Introduction
Filoviruses are responsible for complex hemorrhagic
fevers in humans and primates with case fatality rates of 50–90%
(1). The filovirus family belongs
to the order Mononegavirales and is divided into two genera:
Ebolavirus comprising five species, Zaire ebolavirus
(EBOV), Taï Forest ebolavirus, Reston ebolavirus,
Bundibugyo ebolavirus and Sudan ebolavirus; and
Marburgvirus, which contains one species, Marburg
marburgvirus. A third genus, Cuevavirus has also been
recently proposed (2).
Ebolaviruses have a non-segmented negative-sense (NNS)
single-stranded RNA genome, a characteristic of the order,
Mononegavirales, and most closely resembling the families
Rhabdoviridae and Paramyxoviridae in its genetic
assembly. The EBOV genome is ~19,000 base pairs in length and
contains seven open reading frames that code for seven structural
proteins in order, from 3′ to 5′, nucleoprotein (NP), VP35, VP40,
glycoprotein (GP), VP30, VP24 and the L protein (3).
The L protein is 2,212 amino acids in length and is
the largest protein encoded by the EBOV virus. It does not function
independently but is part of an RNA-dependent RNA polymerase (RdRp)
complex with the NP and viral transcription factors, VP30 and VP35.
This complex is important in the replication and transcription of
the viral genome. The L protein is hypothesized to serve as the
major catalytic subunit of this complex (4). The majority of the data available on
the structure and function of the EBOV L protein is theoretical and
inadequate, relying on comparative analysis and data from the L
proteins of other well-studied NNS RNA viruses of the order
Mononegavirales rather than on experimental
observations.
The current 2014 EBOV epidemic in West Africa is the
largest ever reported Ebola epidemic across multiple West African
countries with unprecedented transmissibility. There are no
marketed antiviral therapeutic agents or vaccines against EBOV and
the continued search for effective and safe therapeutic agents is
required (5). RdRp of other RNA
viruses, such as hepatitis C virus, have been successful targets
for the development of antiviral therapeutic agents and vaccines
due to the presence of various highly conserved motifs and/or
residues (6,7). A previous study identified single
nucleotide polymorphisms that distinguish the 2014 West African
outbreak from previous outbreaks and demonstrated how the virus
spread from Guinea to Sierra Leone in a matter of few months
(8). The authors have identified
16 novel non-synonymous amino acid substitutions in the L protein
that are present in the 2014 outbreak isolates (8).
In the present study, the 2014 EBOV L protein
sequences were compared with other NNS RNA viruses, including
Paramyxoviridae and Rhabdoviridae. The majority of
previously reported L protein specific linear were conserved
(9). Novel conserved regions that
were not previously documented were also identified in the current
study. Notably, all the mutations identified in the 2014 EBOV
isolates were tolerant, they were pathogenic with certain examples
occurring within previously determined functional conserved motifs,
possibly attenuating viral pathogenicity, replication and
virulence. Phylogenetic analysis of L protein sequences from past
EBOV outbreaks was also conducted and was consistent with previous
studies.
Materials and methods
Sequence alignment
In order to align the sequences, 475 amino acid
sequences of the L proteins belonging to the family
Rhabdoviridae (vesicular stomatitis virus and rabies virus)
and Paramyxoviridae (Sendai virus, Newcastle disease virus
and measles virus) were randomly selected from the National Center
for Biotechnology Information (NCBI) Protein Database (http://www.ncbi.nlm.nih.gov/protein). The
sequences were analyzed in the Jalview Desktop Multiple Alignment
Editor (v.2.8.1) (10) with 81
EBOV L protein amino acid sequences from the 2014 outbreak, to draw
a representative consensus sequence for each of the six viruses.
The resulting consensus sequences of all six viruses were then
aligned using the built-in MAFFT multiple sequence alignment
program (v.7.205) (11) with the
L-INS-i algorithm (12).
Phylogenetic tree construction
A phylogenetic tree of all 101 EBOV sequences
reported to date (including the 2014 EBOV sequences) was also
constructed in MEGA.v.6.06 (13),
employing the Neighbor-Joining algorithm with the Poisson
correction method (14). All 81
2014 EBOV sequences were used in constructing the tree; however,
they are presented as a single group in the tree figure for
clarity.
Amino acid substitution analysis
Furthermore, a standard prediction tool, SIFT Blink
was used to predict and analyze the effect of the 16 amino acid
substitutions on the function of the L protein (15).
Results
Consensus sequencing
In the present study, 475 L protein sequences of
vesicular stomatitis virus, rabies virus, Newcastle disease virus,
Sendai virus, measles virus and EBOV were extracted. The sequences
were fed into the Jalview Desktop Multiple Alignment Editor
(v.2.8.1) software to draw consensus sequences of the L protein of
each virus. The consensus sequences of the L proteins were aligned
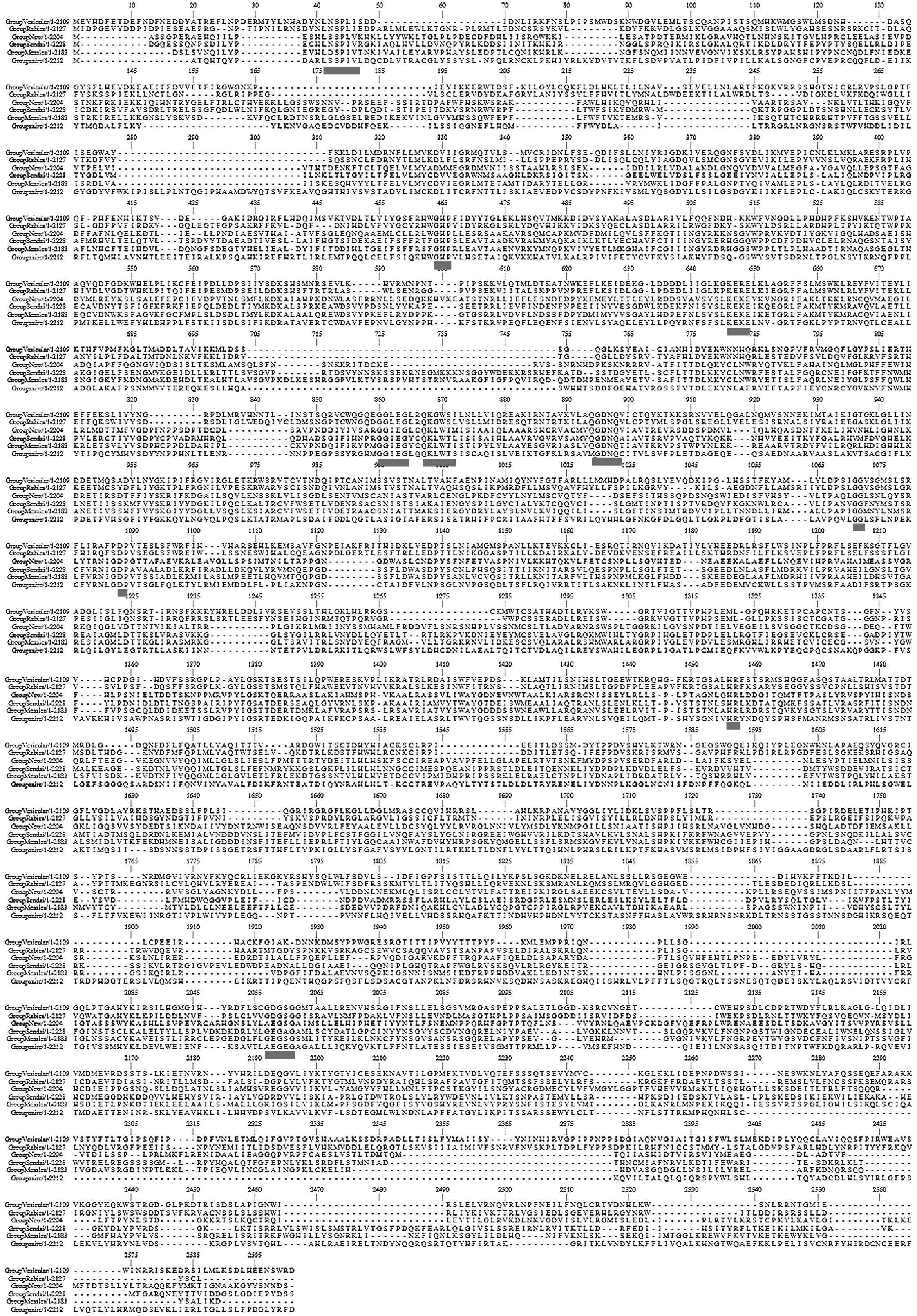
to analyze the sequence variation (Fig. 1). Each row demonstrates a
representative consensus sequence for each virus respectively. The
dashes represent gaps introduced to optimize the alignment. The
six-way alignment established that strong homologies exist over
almost the entire length of the proteins. In addition, the gaps
introduced in order to maximize similarities are typically limited.
The relative frequency of Gly (14.3~) and Trp (3–9%) residues among
the invariant amino acids is markedly higher (2.5–3-fold) compared
with their average abundance in the L proteins (4.5 and 5.5%,
respectively), signifying that these residues are particularly
significant for the L protein structure and/or function, whereas
basic and acidic residues also tend to be more strictly retained
than hydrophobic ones. A similarity profile of the six-way
alignment (Fig. 1) clearly
revealed that conserved residues are not scattered randomly, but
are clustered into six blocks of strong conservation (11) connected by variable regions of low
conservation. The blocks of highest amino acid conservation (II–V)
were located in the central region of the protein (positions
521–1397). In each block there were uninterrupted stretches of
strictly or conservatively maintained amino acids, as highlighted
in the figure. In particular, the highly conserved motifs within
the EBOV share extended similarities with motifs found throughout
all other RNA dependent polymerases. Thus, these may well
constitute the active site for phosphodiester bond formation and/or
template recognition of unsegmented negative-strand RNA virus
polymerases.
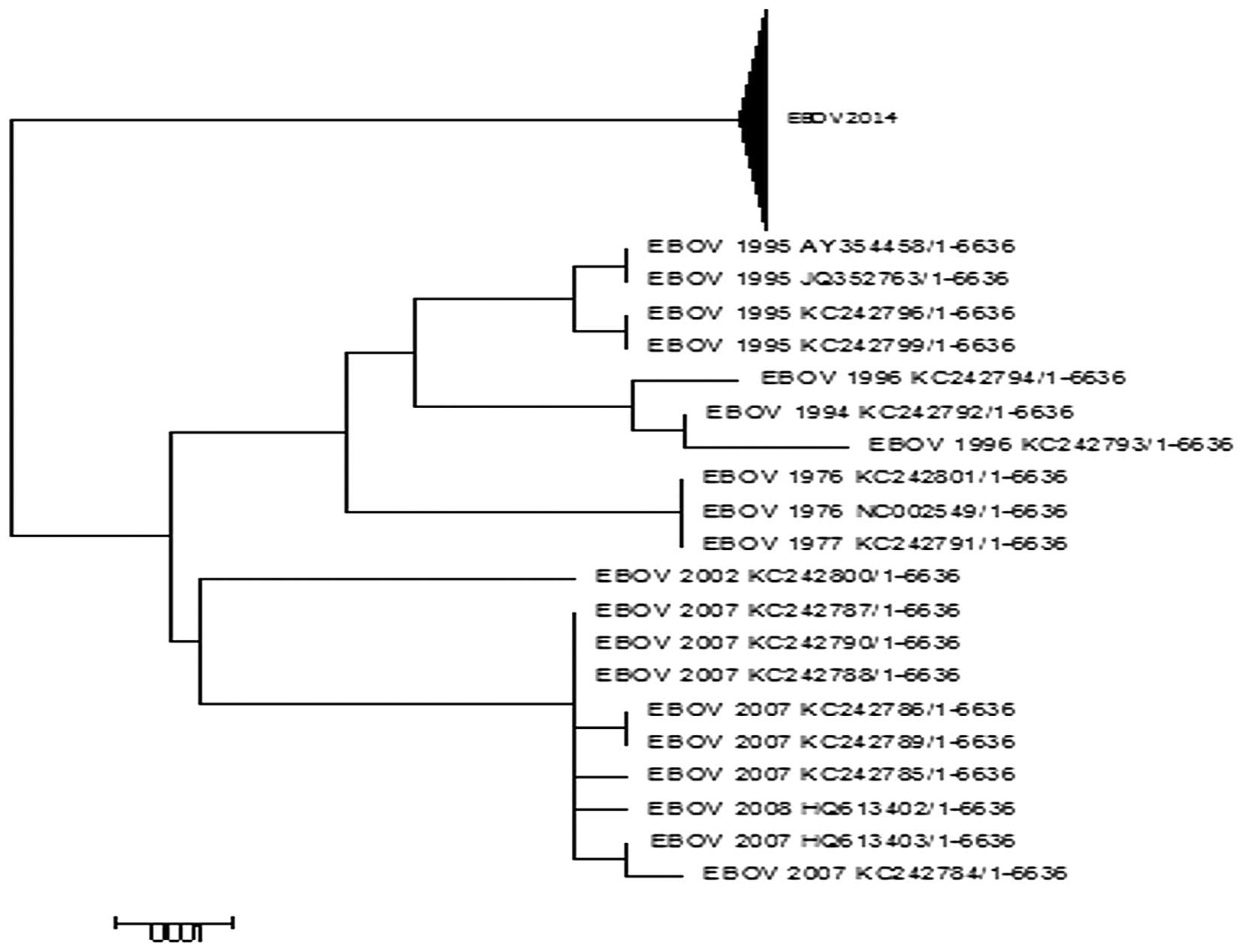
Phylogenetic analysis
The 2014 EBOV isolates were recovered from Sierra
Leone and Guinea, countries with the worst outbreak of Ebola virus
disease (EVD) in history. To determine the evolutionary
relationship between these isolates with those of previous EBOV
outbreaks, a phylogenetic analysis was conducted using the
sequences of the L proteins as presented in Fig. 2. The phylogenetic analysis
indicated that all but the 2014 EBOV sequences were clustered
together. This suggests that previous outbreaks accumulated few
mutations in the L protein and were more closely associated with
each other as the majority of the previous outbreaks exhibit almost
identical sequences (8). A
sequence comparison (not shown) of the L protein from the 2014 EBOV
outbreak and 2007 EBOV outbreak alone demonstrated 26 amino acid
substitutions, confirming the high mutation rate of the virus
(Table I) (8). The branch leading towards the 2014
EBOV outbreak is long compared with others as it is a divergent
lineage that has accumulated multiple mutations.
 | Table IAmino acid differences observed in L
protein between the 2014 and 2007 EBOV outbreak. |
Table I
Amino acid differences observed in L
protein between the 2014 and 2007 EBOV outbreak.
| Amino acid
position | EBOV 2014 | EBOV 2007 |
|---|
| 197 | L | M |
| 202 | T | S |
| 215 | T | A |
| 337 | I | M |
| 342 | P | S |
| 346 | H | Q |
| 692 | N | D |
| 759 | G | D |
| 1405 | Q | R |
| 1607 | H | Q |
| 1610 | F | L |
| 1615 | N | S |
| 1654 | D | Y |
| 1656 | T | A |
| 1658 | N | D |
| 1673 | K | E |
| 1685 | D | G |
| 1690 | S | N |
| 1729 | P | S |
| 1753 | G | D |
| 1774 | Q | K |
| 1826 | N | S |
| 1944 | H | Y |
| 1951 | V | I |
| 2059 | L | F |
| 2085 | V | I |
Amino acid substitution analysis
Table II presents
all 16 amino acid substitutions that, when compared with ancestral
amino acid residues, were fixed within the 2014 EBOV outbreak
according to the SIFT amino acid prediction of substitution. To
assess the effect of a substitution, SIFT assumes that important
positions in a protein sequence have been conserved throughout
evolution and therefore substitutions at these positions may have
an adverse effect on protein function. Thus, by using sequence
homology, SIFT predicts the effects of all possible substitutions
at each position in the protein sequence. All substitutions were
tolerant in the present study; hence, it can be inferred that amino
acid substitutions or insertions/deletions in these regions were
unlikely to affect function.
| Table IIFixed amino acid substitutions
observed in the L proteins of the 2014 EBOV isolates along with
SIFT amino acid predictions. |
Table II
Fixed amino acid substitutions
observed in the L proteins of the 2014 EBOV isolates along with
SIFT amino acid predictions.
| Amino acid
position | Ancestral amino
acid | Substituted amino
acid | SIFT amino acid
predictions |
|---|
| 197 | M | L | Tolerated |
| 215 | A | T | Tolerated |
|
337 | M | I | Tolerated |
|
346 | G | H | Tolerated |
|
692 | A | A | Tolerated |
| 1607 | G | H | Tolerated |
| 1615 | S | A | Tolerated |
| 1654 | T | A | Tolerated |
| 1656 | A | T | Tolerated |
| 1658 | A | A | Tolerated |
| 1673 | G | L | Tolerated |
| 1690 | A | S | Tolerated |
| 1826 | S | A | Tolerated |
| 1944 | T | H | Tolerated |
| 1951 | I | V | Tolerated |
| 2085 | I | V | Tolerated |
Discussion
Sequences of residues ranging in length from four to
seven amino acids, clustered into six domains of strong
conservation and separated by variable regions have been defined
for the families, Rhabdoviridae and Paramyxoviridae
(9,16). These six domains were also observed
to be conserved in previous EBOV L proteins compared with
paramyxoviruses (17). Notably,
these conserved regions, are not retained in the current 2014 EBOV
isolates, except in the mutated regions. The domains are key for
crucial activities conducted by the L proteins.
Three polymerase motifs, motif A (aa 646–665), motif
B (aa 892–898) and motif C (aa 2,034–2,063) identified previously
in other NNS viruses, including EBOV L proteins were also observed
in the 2014 EBOV isolates, but with a number of mutations (17). The invariant tripeptide GHP motif
(aa 464–466), recognized previously as a turn structure exposing
the histidine residue, was identified to be conserved amongst all
the viruses (9). This motif may
potentially serve as a target for future antiviral therapeutic
agents, and site-specific mutagenesis studies may further elucidate
its function.
Another pentapeptide, QGDNQ (aa 894–898), part of
motif B is conserved in all NNS viruses, excluding EBOV. This motif
is crucial for RNA polymerase function and phosphodiester bond
formation and/or template recognition. In EBOV, the first glycine
residue is substituted for a methionine residue (18). This substitution is not specific to
the 2014 EBOV isolates as it has been demonstrated across all other
Ebolavirus species. This substitution was not key in the
2014 outbreak, however, its evolutionary role in the transcription
and the replication process requires further elucidation.
Furthermore, the GDN part of the sequence corresponds to the
strictly conserved residues, GDD in other polymerases (18). It is therefore an ideal target for
antiviral therapeutic agents.
The KEKE tetrapeptide (aa 646–649), part of motif A
that is responsible for the positioning and binding of the RNA
template, was observed to be conserved in all viruses, except
vesicular stomatitis and rabies viruses. In Rhabdoviridae,
the lysine residue was substituted for glutamic acid, consistent
with previous studies indicating that filoviruses are more closely
associated with the paramyxoviruses than to rhabdoviruses (9,16,17).
Another pentapeptide motif GG(I/L)EG (aa 860–865)
was also conserved in the alignment. EBOV contained an isoleucine
rather than a leucine, further suggesting a closer association with
the family Paramyxoviridae (9,19).
Two invariant dipeptides GG (aa 1,071–1,072) and DP (aa
1,088–1,089) were also conserved as reported previously (9).
The HR motif (aa 1,456–1,457), previously indicated
to be important for the PRNTase activity of the L protein at the
enzyme-pRNA intermediate formation step, was also observed to be
conserved across all the virus sequences (16,20).
Furthermore, the GXGXG pentapeptide (aa 2,057–2,061)
part of motif C was demonstrated to be conserved in all members of
the Rhabdoviridae family and two members of the
Paramyxoviridae family (Sendai and measles) with slight
inter-species variations in the X amino acids. This pentapeptide is
hypothesized to be involved in mRNA capping via a
2′-O-ribose-methyltransferase action (21).
To the best of our knowledge, this is the first
study to demonstrate that the L(N/S)SP(L/I)V motif (aa 41–46) is
conserved across the viruses. However, the function of this motif
remains to be elucidated. A QK(G/L)W(S/T) motif (aa 867–871) was
also observed to be conserved, the exact function of this motif
remains to be determined. In addition, the pattern of conservation
itself is notable, beyond the 1,540th amino acid, the conservation
decreases to the 2,024th amino acid, following which there is
another stretch of partially conserved residues to the 2,241th
amino acid. Beyond this, there is a highly non-conserved region
filled with large gaps. This demonstrates that the conserved
residues predominantly lie in the mid-region of the protein,
consistent with the results of a previous study (9).
Furthermore, the phylogenetic tree of the EBOV
sequences is similar to trees drawn previously on the basis of GP
and NP sequences, demonstrating the same pattern of clustering
(22). Though the previously drawn
trees did not take into account the latest 2014 sequences, the
pattern of clustering remains the same, predominantly due to
minimal mutations acquired by the viruses in previous outbreaks.
The same findings were concluded in a recent study using whole
genome analysis (8).
As presented in Table
II, it is notable that all the substitutions are tolerated and
pathogenic. Of the first four mutations, two are located within
domain I that spans aa 313–515 of the EBOV L protein in the
alignment in the present study (aa 222–426 without the gaps for
improved alignment). As demonstrated previously, this domain
contains stretches of highly conserved residues and motifs,
including GHP (9). In a previous
study, it was demonstrated that the binding domain for the
polymerase cofactor VP35, is located within the first 370 amino
acids of the L protein towards the N-terminal, while the core
binding domain is located between aa 280–370. This binding domain
overlaps with part of domain I (23). Previous site-directed mutagenesis
studies into the Sendai virus L protein have demonstrated that
mutation in this domain leads to a defective replication and
transcription mechanism and improper P protein binding (24). The first four mutations presented
in Table II are located within
this binding domain, with the M337I and Q346H mutations
specifically in the core binding region. However, such mutations
appear to be advantageous to the 2014 EBOV and may have enhanced
genome replication efficiency. The fourth substitution is isolated,
it does not lie within any known functional motif, thus, its role,
if any, in the virulence of the virus requires elucidation by
site-directed mutagenesis studies.
The next five mutations are also notable. All the
mutations lie close to each other within a span of 90 amino acids.
These mutations occur outside the conserved domains, within a
region of high variability also observed within previous filovirus
L protein sequences by comparative analysis (17). Further research regarding this
region is required prior to drawing any conclusions about these
mutations.
Notably, the S1826D mutation lies within the
conserved motif C identified earlier (17). The mutation is close to the GXGXG
pentapeptide. Earlier mutation studies within this region in the
Sendai virus have led to reduced replication and transcription
activities, however, it did not completely block the polymerase
function (25). This mutation is
tolerated and appears to be a gain of function mutation.
The final three mutations lie within a highly
unconserved region, for which no functional domains or motifs have
been defined or hypothesized. However, the I1951V substitution is
notable. The amino acid isoleucine is conserved at position 1,951
in all other Ebolavirus species. It is only substituted to
valine in the 2014 EBOV isolates. The impact of this substitution
remains to be elucidated and requires further research.
In conclusion, the present study provided an
overview of the L protein amino acid sequences of the 2014 EBOV
isolates. The sequence and mutation analysis of L polymerase will
aid in advancing towards confinement of future outbreaks. The
current study may guide future site-directed mutagenesis studies,
and design of site-specific direct targeting antiviral therapeutic
agents. It may also help with the development of future diagnostic
studies.
References
|
1
|
Sanchez A, Geisbert TW and Feldmann H:
Filoviridae: Marburg and ebola viruses. Fields Virology. Knipe DM
and Howley PM: Lippincott Williams and Wilkins; Philadelphia, PA:
pp. 1409–1448. 2007
|
|
2
|
Kuhn JH, Becker S, Ebihara H, Geisbert TW,
Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol
ST, et al: Proposal for a revised taxonomy of the family
Filoviridae: Classification, names of taxa and viruses and virus
abbreviations. Arch Virol. 155:2083–2103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanchez A, Kiley MP, Holloway BP and
Auperin DD: Sequence analysis of the Ebola virus genome:
Organization, genetic elements, and comparison with the genome of
Marburg virus. Virus Res. 29:215–240. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boehmann Y, Enterlein S, Randolf A and
Mühlberger E: A reconstituted replication and transcription system
for Ebola virus Reston and comparison with Ebola virus Zaire.
Virology. 332:406–417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Waheed Y: Ebola in West Africa: An
international medical emergency. Asian Pac J Trop Biomed.
4:673–674. 2014. View Article : Google Scholar
|
|
6
|
Waheed Y, Bhatti A and Ashraf M: RNA
dependent RNA polymerase of HCV: A potential target for the
development of antiviral drugs. Infect Genet Evol. 14:247–257.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Waheed Y, Saeed U, Anjum S, Afzal MS and
Ashraf M: Development of global consensus sequence and analysis of
highly conserved domains of the HCV NS5B prote in. Hepat Mon.
12:e61422012.PubMed/NCBI
|
|
8
|
Gire SK, Goba A, Andersen KG, Sealfon RS,
Park DJ, Kanneh L, Jalloh S, Momoh M, Fullah M, Dudas G, et al:
Genomic surveillance elucidates Ebola virus origin and transmission
during the 2014 outbreak. Science. 345:1369–1372. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poch O, Blumberg BM, Bougueleret L and
Tordo N: Sequence comparison of five polymerases (L proteins) of
unsegmented negative-strand RNA viruses: Theoretical assignment of
functional domains. J Gen Virol. 71:1153–1162. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clamp M, Cuff J, Searle SM and Barton GJ:
The Jalview Java alignment editor. Bioinformatics. 20:426–427.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Katoh K and Standley DM: Improvements in
performance and usability. Mol Biol Evol. 30:772–780. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nuin PA, Wang Z and Tillier ER: The
accuracy of several multiple sequence alignment programs for
proteins. BMC bioinformatics. 7:4712006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tamura K, Stecher G, Peterson D, Filipski
A and Kumar S: MEGA6: Molecular evolutionary genetics analysis
version 6.0. Mol Biol Evol. 30:2725–2729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saitou N and Nei M: A new method for
reconstructing phylogenetic trees. Mol Biol Evol. 4:406–425.
1987.PubMed/NCBI
|
|
15
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marston DA, McElhinney LM, Johnson N,
Müller T, Conzelmann KK, Tordo N and Fooks AR: Comparative analysis
of the full genome sequence of European bat lyssavirus type 1 and
type 2 with other lyssaviruses and evidence for a conserved
transcription termination and poly-adenylation motif in the G-L 3′
non-translated region. J Gen Virol. 88:1302–1314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Volchkov VE, Volchkova VA, Chepurnov AA,
Blinov VM, Dolnik O, Netesov SV and Feldmann H: Characterization of
the L gene and 5′ trailer region of Ebola virus. J Gen Virol.
80:355–362. 1999. View Article : Google Scholar
|
|
18
|
Schnell MJ and Conzelmann KK: Polymerase
activity of in vitro mutated rabies virus L protein. Virology.
214:522–530. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blumberg BM, Crowley JC, Silverman JI,
Menonna J, Cook SD and Dowling PC: Measles virus L protein
evidences elements of ancestral RNA polymerase. Virology.
164:487–497. 1998. View Article : Google Scholar
|
|
20
|
Ogino T and Banerjee AK: The HR motif in
the RNA-dependent RNA polymerase L protein of Chandipura virus is
required for unconventional mRNA-capping activity. J Gen Virol.
91:1311–1314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferron F, Longhi S, Henrissat B and Canard
B: Viral RNA-polymerases-a predicted 2′-O-ribose methyltransferase
domain shared by all Mononegavirales. Trends Biochem Sci.
27:222–224. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grard G, Biek R, Tamfum JJ, Fair J, Wolfe
N, Formenty P, Paweska J and Leroy E: Emergence of divergent Zaire
Ebola virus strains in democratic republic of the Congo in 2007 and
2008. J Infect Dis. 204(Suppl 3): S776–S784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Trunschke M, Conrad D, Enterlein S,
Olejnik J, Brauburger K and Mühlberger E: The L-VP35 and L-L
interaction domains reside in the amino terminus of the Ebola virus
L protein and are potential targets for antivirals. Virology.
441:135–145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chandrika R, Horikami SM, Smallwood S and
Moyer SA: Mutations in conserved domain I of the Sendai virus L
polymerase protein uncouple transcription and replication.
Virology. 213:352–363. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feller JA, Smallwood S, Horikami SM and
Moyer SA: Mutations in conserved domains IV and VI of the large (L)
subunit of the Sendai virus RNA polymerase give a spectrum of
defective RNA synthesis phenotypes. Virology. 269:426–439. 2000.
View Article : Google Scholar
|