA has increased by 22%, and
iCCA is associated with high rates of mortality with <10% of
patients surviving 5 years following diagnosis (2) due to its rapid growth and tendency to
invade adjacent organs and metastasize. However, the molecular
pathogenesis of iCCA remains to be elucidated.
Genome-wide analysis of gene expression with
microarray and RNA-Seq technology has enabled an overall insight
into iCCA molecular processes, and will enable characterization of
the complex mechanism underlying the development of iCCA. Numerous
microarray studies have identified hundreds of differentially
expressed genes (DEGs) in tumor tissues or cell lines of iCCA when
compared with normal intrahepatic bile duct epithelia or normal
liver tissues (3,4), and a series of genes have been
revealed as potential biomarkers or therapeutic targets for iCCA. A
recent RNA-Seq study determined fusion and mutation in iCCA
para-tumor/tumor sample pairs (5),
allowing the comparison of gene expression in tumor samples of
iCCA.
The present study analyzed RNA-Seq data from seven
para-tumor and tumor sample pairs of iCCA. In order to clarify the
transcriptomic difference between tumor and para-tumor tissue of
iCCA, DEG analysis was performed. The RPL41 gene has two ensemble
ID (ENSG00000279483 and ENSG00000229117) in the latest release of
the Ensembl GRCh38 human reference genome. In the present analysis,
only the differential expression of ENSG00000279483 was identified.
It was identified that the expression of PDZK1IP1, EEF1A2 and RPL41
genes was significantly upregulated in the iCCA samples, this may
indicate that PDZK1IP1, EEF1A2 and RPL41 are key molecular markers
associated with the tumorigenesis and progression of iCCA. Notably,
RPL41 has been annotated in the Genome Reference Consortium
(GRC)h38 database for the first time and is also identified to be
overexpressed in iCCA. Furthermore, genes associated with the
immune system, metabolism and metabolic energy were significantly
downregulated in iCCA tumor tissues, which indicates that the basic
functions of the liver and cholangiole was affected in iCCA. The
present study provided a global gene expression view of the
tumorigenesis of iCCA.
Materials and methods
RNA-Seq data processing and expression
profiling
Seven para-tumor/tumor pairs of iCCA RNA-Seq data
were downloaded from the NCBI Gene Expression Omnibus (GEO;
accession no. GSE63420; http://www.ncbi.nlm.nih.gov/geo/) and subsequently
reanalyzed. These data were obtained from fresh frozen tumour
tissue and corresponding normal tissue from 7 resected patients
with iCCA, which were collected from the Biorepository Tissue Bank
at the Icahn School of Medicine at Mount Sinai (New York, NY, USA)
(5). Data were generated by the
Illumina HiSeq 2500 system with 100 nucleotide single-end reads.
High-quality sequences were aligned to Ensembl human genome GRCh38
(released on Jul 2014), which contain 20,364 coding genes and
196,345 gene transcripts, using a TopHat (v2.0.13) alignment tool
with default parameters (6).
Gene/transcripts expression levels were quantified by Cufflinks
(v2.0.2) (7), yielding raw read
counts and normalized FPKM values (reads per kilobase per million
reads) which enables the comparison between para-tumor and tumor
samples. Then, the gene/transcript expression values were log2
transformed and genes with null values were removed in all 7 pairs
of samples. Pearson correlation coefficient and hierarchical
clustering analysis (HCA), calculated by the Ward method (8) based on a distance matrix of the
Pearson correlation of the samples, were performed in the R
(www.r-project.org/) environment using
its 'base' function and 'stat' packages. Samples with similar gene
expression profiles were clustered together.
Differential expression analysis
Differential expression of genes between tumor
samples and matched para-tumor samples was identified. Cuffdiff
(v2.0.2) was applied to calculate fold changes using the FPKM value
of each gene between tumor and matched para-tumor samples, and
statistical significance of DEGs was presented by calculating a
P-value according to Poisson distribution. Then, significance of a
DEG/transcript between two samples was determined according the
threshold of |log2 (fold change)|>5 and false discovery rate
(FDR) adjusted to P<0.05.
Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analysis of DEGs
Upregulated genes and downregulated genes identified
by comparing gene expression between tumor and para-tumor samples
were analyzed using the KEGG pathway database, as previously
described (9), in order to
determine the biological function of these DEGs. Enriched pathway
was determined by significant fisher exact test (P<0.05), and at
least 3 DEGs were involved in the pathway. The pathway enrichment
analysis was performed using 'KEGG.db' and 'KEGGprofile' packages
from R project.
Results
Overall gene expression profiling of iCCA
samples
In order to characterize the global gene expression
changes of iCCA samples, all the sequenced reads were aligned to
the Ensembl GRCh38 human genome. Expression level values (FPKM) for
64,232 genes (including coding and non-coding genes) were obtained,
and 20,880 genes with zero FPKM in all the 14 samples were
excluded, finally, ~43,352 genes remained. After log2
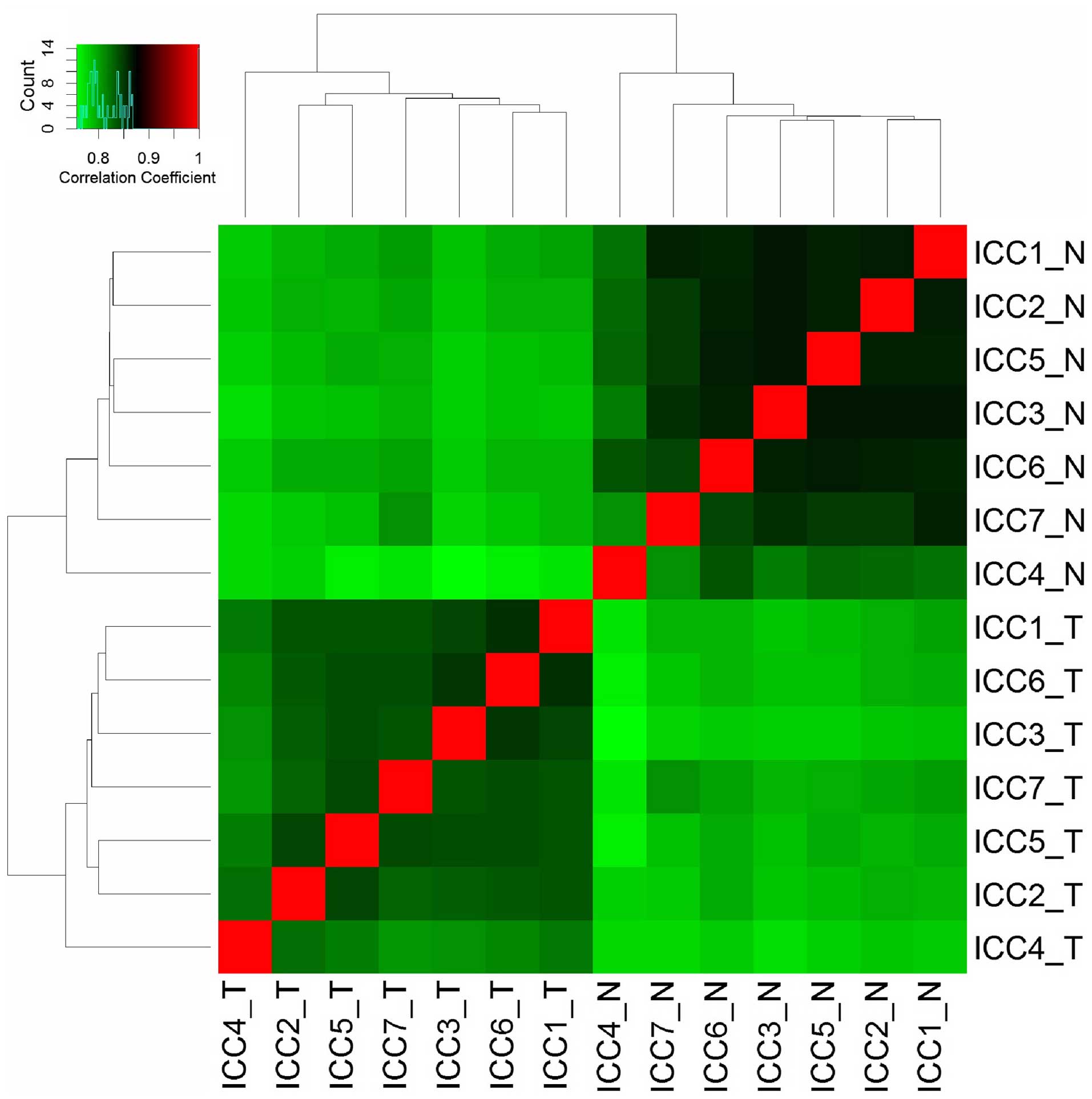
transformation of FPKM values, HCA revealed the variability of each
sample in terms of their gene expression levels. Samples were
clearly clustered into two groups according to tumor and para-tumor
samples (Fig. 1). The results of
HCA showed marked gene expression alteration in iCCA tumor tissues.
In addition, high correlation coefficient within the tumor and
para-tumor groups indicates a satisfactory reproducibility and low
variability in iCCA biological samples.
DEG analysis
DEGs were identified by comparing the gene
expression profiles of 7 tumor samples and matched para-tumor
tissues. The same cutoffs of P<0.05 and fold change >2 were
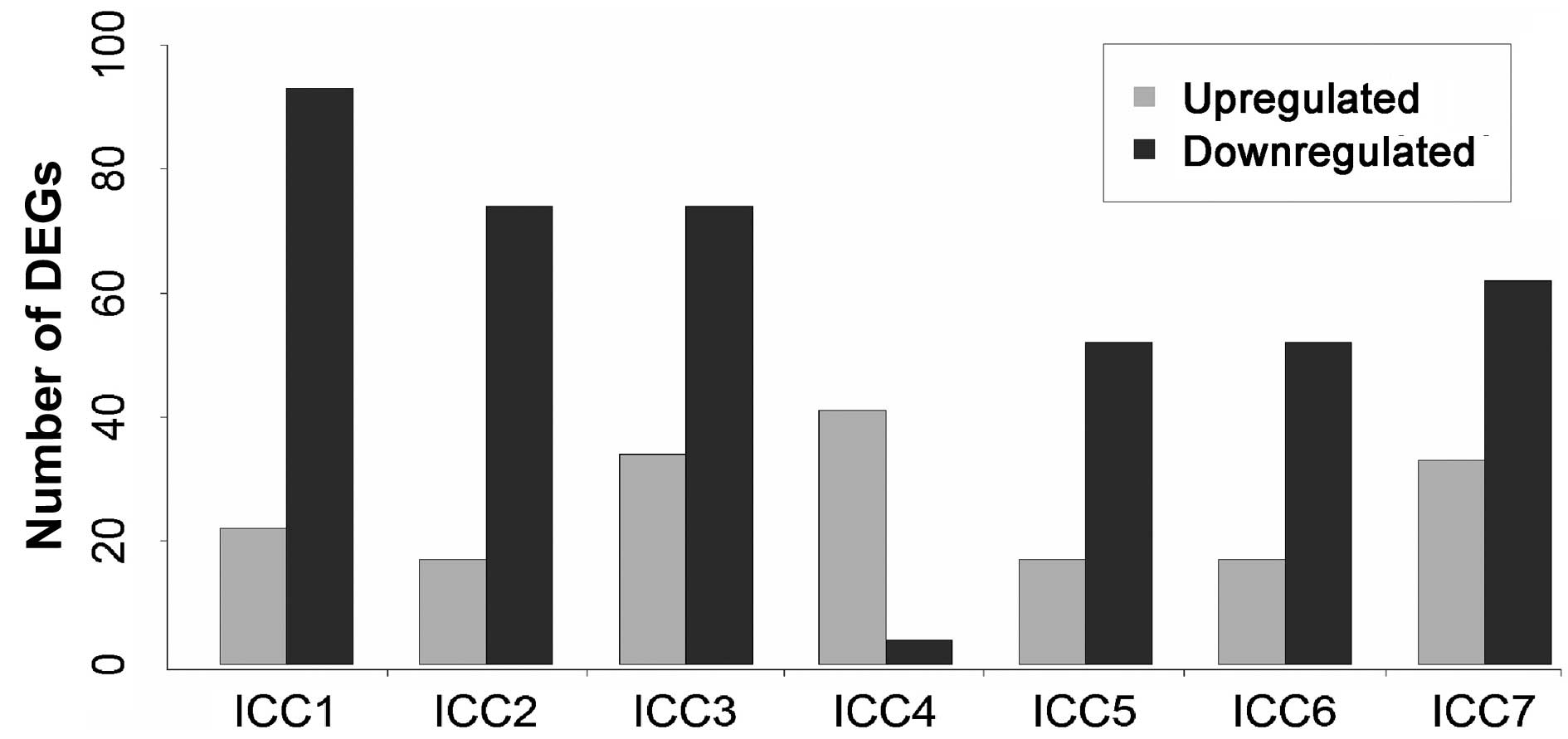
applied to select DEGs for all tumor samples. As shown in Fig. 2, the number of DEGs in 7 tumor
samples varied from 45 to 115, and 284 unique genes were identified
to be DEGs in at least one iCCA patient. No overlapping DEGs were
identified between the 7 iCCA samples. In addition, except for the
iCCA4 sample, the number of downregulated genes was at least double
that of the upregulated genes in each sample. This may indicate an
overall downregulation transcriptomic expression in iCCA tumors.
Differential expression of genes specific to iCCA were identified
as deregulated genes in ≥4 of the iCCA samples.
Pathway enrichment analysis of DEGs
To further clarify the biological function of
identified DEGs, pathway enrichment analysis was performed by
analyzing the DEGs using the KEGG pathway database. Pathways with a
fisher's exact P<0.05 and with at least three genes involved
were determined to be significantly enriched. The results of the
enriched pathways of the 140 downregulated genes are listed in
Table I. The top enriched pathway
was associated with fat digestion and absorption; other pathways
were associated with the basic biological processes of the liver
and gallbladder. The downregulation of these key metabolism genes
may indicate the loss of these functions in iCCA tumors.
 | Table IEnriched pathways of downregulated
genes. |
Table I
Enriched pathways of downregulated
genes.
| Pathway | DEGs | Genes | DEGs/Genes (%) | P-value |
|---|
| Fat digestion and
absorption | 8 | 46 | 0.17 | 6.65E-10 |
| Bile secretion | 8 | 71 | 0.11 | 3.54E-08 |
| Caffeine
metabolism | 3 |
7 | 0.43 | 3.79E-07 |
| Steroid hormone
biosynthesis | 5 | 57 | 0.09 | 2.41E-05 |
| Complement and
coagulation cascades | 5 | 69 | 0.07 | 7.14E-05 |
| Metabolism of
xenobiotics by cytochrome P450 | 5 | 71 | 0.07 | 8.38E-05 |
| Drug
metabolism-cytochrome P450 | 5 | 73 | 0.07 | 9.79E-05 |
| Linoleic acid
metabolism | 3 | 30 | 0.1 | 2.44E-04 |
| Retinol
metabolism | 4 | 64 | 0.06 | 4.95E-04 |
| ABC transporters | 3 | 44 | 0.07 | 1.07E-03 |
| Drug metabolism-other
enzymes | 3 | 52 | 0.06 | 2.00E-03 |
| Starch and sucrose
metabolism | 3 | 54 | 0.06 | 2.30E-03 |
| Pancreatic
secretion | 4 | 101 | 0.04 | 3.73E-03 |
| Cytokine-cytokine
receptor interaction | 7 | 265 | 0.03 | 4.78E-03 |
| PPAR signaling
pathway | 3 | 70 | 0.04 | 5.83E-03 |
| Vascular smooth
muscle contraction | 4 | 116 | 0.03 | 6.64E-03 |
Following analysis of the 107 upregulated genes,
only three pathways were enriched (Table II). The top enriched pathway was
salivary secretion, and no metabolic pathways were identified to be
enriched. Notably, 4 upregulated genes were assigned to the
significantly enriched Wnt signaling pathway and cytokine-cytokine
receptor interaction pathway, respectively. The relevance of these
pathways in cholangiocarcinoma or hepatocellular carcinoma has
previously been confirmed by several studies (10,11).
| Table IIEnriched pathways of upregulated
genes. |
Table II
Enriched pathways of upregulated
genes.
| Pathway | DEGs | Genes | DEGs/Genes (%) | P-value |
|---|
| Salivary
secretion | 4 | 89 | 0.04 | 8.17E-05 |
| Wnt signaling
pathway | 4 | 151 | 0.03 | 9.18E-04 |
| Cytokine-cytokine
receptor interaction | 4 | 265 | 0.02 | 9.65E-03 |
Identification of iCCA specific DEGs
Whole-transcriptome sequencing allows the
determination of key alterations in gene expression profiles that
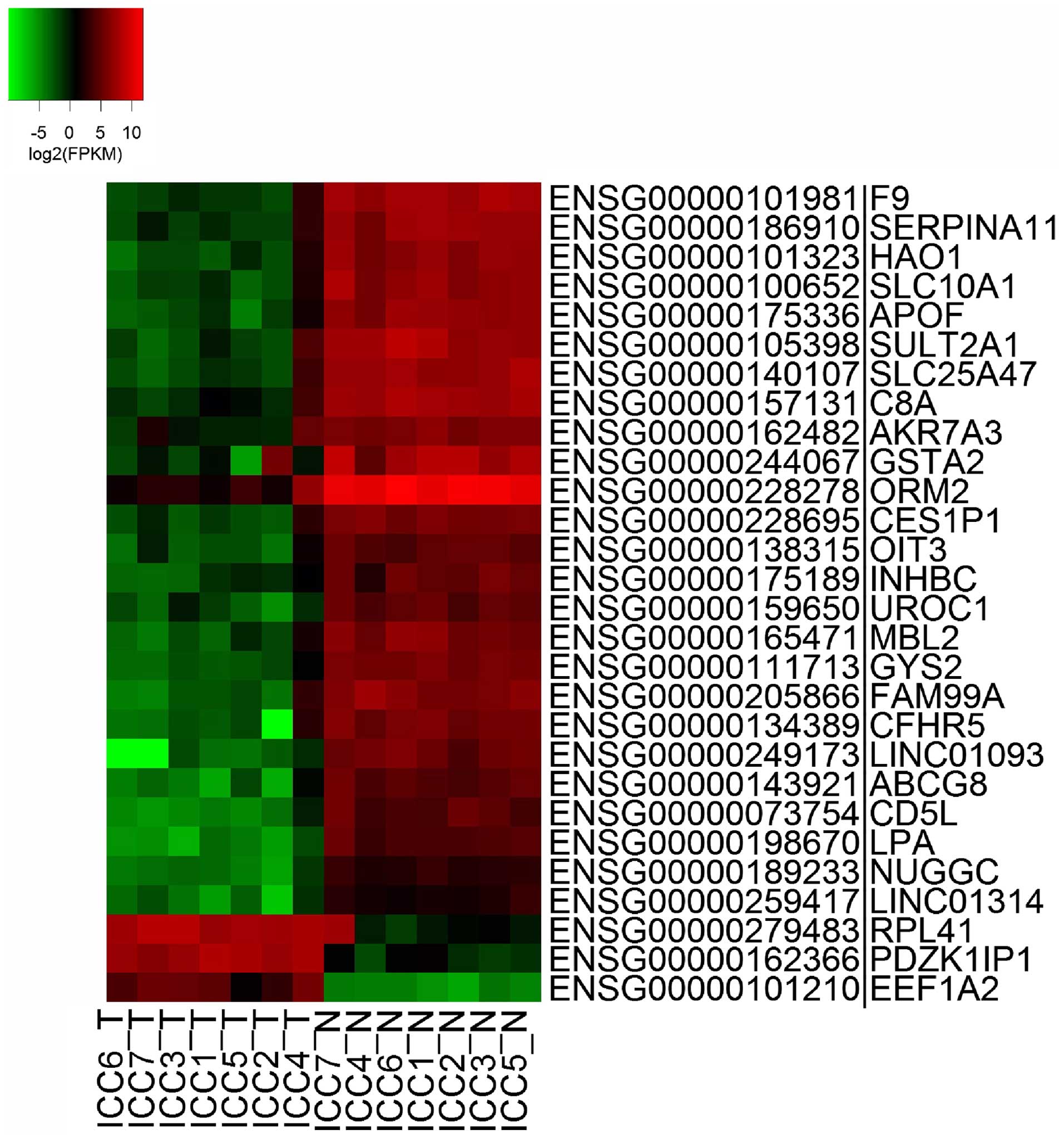
are associated with the pathogenesis of iCCA. As shown in Fig. 3, genes which were differentially
expressed in at least 4 tumor samples were identified as
specifically upregulated or downregulated in iCCA. The genes
PDZK1IP1 (also termed MAP17), RPL41 and EEF1A2 were significantly
upregulated in iCCA tumor samples.
Furthermore, several genes associated with the
immune system and metabolism were significantly downregulated
(Table III). MBL2, ORM2, C8A and
CD5L are important elements in the innate immune system. The
downregulation of these genes may indicate a defect in immune
function contributing to the tumorigenesis of iCCA. LPA, APOF,
HAO1, SLC10A1, GYS2 and SULT2A1 involved in the lipid metabolism
and circulation of bile acids were also found to be downregulated
in iCCA. Furthermore, genes involved in metabolic energy and
glycogen synthesis, such as GYS2, ABCG8 and NUGGC are negatively
regulated in iCCA tumor tissues. Thus, the basic biological
functions of the liver, including glycogen synthesis, are
influenced by iCCA tumors.
| Table IIIGene ontology annotation of key
intrahepatic cholangiocarcinoma-related genes. |
Table III
Gene ontology annotation of key
intrahepatic cholangiocarcinoma-related genes.
| Category | Term | Term name | Genes |
|---|
| Immune↓ | GO:0006950 | Response to
stress | C8A, MBL2, LPA, F9,
CD5L, ORM2 |
| GO:0006952 | Defense response | C8A, MBL2, CD5L,
ORM2 |
| GO:0006954 | Inflammatory
response | C8A, MBL2, ORM2 |
| GO:0006629 | Lipid metabolic
process | HAO1, LPA, SULT2A1,
APOF |
| GO:0002682 | Regulation of immune
system process | C8A, MBL2, ORM2 |
| Metabolism↓ | GO:0006869 | Lipid transport | ABCG8, LPA, APOF |
| GO:0007586 | Digestion | ABCG8, SULT2A1 |
| GO:0046395 | Carboxylic acid
catabolic process | HAO1, SULT2A1 |
| Cancer↓ | GO:0006414 | Translational
elongation | RPL41, EEF1A2 |
Discussion
iCCA is an aggressive primary liver tumor arising
from the epithelial cells of intrahepatic bile ducts and the
incidence of this disease has increased over the past two decades
(2). Dioxin exposure, inflammatory
disease, and parasitic liver diseases may be predominant risk
factors of iCCA. However, the molecular mechanism of iCCA
tumorigenesis remains unclear. The genome-wide gene expression of
iCCA was investigated in order to demonstrate the molecular
pathogenesis of iCCA. The expression of 43,352 genes was quantified
using RNA-Seq data. Differential gene expression analysis and
functional annotation tools were employed to clarify the changes in
expression profiles, and further demonstrated the molecular
mechanism underlying the tumorigenesis and progression of iCCA. In
this study, PDZK1IP1, RPL41 and EEF1A2 were identified to be
significantly overexpressed in iCCA.
RPL41, which is a novel protein coding gene
annotated in the GRCh38 human genome database (12), was upregulated in the present
study. To the best of our knowledge, this is the first annotation
of RPL41 overexpression in iCCA. As a small ribosomal peptide
deregulated in tumors, RPL41 is essential for mitosis and
centrosome integrity (13), and
also functions as a tumor suppressor and deregulates the
bioactivity of tumor cells (13,14).
The correlation between RPL41 and iCCA requires further
investigation.
PDZK1IP1, PDZK1-interacting protein 1, is a small 17
kDa non-glycosylated membrane protein, researchers have revealed
the overexpression of PDZK1IP1 in a variety of human carcinomas
(15). As a common characteristic
of carcinoma (15) overexpression
of PDZK1IP1 enhances malignant behavior such as aggression and
invasion of tumor cells (16).
Overexpression of this gene was also observed in the present study,
demonstrating its important role in the progression of iCCA.
Upregulation of PDZK1IP1 has been demonstrated to enhance reactive
oxygen species (ROS) production and tumorigenesis (16) as well as inhibit Myc-induced
apoptosis through PI3K/AKT pathway activation (17). Therefore, PDZK1IP1 may be a key
element in the tumorigenesis and progression of iCCA and may be
associated with a poor prognosis.
EEF1A2, Elongation factor 1-α2, is upregulated in
numerous cancer types and is essential for cell migration, invasion
and metastasis in cancer (18,19).
As a translation elongation factor, EEF1A2 binds to amino acylated
tRNA directly and guides its connection with the ribosome and mRNA
codon (20). Previous studies have
identified the overexpression of EEF1A2 in various tumors,
including breast cancer, ovarian cancer and prostate cancer
(18,21–23).
In addition, it was demonstrated that the upregulation of EEF1A2
can promote cancer cell migration, invasion and metastasis
(19); therefore, as the present
study identified EEF1A2 overexpression in iCCA, EEF1A2 may be a
useful prognostic factor for iCCA.
In addition, the SLC25A47 gene is significantly
downregulated in iCCA, and its encoded protein has the hallmark
features of mitochondrial carrier proteins in hepatocellular
carcinoma (24). Furthermore,
downregulation of a solute carrier family gene SLC25A47 (also
termed HDMCP) and a serpin peptidase inhibitor SERPINA11 was also
associated with iCCA pathogenesis. Consistently, downregulation of
SLC25A47 has been shown to induce the dissipation of the
mitochondrial membrane potential in hepatocellular carcinoma, a
common phenomenon observed in cancer cells (24). SERPINA11 is correlated with
pathological stage of hepatocellular carcinoma, and is
downregulated in oral squamous cell carcinomas (25). Thus, these genes could be a
potential therapeutic target and biomarker in the treatment of
iCCA.
Overall expression profiling identified a difference
between para-tumor and tumor tissues, however, consistency was
observed among the profiles of tumor tissues from different
individuals. In total, 164 downregulated genes and 121 upregulated
genes were identified in at least one tumor sample. KEGG pathway
enrichment analysis of the DEGs demonstrated that dysregulated
genes were predominantly associated with fat digestion and
absorption, bile secretion and other metabolism pathways, which may
indicate that the basic biological functions of the liver and
cholangiole were altered in iCCA. In addition, 4 genes associated
with the Wnt signaling pathway were significantly upregulated. Thus
tumorigenesis and progression of iCCA may lead to the dysfunction
of liver metabolism. Consistent with the present results, the
expression of the SLC10A1 gene (a member of the sodium/bile acid
cotransporter family which participates in the enterohepatic
circulation of bile acids) was decreased in cholangiocarcinoma
(26). SULT2A1, encoding a protein
which catalyzes the sulfation of steroids and bile acids in the
liver and adrenal glands, and the ABCG8 gene, which is associated
with cholesterol absorption, were downregulated in iCCA. SULT2A1
and ABCG8 are important role in the bile secretion pathway. LPA
(also termed APOA) and APOF involved in transport and/or
esterification of cholesterol were also decreased in iCCA. This may
indicate the dysfunction in lipid metabolism and the influence of
iCCA on the biological functions of the liver.
In conclusion, PDZK1IP1, RPL41 and EEF1A2 were
significantly upregulated in iCCA and may be associated with the
poor prognosis of iCCA. Metabolism and immune function-related
genes, MBL2, ORM2, C8A, CD5L and LPA, APOF, HAO1, SLC10A1, GYS2
were significantly downregulated and influenced in iCCA patients
respectively. Therefore, the present study elucidated the gene
expression patterns associated with the tumorigenesis of iCCA,
which may be novel diagnostic factors for iCCA
Acknowledgments
This study was funded by the Outstanding Leaders
Training Program of Pudong Health Bureau of Shanghai (grant no.
PWR12015-04) and the Scientific Research Start-up Funding of the
Seventh People's Hospital Affiliated to Shanghai University of
Traditional Chinese Medicine.
References
|
1
|
Bosman FT, Carneiro F, Hruban RH and
Theise ND: WHO classification of tumours of the digestive system.
World Health Organization; 2010
|
|
2
|
Everhart JE and Ruhl CE: Burden of
digestive diseases in the United States part II: Lower
gastrointestinal diseases. Gastroenterology. 136:741–754. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang AG, Yoon SY, Oh JH, Jeon YJ, Kim M,
Kim JM, Byun SS, Yang JO, Kim JH, Kim DG, et al: Identification of
intrahepatic cholangiocarcinoma related genes by comparison with
normal liver tissues using expressed sequence tags. Biochem Biophys
Res Commun. 345:1022–1032. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Obama K, Ura K, Li M, Katagiri T, Tsunoda
T, Nomura A, Satoh S, Nakamura Y and Furukawa Y: Genome-wide
analysis of gene expression in human intrahepatic
cholangiocarcinoma. Hepatology. 41:1339–1348. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sia D, Losic B, Moeini A, Cabellos L, Hao
K, Revill K, Bonal D, Miltiadous O, Zhang Z, Hoshida Y, et al:
Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion
and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun.
6:60872015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biology. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ward JH Jr: Hierarchical grouping to
optimize an objective function. J Am Statist Assoc. 58:236–244.
1963. View Article : Google Scholar
|
|
9
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
10
|
Patel T: New insights into the molecular
pathogenesis of intrahepatic cholangiocarcinoma. J Gastroenterol.
49:165–172. 2014. View Article : Google Scholar :
|
|
11
|
DeMorrow S, Francis H, Gaudio E, Venter J,
Franchitto A, Kopriva S, Onori P, Mancinelli R, Frampton G, Coufal
M and Mitchell B: The endocannabinoid anandamide inhibits
cholangiocarcinoma growth via activation of the noncanonical Wnt
signaling pathway. Am J Physiol Gastrointest Liver Physiol.
295:G1150–G1158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cunningham F, Amode MR, Barrell D, Beal K,
Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G,
Fitzgerald S, et al: Ensembl 2015. Nucleic Acids Res. 43(Database
issue): D662–D669. 2015. View Article : Google Scholar :
|
|
13
|
Wang S, Huang J, He J, Wang A, Xu S, Huang
SF and Xiao S: RPL41, a small ribosomal peptide deregulated in
tumors, is essential for mitosis and centrosome integrity.
Neoplasia. 12:284–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang A, Xu S, Zhang X, He J, Yan D, Yang Z
and Xiao S: Ribosomal protein RPL41 induces rapid degradation of
ATF4, a transcription factor critical for tumour cell survival in
stress. J Pathol. 225:285–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guijarro MV, Leal JF, Fominaya J, Lleonart
M, Castellvi J, Ruiz L, Ramon Y, Cajal S and Carnero A: MAP17
overexpression is a common characteristic of carcinomas.
Carcinogenesis. 28:1646–1652. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guijarro MV, Leal JF, Blanco-Aparicio C,
Alonso S, Fominaya J, Lleonart M, Castellvi J, Ramon y Cajal S and
Carnero A: MAP17 enhances the malignant behavior of tumor cells
through ROS increase. Carcinogenesis. 28:2096–2104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guijarro MV, Link W, Rosado A, Leal JF and
Carnero A: MAP17 inhibits Myc-induced apoptosis through PI3K/AKT
pathway activation. Carcinogenesis. 28:2443–2450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun Y, Du C, Wang B, Zhang Y, Liu X and
Ren G: Up-regulation of eEF1A2 promotes proliferation and inhibits
apoptosis in prostate cancer. Biochem Biophys Res Commun. 450:1–6.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu C, Hu DM and Zhu Q: eEF1A2 promotes
cell migration, invasion and metastasis in pancreatic cancer by
upregulating MMP-9 expression through Akt activation. Clin Exp
Metastasis. 30:933–944. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Browne GJ and Proud CG: Regulation of
peptide-chain elongation in mammalian cells. Eur J Biochem.
269:5360–5368. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tomlinson VA, Newbery HJ, Wray NR, Jackson
J, Larionov A, Miller WR, Dixon JM and Abbott CM: Translation
elongation factor eEF1A2 is a potential oncoprotein that is
overexpressed in two-thirds of breast tumours. BMC Cancer.
5:1132005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kulkarni G, Turbin DA, Amiri A, Jeganathan
S, Andrade-Navarro MA, Wu TD, Huntsman DG and Lee JM: Expression of
protein elongation factor eEF1A2 predicts favorable outcome in
breast cancer. Breast Cancer Res Treat. 102:31–41. 2007. View Article : Google Scholar
|
|
23
|
Pinke DE, Kalloger SE, Francetic T,
Huntsman DG and Lee JM: The prognostic significance of elongation
factor eEF1A2 in ovarian cancer. Gynecol Oncol. 108:561–8. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tan MG, Ooi LL, Aw SE and Hui KM: Cloning
and identification of hepatocellular carcinoma down-regulated
mitochondrial carrier protein, a novel liver-specific uncoupling
protein. J Biol Chem. 279:45235–45244. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shiiba M, Nomura H, Shinozuka K, Saito K,
Kouzu Y, Kasamatsu A, Sakamoto Y, Murano A, Ono K, Ogawara K, et
al: Down-regulated expression of SERPIN genes located on chromosome
18q21 in oral squamous cell carcinomas. Oncol Rep. 24:241–249.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yeh CN, Weng WH, Lenka G, Tsao LC, Chiang
KC, Pang ST, Chen TW, Jan YY and Chen MF: cDNA microarray profiling
of rat cholangiocarcinoma induced by thioacetamide. Mol Med Rep.
8:350–360. 2013.PubMed/NCBI
|