Introduction
β-arrestins, including β-arrestin1 and β-arrestin2,
two ubiquitously expressed members of the arrestin family in
various types of tissues, localize in the cytoplasm and plasma
membrane, and modulate the desensitization and trafficking of seven
transmembrane receptors (1,2).
Recent evidence revealed that β-arrestins also served as
multi-functional adaptors that contribute to regulating multiple
signaling molecules. For example, β-arrestins associate with c-Src
and mitogen-activated protein kinase (MAPK) family members,
including extracellular signal-regulated kinase (ERK), p38 and
c-Jun N-terminal kinase (3,4). The
binding of β-arrestins with these signaling molecules modulates
phosphorylation, ubiquitination and/or subcellular distribution of
their binding partners (5,6). The biological and clinical behaviors
of numerous types of tumor are largely determined by multiple
molecular signaling pathways. In addition, it was recently
established that β-arrestins were involved in signaling events
responsible for tumor viability and metastasis (7). It has also been reported that
β-arrestins are involved in the anti-apoptosis pathway by
associating with kinases such as Akt and ERK and altering their
activities (4,8,9).
However, the role of β-arrestins in tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL)-induced apoptosis remains
unclear.
TRAIL induces apoptosis in a variety of cancer cell
lines by interacting with their death receptors (DRs) while causing
minimal or no toxicity to normal cells, establishing it as an
attractive agent for cancer therapy (10). However, numerous types of cancer
cell have been shown to be resistant to TRAIL-induced apoptosis,
including hepatic carcinoma and human breast cancer cells (11,12).
TRAIL signaling involves the activation of effector caspases and
initiation of apoptosis, and thus the activation of pro-survival
signaling pathways involving nuclear factor-κB, Akt and MAPKs may
have contributed to the development of TRAIL resistance in tumor
cells (13,14).
In the present study, the role and molecular
mechanisms underlying the modulation of TRAIL-mediated apoptosis by
β-arrestin2 in HepG2 cells was investigated.
Materials and methods
Antibodies and reagents
Monoclonal rabbit antibodies against poly ADP ribose
polymerase (PARP; 9532s, 1:1,000), pro-caspase-3 (9665s; 1:1,000),
cleaved caspase-3 (9664s; 1:500), Src (2109s; 1:1,000),
phosphorylated (p)-Src (Tyr416; 6943s; 1:1,000), ERK (9102s;
1:1,000), p-ERK (Thr202/Tyr204; 4376s; 1:1,000), DR5 (8074s;
1:1,000), β-arrestin2 (3857s; 1:1000) and β-actin (4970s; 1:1,000)
were obtained from Cell Signaling Technology, Inc. (Danvers, MA,
USA). In addition, mouse monoclonal antibody against DR4 (sc-8411;
1:500) and monoclonal rabbit antibody against β-arrestin1
(sc-53780; 1:500) were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Polyclonal rabbit anti-GAPDH antibody
(AP0063; 1:1,000) was purchased from Bioworld Technology, Inc. (St.
Louis Park, MN, USA). Anti-green fluorescent protein (GFP) antibody
(11814460001; 1:1,000) was obtained from Roche Diagnostics
(Indianapolis, IN, USA). Recombinant human TRAIL was produced by
PeproTech, Inc. (Rocky Hill, NJ, USA), and PP2, PP3 and U0126 were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
DNA constructs
pcDNA3.0-GFP-arrestin1/2 and pBS-U6-β-arrestin1/2
were provided by Dr. Gang Pei (Chinese Academy of Sciences,
Shanghai, China). All expression vectors were sequenced and
purified using the EndoFree Plasmid Preparation kit (Qiagen China
Co., Ltd., Shanghai, China).
Cell culture and transfection
Hepatic carcinoma (HepG2) cells were obtained from
the American Type Culture Collection (Manassas, VA, USA). The cells
were cultured in Invitrogen Dulbecco's modified Eagles's medium
(DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% (v/v) fetal bovine serum (Hyclone; GE
Healthcare Life Sciences, Logan, UT, USA) and 1% antibiotics (100
U/ml penicillin and 100 µg/ml streptomycin; Hyclone) at 37°C
and 5% CO2 in a fully humidified incubator. Transient
transfection was performed using the Fugene® HP
Transfection Reagent (Roche) according to the manufacturer's
instructions. The total quantity of DNA was normalized to empty
control plasmids.
Co-immunoprecipitation and immunoblotting
analysis
Cells were rinsed twice with ice-cold
phosphate-buffered saline (PBS) and lysed on ice in a lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China) containing 20
mM Tris (pH 7.5), 2 mM EDTA, 135 mM NaCl, 2 mM dithiothreitol, 25
mM β-glycerophosphate, 2 mM sodium pyrophosphate, 10% glycerol, 1
mM Na3VO4, 1% Triton X-100, 10 mM NaF, 10
µg/ml leupeptin, 10 µg/ml aprotinin, and 1 mM
phenylmethanesulfonyl fluoride supplemented with 0.01% complete
protease inhibitor cocktail (Roche) for 30 min. Lysates were
centrifuged at 12,500 × g for 15 min at 4°C. Equal quantities of
proteins were immunoprecipitated overnight with β-arrestin2
monoclonal antibody at 4°C. The precleared Protein A/G PLUS-Agarose
beads (20 µl; Santa Cruz Biotechnology, Inc.) were
co-incubated with immunocomplexes for an additional 2 h, then
washed four times with the cold lysis buffer. The
immunoprecipitates were electrophoresed on 12% SDS-PAGE (135 V; 2
h) and transferred onto nitrocellulose membranes (GE Healthcare
Life Sciences). Immunoblotting was subsequently performed. The
LI-COR Odyssey Infrared Imaging system and IRDye® 800
flurophore-conjugated antibody (LI-COR Biosciences, Lincoln, NE,
USA) were used to visualize the antibody-antigen complexes. Band
intensity quantification was directly performed on the blot using
LI-COR Odyssey Analysis software 1.2. Aliquots of whole cell
lysates were subjected to immunoblotting analysis to confirm
appropriate protein expression.
RNA interference
HepG2 cells were transfected with short hairpin
(sh)RNA constructs against β-arrestin1 or β-arrestin2
(pBS-U6-β-arrestin1 or pBS-U6-β-arrestin2), or a negative control
vector using Fugene® HP Transfection Reagent according
to the manufacturer's instructions. Interference efficiency was
confirmed by immunoblot analysis following a 72-h transfection
using β-arrestin1 or β-arrestin2 antibodies.
Flow cytometry
Cell apoptosis was determined using the Annexin
V/propidium iodide (PI) double staining assay (Kaiji Materials Co.,
Ltd., Nanjing, China). Briefly, following transfection with shRNA
or a negative control vector, the cells were washed with PBS,
harvested by trypsinization, precipitated by centrifugation (2,000
× g for 5 min), rinsed with PBS again, resuspended with 500
µl binding buffer, and stained with Annexin V and PI.
Apoptotic cells were detected directly using the Guava Easy Cyte™
system, and the data were analyzed using Guava TUNEL Software
(Guava Technologies, Inc., Hayward, CA, USA).
Cell viability assay
Cell viability was determined using a Cell Counting
Kit-8 (CCK-8; Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Briefly, HepG2 cells were seeded at a
density of 1×104 into 96-well plates 24 h before
treatment. Cells were pretreated with PP2 (5 µM), PP3 (5
µM) or 0.1% dimethyl sulfoxide (DMSO) for 2 h, then exposed
to 200 ng/ml TRAIL for 24 h, followed by incubation with 10
µl CCK-8 working solution at 37°C for 2 h. The absorbance of
each well at a wavelength of 450 nm was measured using a Synergy2
multi-mode microplate reader (Bio-Tek, Inc.). Three experiments
were performed for each of the different treatments.
Immunofluorescence microscopy and
4′,6-diamidino-2-phenylindole (DAPI) staining
HepG2 cells were preincubated with PP2 (5
µM), PP3 (5 µM) or 0.1% DMSO for 2 h, then stimulated
with TRAIL; the cells were subsequently fixed with 4%
paraformaldehyde (JRDun Biotechnology, Co., Ltd., Shanghai, China)
and permeabilized with 0.2% Triton X-100. Following incubation with
1 µg/ml DAPI for 5 min at room temperature, the cells were
washed with PBS again. The immunofluorescence images were captured
using a fluorescence microscope (Leica TCS SP8; Leica Microsystems
GmbH, Wetzlar, Germany). The number of cells exhibiting nuclear
condensation and fragmentation were counted in randomly selected
fields, and the ratio of these cells to the total number of cells
was calculated.
Statistical analysis
Data are presented as the mean ± standard deviation.
One-way analysis of variance was used to determine significant
differences between two groups. P<0.05 was considered to
indicate a statistically significant difference. Statistical
calculations were performed using SPSS 17.0 (SPSS, Inc., Chicago,
IL, USA).
Results
β-arrestin2 inhibited TRAIL-induced
apoptosis in HepG2 cells
A previous study demonstrated that β-arrestins
prevented cell apoptosis via the ERK, p38 and Akt signaling
pathways (4), which are involved
in cancer progression (7,15). Furthermore, TRAIL has previously
demonstrated marked anticancer effects in numerous tumor types, but
not in normal cells (10).
Conversely, previous studies reported that certain human cancers
were resistant to TRAIL (11,12,16,17),
although the underlying molecular mechanisms were unclear. The
present study hypothesized that β-arrestins are involved in
TRAIL-resistance by combining with pro-survival signaling molecules
and regulating their activity.
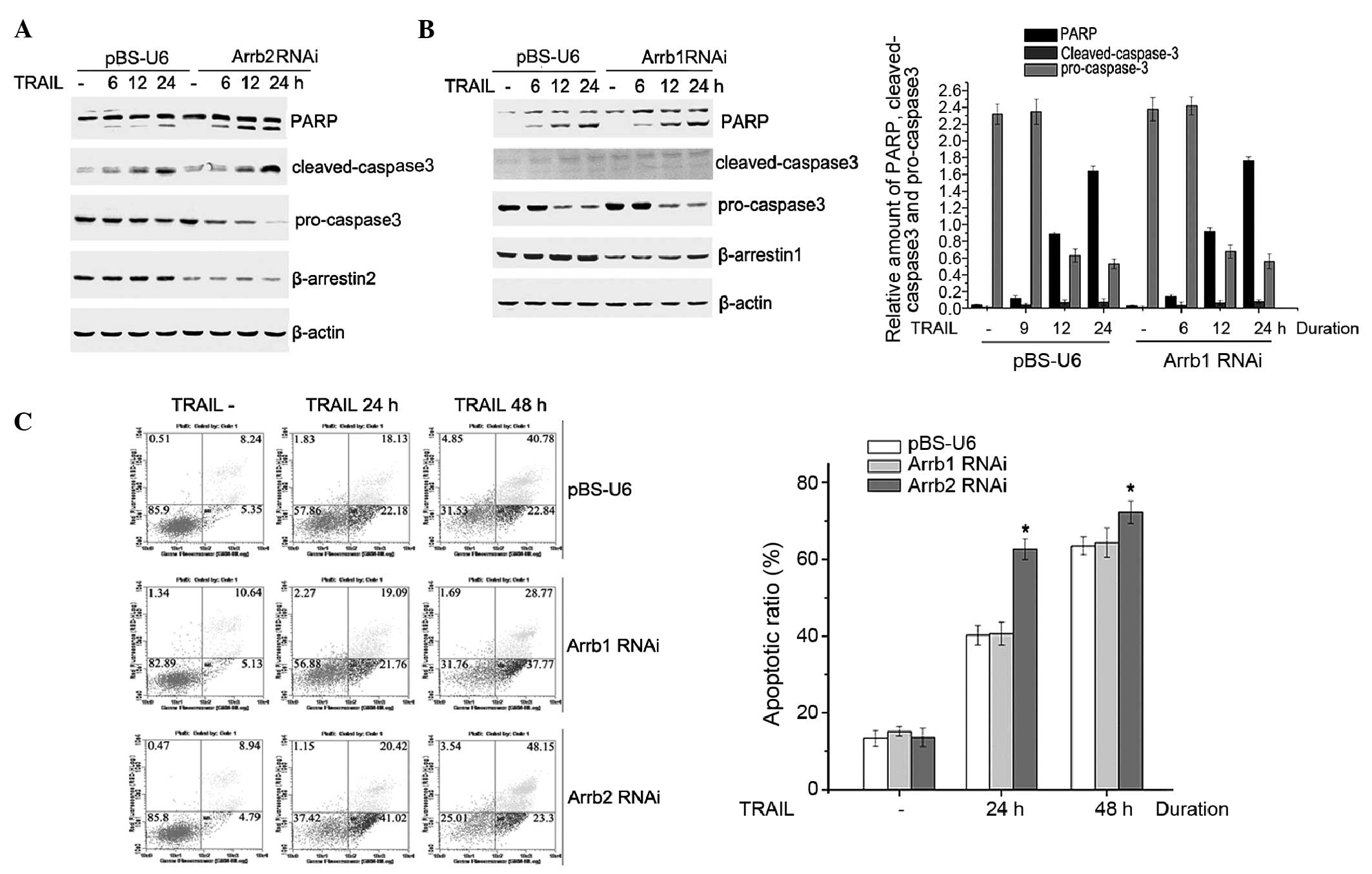
In the present study, the role of β-arrestins in the
TRAIL-induced apoptosis of HepG2 cells was evaluated. HepG2 cells
were transiently transfected with pBS-U6-β-arrestin1 and 2 or
control plasmid. Following a 72-h transfection, HepG2 cells were
treated with 200 ng/ml TRAIL for 6, 12 or 24 h, and cell lysates
were analyzed by western blotting. Following TRAIL stimulation,
cleaved PARP and cleaved caspase-3 expression levels were
increased, whereas the protein expression level of pro caspase-3
was decreased, in a time-dependent manner. In addition, as compared
with the control vector, the cleaving of PARP and caspase-3 was
markedly enhanced, whereas the pro-caspase-3 expression level was
significantly reduced, in pBS-U6-β-arrestin2 transfected cells.
However, there was no significant difference identified between the
level of apoptosis-associated proteins in the pBS-U6-β-arrestin1
and the control vector transfected cells (Fig. 1A and B). These results suggest that
β-arrestin2, but not β-arrestin1, is involved in TRAIL-induced
HepG2 cell apoptosis.
To further investigate the role of β-arrestins in
modulating TRAIL-induced HepG2 cell apoptosis, the apoptotic ratio
was detected using the AnnexinV/PI assay. The TRAIL-induced
apoptotic ratio in β-arrestin2 RNA interference (RNAi) cells was
62.12% at 24 h, whereas the apoptotic ratio was 40.24% in the
control plasmid transfected cells; however, no significant
difference was observed between the apoptotic ratio of β-arrestin1
RNAi cells and control plasmid transfected cells (Fig. 1C).
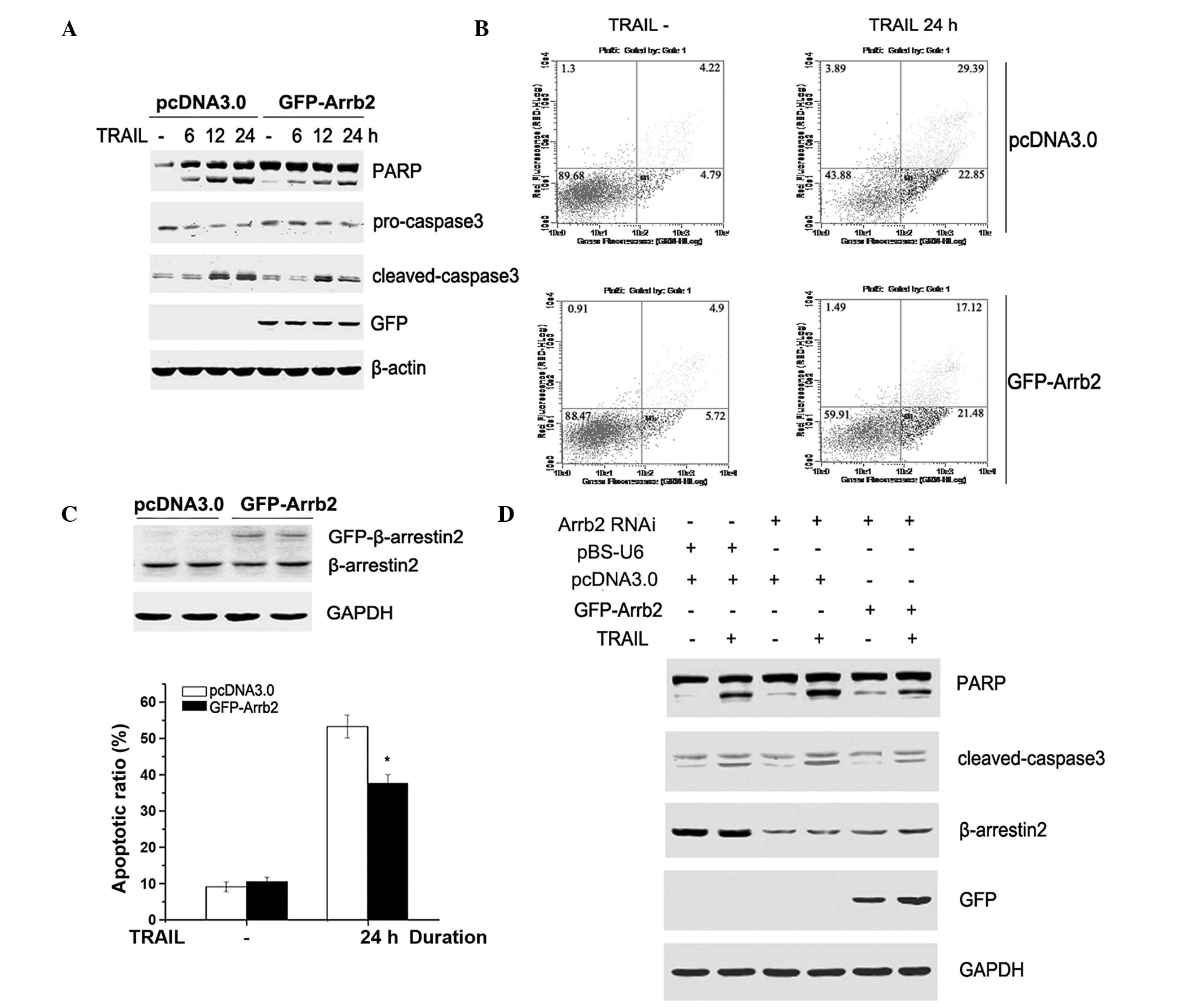
To further clarify the role of β-arrestin2 in
TRAIL-induced apoptosis, GFP-tagged β-arrestin2 (GFP-Arrb2) was
overexpressed in HepG2 cells. Following a 48-h transfection, the
cells were stimulated with TRAIL for 6, 12 or 24 h. As presented in
Fig. 2A–C, caspase-3 and PARP
cleavage, and the apoptotic ratio induced by TRAIL, were attenuated
by overexpression of β-arrestin2 in HepG2 cells. To further
demonstrate the role of β-arrestin2 in TRAIL-induced apoptosis,
β-arrestin2 shRNA was transiently transfected into HepG2 cells for
72 h, followed by transfection with GFP-Arrb2 or empty vectors for
48 h. As shown in Fig. 2D, the
levels of cleaved caspase-3 and PARP in β-arrestin2 RNAi cells were
reversed following overexpression with β-arrestin2 plasmid. These
results demonstrate that β-arrestin2 exerts an anti-apoptotic role
in TRAIL-induced HepG2 cell apoptosis.
β-arrestin2 mediated activation of the
Src-ERK signaling pathway in response to TRAIL
It has been reported that TRAIL activates apoptosis
signaling pathways, as well as survival signaling pathways, which
are involved in the development of TRAIL resistance. The Src-ERK
signaling pathway was demonstrated to counteract TRAIL toxicity in
tumor cells (18). Therefore, the
activation of Src-ERK signaling upon TRAIL stimulation was
investigated in the present study by western blotting. HepG2 cells
were treated with 200 ng/ml TRAIL for 0.25, 0.5, 1, 2 or 4 h, and
the levels of p-Src, p-ERK, total Src and ERK were detected by
immunoblotting. As presented in Fig.
3A, Src and ERK were activated as a result of TRAIL
stimulation. To investigate the potential role of β-arrestin2 in
TRAIL-induced activation of Src and ERK, β-arrestin2 was knocked
down in HepG2 cells via transfection of pBS-U6-β-arrestin2. Upon
TRAIL stimulation, the downregulated expression of β-arrestin2
markedly reduced the phosphorylation of Src and ERK, although did
not affect the total Src and ERK expression (Fig. 3B). To further evaluate the effect
of β-arrestin2 on the TRAIL-induced Src-ERK signaling pathway,
GFP-Arrb2 plasmids and control vectors were transfected into HepG2
cells. Compared with the empty vector group, β-arrestin2
overexpression markedly facilitated the TRAIL-induced activation of
Src and ERK (Fig. 3C). These
results indicate that β-arrestin2 mediated activation of the
Src-ERK signaling pathway upon TRAIL stimulation.
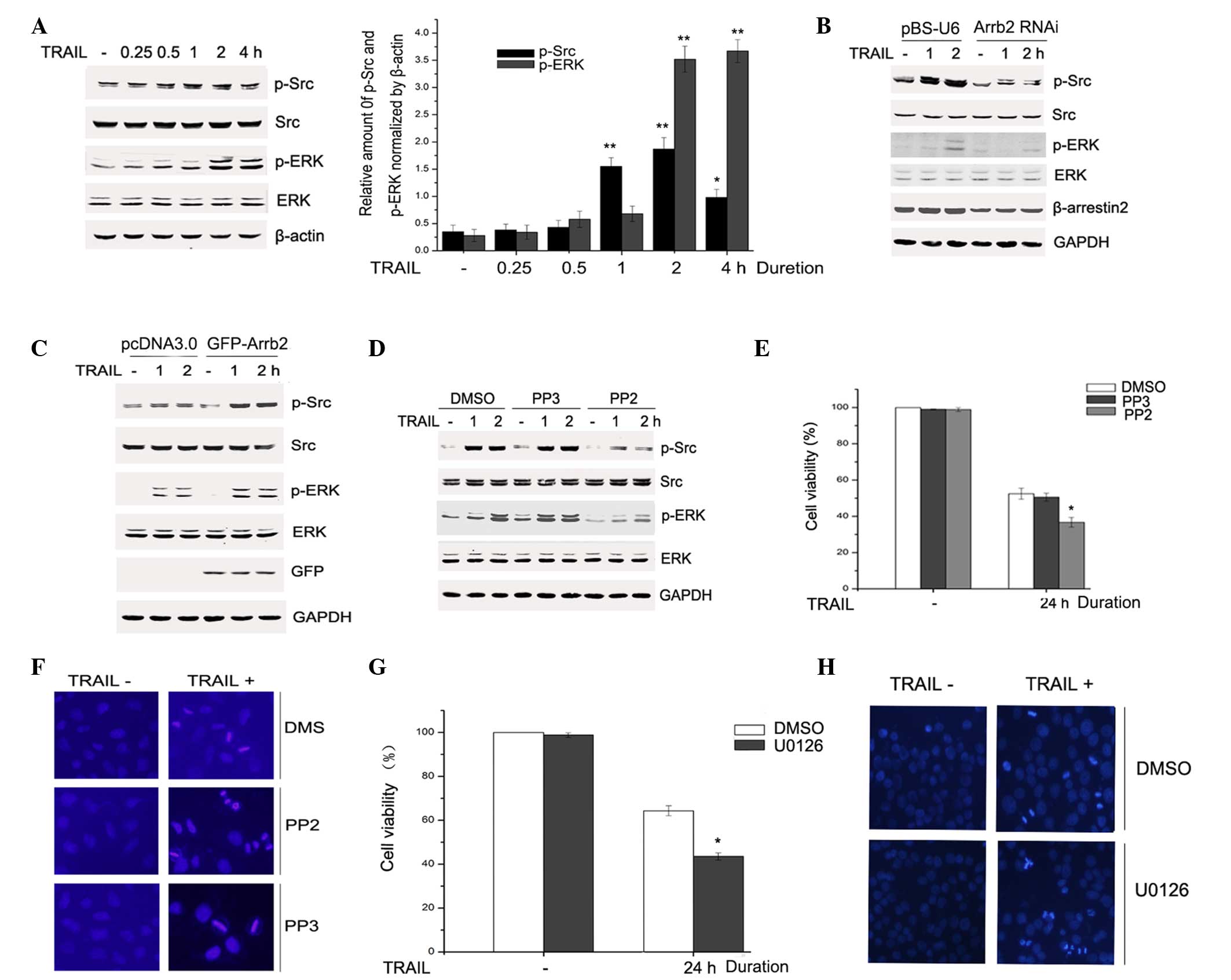 | Figure 3β-arrestin2 mediated activation of the
TRAIL-induced Src-ERK signaling pathway. (A) HepG2 cells were
treated with TRAIL for 0.25, 0.5, 1, 2 or 4 h and the cell lysates
were subjected to immunoblotting with the indicated antibodies. The
band intensities were quantified by densitometry using LI-COR
Odyssey Analysis software 1.2. *P<0.05,
**P<0.01 vs. the TRAIL− group. HepG2 cells
were transfected with (B) pBS-U6-β-arrestin2 or control plasmid for
72 h or (C) pcDNA3.0 and GFP-Arrb2 plasmids for 48 h. (B and C)
Cells were stimulated with TRAIL for 1 or 2 h and the levels of p-,
total Src and ERK were detected by western blotting. Equal protein
loading was confirmed by GAPDH. (D) Cells were pretreated with 0.1%
DMSO or 5 µM PP2 or 5 µM PP3 for 2 h, then treated
with 200 ng/ml TRAIL. Cell lysates were prepared and subjected to
western blotting using the indicated antibodies. (E) Cell viability
was detected using a CCK-8 and experiments were independently
repeated three times. *P<0.05 vs. DMSO. (F) Cells
were stained with DAPI and the nuclear morphology was observed by
immunofluorescent microscopy (magnification, ×200). (G) Cells were
pretreated with 0.1% DMSO or 20 µM U0126 for 2 h, then
exposed to 200 ng/ml TRAIL for 24 h. Cell viability was detected by
CCK-8 and experiments were independently repeated for three times.
*P<0.05 vs. DMSO. (H) Cells were stained with DAPI
and the nuclear morphology was observed by immunofluorescent
microscopy (magnification, ×200). TRAIL, tumor necrosis
factor-related apoptosis-inducing ligand; p, phosphorylated; RNAi,
RNA interference; Arrb, β-arrestin; GFP, green fluorescent protein;
ERK, extracellular signal-regulated kinase; DMSO, dimethyl
sulfoxide; CCK-8, Cell Counting Kit-8; DAPI,
4′,6-diamidino-2-phenylindole. |
Src-ERK signaling pathway is involved in
TRAIL-induced HepG2 apoptosis
It has been recognized that Src may mediate ERK
activation and is an upstream kinase in various stimulus-induced
signaling cascades (19,20). Therefore, PP2 was used to evaluate
the effect of Src on the activation of ERK upon TRAIL stimulation.
Blocking Src activity using PP2 prevented TRAIL-induced
phosphorylation of ERK (Fig. 3D).
These results verified that, in HepG2 cells, Src serves as an
upstream kinase in TRAIL-induced ERK pro-survival molecule
activation. In order to further detect the role of the Src-ERK
signaling pathway in TRAIL-induced HepG2 cell apoptosis, cell
viability was examined with the CCK-8 and the nuclear morphology
was detected by DAPI staining. HepG2 cells were pretreated with PP2
(5 µM) or PP3 (5 µM) for 2 h, then stimulated with
TRAIL (200 ng/ml) for 24 h, and cell viability was detected.
Fig. 3E demonstrates that PP2
inhibition of the activation of Src-ERK signaling reduced HepG2
cell viability markedly upon TRAIL stimulation. The DAPI-stained
cells were detected by immunofluorescent microscopy. Furthermore,
PP2 pretreated cells exhibited typical nuclear morphological
changes of apoptotic cells, such as nuclear condensation and
nuclear fragmentation, as compared with cells that were pretreated
with PP3 (Fig. 3F).
U0126, an ERK inhibitor, was used to evaluate the
effect of ERK in TRAIL-induced apoptosis. HepG2 cells were
pretreated with 20 µM U0126 or 0.1% DMSO for 2 h, and
subsequently exposed to TRAIL (200 ng/ml) for 24 h. Cell viability
was determined by the CCK-8 assay and cell nuclear morphology was
observed by DAPI staining. Fig. 3G
indicated that suppression of ERK activation also reduced cell
survival, which was mediated by TRAIL. In Fig. 3H, U0126 pretreated cells exhibited
marked nuclear condensation and nuclear fragmentation, as compared
with cells that were pretreated with DMSO. These results indicate
that Src-ERK signaling pathway activation is involved in
TRAIL-induced HepG2 cell apoptosis.
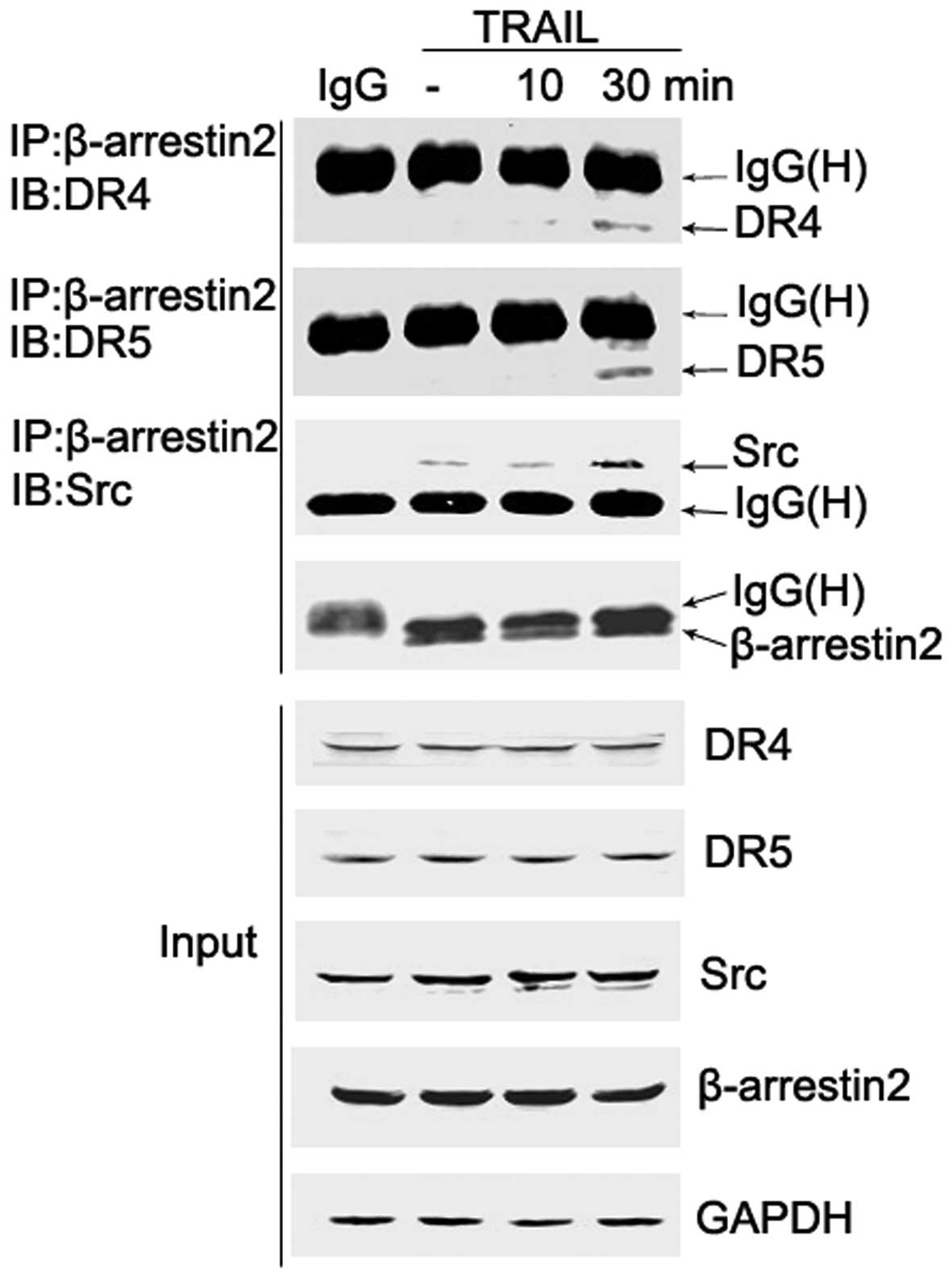
β-arrestin2, and Src and DR formed
ternary complexes upon TRAIL stimulation in HepG2 cells
It has been reported that β-arrestins recruit
signaling proteins, such as c-Src to ligand-bound G protein-coupled
receptors (GPCRs) (21). However,
the role of β-arrestins in DR signaling pathways remains unclear.
To evaluate whether β-arrestin2 recruits Src to the
TRAIL-associated DR, co-immunoprecipitation experiments were
conducted in HepG2 cells. Cells were treated with TRAIL (200 ng/ml)
for 0, 10 and 30 min, and β-arrestin2/Src/DR complexes were
immunoprecipitated with the β-arrestin2 antibody. In addition, a
portion of the cell lysate was analyzed prior to
immunoprecipitation and served as a control (input).
Co-immunoprecipitation analyses revealed that β-arrestin2
physically combined with Src, and TRAIL stimulation resulted in an
increased quantity of the β-arrestin2-Src complex. Whether DRs were
associated with the complex was subsequently investigated by
detecting the presence of DRs in β-arrestin2 immunoprecipitates.
Notably, the association of β-arrestin2 and DR4/5 following TRAIL
stimulation was also observed (Fig.
4). These results indicate that DR/β-arrestin2/Src ternary
complexes are formed following TRAIL treatment. Employing other
techniques, such as protein purification/co-elution experiments may
validate this finding, and fluorescence resonance energy transfer
(FRET)-based technology may aid with evaluating the mechanism of
β-arrestin2-mediated Src-ERK signaling pathway activation. Further
investigations are required to elucidate whether the mechanism is
cell-type specific or universal, and establish the role of the
ternary complex in TRAIL-induced HepG2 cell apoptosis.
Discussion
Recent studies have demonstrated that β-arrestins
contribute to anti-apoptotic effects in apoptosis that is induced
by a variety of stimuli (5,8,22).
However, the role of β-arrestins in TRAIL-induced apoptosis remains
unclear. In the present study, knockdown of β-arrestin2 in HepG2
cells increased cell apoptosis, and reduced activation of the
Src-ERK signaling pathway upon TRAIL stimulation. Notably, TRAIL
treatment enhanced the quantity of β-arrestin2/Src complexes and
the association between β-arrestin2 and DR4/5 was observed only in
the presence of TRAIL. These results indicate that β-arrestin2 acts
as a negative regulator in TRAIL-induced HepG2 cell apoptosis via
formation of ternary complexes and mediating activation of the
Src-ERK signaling pathway.
TRAIL activates apoptosis signaling pathways and
also survival signaling pathways (13), which may contribute to the
development of TRAIL resistance (23). The Src-ERK signaling pathway is an
important pro-survival signaling pathway and responds to different
stimuli (20,24). In addition, recent studies revealed
that β-arrestins serve as adaptors for scaffolding intracellular
signaling networks to modulate downstream kinase activity (3). Therefore, the present study
hypothesized that β-arrestins mediate Src recruitment, which could
be involved in the effect of TRAIL on HepG2 cell apoptosis.
A cytoprotective role of β-arrestins was initially
verified in HepG2 cells upon TRAIL stimulation. RNAi of
β-arrestin2, but not β-arrestin1, potentiated TRAIL-induced
apoptosis. Recent studies identified that β-arrestin1 exerted a
significant role in the proliferation and anti-apoptotic activity
of nicotine-induced human non-small cell lung cancer (25,26);
the explanations were associated with differences in cell types and
the specificity of the action of different arrestin subtypes.
Recently, Kook et al (27)
reported that β-arrestin1 was cleaved by caspases during apoptosis,
which was not consistent with the present study. Kook et al
(27) used mouse embryonic
fibroblasts (MEFs) cells whereas the current study adopted HepG2
cells. β-arrestin has been demonstrated to have diverse roles via
distinct mechanisms in various experimental models (22). In the present study, overexpression
of β-arrestin2 using GFP-Arrb2 plasmids demonstrated that
β-arrestin2 exerted anti-apoptotic role in HepG2 cells. Using
β-arrestin2 RNAi and β-arrestin2 overexpression, the current
results verified that β-arrestin2 has a significant role in
TRAIL-induced HepG2 cell apoptosis. The contribution of β-arrestin2
to TRAIL-induced Src-ERK signaling activation was also
demonstrated. Downregulating β-arrestin2 markedly attenuated the
phosphorylation of Src and ERK upon TRAIL stimulation, and
β-arrestin2 overexpression enhanced Src-ERK signaling pathway
activation. To identify the role of Src activation in TRAIL-induced
HepG2 cell apoptosis, PP2 was used to block Src phosphorylation,
and the results demonstrated that suppression of Src activation
following TRAIL treatment reduced the activation of ERK, and
enhanced TRAIL-induced HepG2 cell apoptosis. In addition, U0126, an
inhibitor of ERK, was used to block ERK phosphorylation, and a
CCK-8 assay and DAPI staining revealed that inhibiting ERK
activation prevented TRAIL-mediated cell survival. As a
multifunctional scaffold, β-arrestins regulate various key
signaling molecules through protein-protein interactions (28), thus, the association between
β-arrestin2 and Src was examined further. The present study
demonstrated that β-arrestin2 physically combined with Src, and the
quantity of β-arrestin2 and Src complexes was enhanced upon TRAIL
stimulation. It is proposed that β-arrestin2 regulated the
activation of Src by increasing the number of β-arrestin2/Src
complexes in response to TRAIL stimulation. It was reported that
β-arrestins bind to Src family kinases and recruit them to
activated GPCRs (29,30), which results in numerous
physiological effects, including the generation of signal complexes
where β-arrestins scaffold various proteins to potentiate distinct
downstream signaling events (31).
Using β-arrestin2 RNAi, β-arrestin2 was demonstrated to be
important in regulating Src-ERK activation. Further investigation
was performed to evaluate whether β-arrestin2 recruited Src to the
TRAIL-associated DR. The present study demonstrated that TRAIL
stimulation induced the formation of DR/β-arrestin2/Src ternary
complexes. However, the formation of ternary complexes could be
validated more effectively with protein purification/co-elution
experiments, or with FRET-based technology; therefore, further
studies are required. The current data indicates that TRAIL may
induce activation of the Src-ERK signaling pathway via formation of
DR/β-arrestin2/Src complexes. However, this requires further
investigation. In addition, further studies are required to
establish whether the ternary complex was necessary in
β-arrestin2-mediated, TRAIL-induced HepG2 cell apoptosis. In future
studies, whether the formation of ternary complexes is cell-type
specific or universal and the mechanism by which β-arrestin2
exerted its action on activation of the Src-ERK signaling pathway
requires investigation.
In conclusion, the present study elucidated that
β-arrestin2 protects HepG2 cells from TRAIL-induced apoptosis by
facilitating the activation of Src-ERK pro-survival signaling. This
may have been achieved by the recruitment of Src to DR by
β-arrestin2 and the formation of DR/β-arrestin2/Src ternary
complexes in response to TRAIL stimulation. These findings provide
novel insight into the mechanism by which β-arrestin2 protects
cells against TRAIL-induced apoptosis.
Acknowledgments
The present study was supported by grants from the
Natural Science Research Project of Anhui Colleges and Universities
(grant no. KJ2016SD59), College Outstanding Young Talent Support
Program Key Projects (grant no. gxyqZD2016173), the Natural Science
Research Project of Anhui Provincial Education Department (grant
no. KJ2013B311), the National Nature Science Foundation of China
(grant no. 31301171) and the Anhui Province Key Laboratory of
active biological macromolecules (grant no. 1306C083008). The
authors would like to thank Dr. Zhimin Yin for assisting with the
experiment.
Abbreviations:
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
PBS
|
phosphate-buffered saline
|
|
TRAIL
|
tumor necrosis factor-related
apoptosis-inducing ligand
|
|
DR
|
death receptor
|
|
RNAi
|
RNA interference
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
GFP-ARRB2
|
green fluorescent protein-tagged
β-arrestin2
|
References
|
1
|
Shenoy SK and Lefkowitz RJ:
β-Arrestin-mediated receptor trafficking and signal transduction.
Trends Pharmacol Sci. 32:521–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Violin JD and Lefkowitz RJ:
Beta-arrestin-biased ligands at seven-transmembrane receptors.
Trends Pharmacol Sci. 28:416–422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DeFea KA: Beta-arrestins as regulators of
signal termination and transduction: How do they determine what to
scaffold? Cellular signalling. 23:621–629. 2011. View Article : Google Scholar
|
|
4
|
Yang X, Zhou G, Ren T, Li H, Zhang Y, Yin
D, Qian H and Li Q: β-Arrestin prevents cell apoptosis through
pro-apoptotic ERK1/2 and p38 MAPKs and anti-apoptotic Akt pathways.
Apoptosis. 17:1019–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Z, Hao J, Zhao Z, Ben P, Fang F, Shi
L, Gao Y, Liu J, Wen C, Luo L and Yin Z: Beta-Arrestins facilitate
ubiquitin-dependent degradation of apoptosis signal-regulating
kinase 1 (ASK1) and attenuate H O 2009. 2 2-induced apoptosis. Cell
Signal. 21:1195–1206
|
|
6
|
Wang Y, Tang Y, Teng L, Wu Y, Zhao X and
Pei G: Association of beta-arrestin and TRAF6 negatively regulates
Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol.
7:139–147. 2006. View
Article : Google Scholar
|
|
7
|
Hu S, Wang D, Wu J, Jin J, Wei W and Sun
W: Involvement of β-arrestins in cancer progression. Mol Biol Rep.
40:1065–1071. 2013. View Article : Google Scholar
|
|
8
|
Ahn S, Kim J, Hara MR, Ren XR and
Lefkowitz RJ: {beta}-Arrestin-2 mediates anti-apoptotic signaling
through regulation of BAD phosphorylation. J Biol Chem.
284:8855–8865. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun X, Zhang Y, Wang J, Wei L, Li H,
Hanley G, Zhao M, Li Y and Yin D: Beta-arrestin 2 modulates
resveratrol-induced apoptosis and regulation of Akt/GSK3β pathways.
Biochim Biophys Acta. 1800:912–918. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu GS: TRAIL as a target in anti-cancer
therapy. Cancer Lett. 285:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moon DO, Park SY, Choi YH, Ahn JS and Kim
GY: Guggulsterone sensitizes hepatoma cells to TRAIL-induced
apoptosis through the induction of CHOP-dependent DR5: Involvement
of ROS-dependent ER-stress. Biochem Pharmacol. 82:1641–1650. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo SY, Liu SG, Liu L, Zhou XJ and Gu Y:
RNAi silencing of the MEKK3 gene promotes TRAIL-induced apoptosis
in MCF-7 cells and suppresses the transcriptional activity of
NF-κB. Oncol Rep. 27:441–446. 2012.
|
|
13
|
Falschlehner C, Emmerich CH, Gerlach B and
Walczak H: TRAIL signalling: Decisions between life and death. Int
J Biochem Cell Biol. 39:1462–1475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
LeBlanc HN and Ashkenazi A: Apo2 L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Álvarez CJ, Lodeiro M, Theodoropoulou M,
Camiña JP, Casanueva FF and Pazos Y: Obestatin stimulates Akt
signalling in gastric cancer cells through beta-arrestin-mediated
epidermal growth factor receptor transactivation. Endocr Relat
Cancer. 16:599–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Im SR and Jang YJ: Aspirin enhances
TRAIL-induced apoptosis via regulation of ERK1/2 activation in
human cervical cancer cells. Biochem Biophys Res Commun. 424:65–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao J, Lu Y and Shen HM: Targeting p53 as
a therapeutic strategy in sensitizing TRAIL-induced apoptosis in
cancer cells. Cancer Lett. 314:8–23. 2012. View Article : Google Scholar
|
|
18
|
Qi S, Xin Y, Qi Z, Xu Y, Diao Y, Lan L,
Luo L and Yin Z: HSP27 phosphorylation modulates TRAIL-induced
activation of Src-Akt/ERK signaling through interaction with
β-arrestin2. Cell Signal. 26:594–602. 2014. View Article : Google Scholar
|
|
19
|
Bareford MD, Hamed HA, Allegood J,
Cruickshanks N, Poklepovic A, Park MA, Ogretmen B, Spiegel S, Grant
S and Dent P: Sorafenib and pemetrexed toxicity in cancer cells is
mediated via SRC-ERK signaling. Cancer Biol Ther. 13:793–803. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park EJ, Chung HJ, Park HJ, Kim GD, Ahn YH
and Lee SK: Suppression of Src/ERK and GSK-3/β-catenin signaling by
pinosylvin inhibits the growth of human colorectal cancer cells.
Food Chem Toxicol. 55:424–433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pierce KL and Lefkowitz RJ: Classical and
new roles of beta-arrestins in the regulation of G-protein-coupled
receptors. Nat Rev Neurosci. 2:727–733. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kook S, Gurevich VV and Gurevich EV:
Arrestins in apoptosis. Arrestins-Pharmacology and Therapeutic
Potential. Gurevich VV: 219. 1st edition. Springer-Verlag; Berlin,
Heidelberg: pp. 309–339. 2014, View Article : Google Scholar
|
|
23
|
Maksimovic-Ivanic D, Stosic-Grujicic S,
Nicoletti F and Mijatovic S: Resistance to TRAIL and how to
surmount it. Immunol Res. 52:157–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim SR, Jung YR, Kim DH, An HJ, Kim MK,
Kim ND and Chung HY: Caffeic acid regulates LPS-induced NF-κB
activation through NIK/IKK and c-Src/ERK signaling pathways in
endothelial cells. Arch Pharm Res. 37:539–547. 2014. View Article : Google Scholar
|
|
25
|
Kim JI, Lakshmikanthan V, Frilot N and
Daaka Y: Prostaglandin E2 promotes lung cancer cell migration via
EP4-βArrestin1-c-Src signalsome. Mol Cancer Res. 8:569–577. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dasgupta P, Rizwani W, Pillai S, Davis R,
Banerjee S, Hug K, Lloyd M, Coppola D, Haura E and Chellappan SP:
ARRB1-mediated regulation of E2F target genes in nicotine-induced
growth of lung tumors. J Natl Cancer Inst. 103:317–333. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kook S, Zhan X, Cleghorn WM, Benovic JL,
Gurevich VV and Gurevich EV: Caspase-cleaved arrestin-2 and BID
cooperatively facilitate cytochrome C release and cell death. Cell
Death Differ. 21:172–184. 2014. View Article : Google Scholar
|
|
28
|
Lodeiro M, Theodoropoulou M, Pardo M,
Casanueva FF and Camiña JP: C-Src regulates Akt signaling in
response to ghrelin via beta-arrestin signaling-independent
and-dependent mechanisms. PLoS One. 4:e46862009. View Article : Google Scholar
|
|
29
|
Marchese A, Chen C, Kim YM and Benovic JL:
The ins and outs of G protein-coupled receptor trafficking. Trends
Biochem Sci. 28:369–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laporte SA, Oakley RH, Holt JA, Barak LS
and Caron MG: The interaction of beta-arrestin with the AP-2
adaptor is required for the clustering of beta 2-adrenergic
receptor into clathrin-coated pits. J Biol Chem. 275:23120–23126.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prossnitz ER: Novel roles for arrestins in
the post-endocytic trafficking of G protein-coupled receptors. Life
Sci. 75:893–899. 2004. View Article : Google Scholar : PubMed/NCBI
|