Introduction
Marfan syndrome (MFS, OMIM: 154700) is a fatal
autosomal dominant disorder of the connective tissue with an
incidence rate of ~1 per 5,000 individuals (1). Characteristic manifestations of MFS
include a series of features that involve the cardiovascular
(aortic dissection, aneurysm, mitral valve prolapse, aortic root
dilatation), ocular (ectopia lentis) and skeletal (special face,
disproportionate slender stature, thorax deformities, scoliosis,
arachnodactyly, joint hypermobility, and flat feet) systems
(2). Additionally, the lungs, skin
and dura may also be affected in patients with MFS. The key lethal
cardiovascular symptom of patients with MFS is the development of
an aneurysm or thoracic aortic dissection, particularly at an early
age (<50 years of age) (3).
Previous studies revealed that 91% of cases of MFS were associated
with fibrillin-1 (FBN1) gene mutations (MFS-1, OMIM 134797)
(4), whereas a further small
percentage of the patients had an underlying mutation in the
transforming growth factor β receptor 2 (TGFBR2) gene
(MFS-2, OMIM 190182) (5).
Fibrillin-1, encoded by FBN1, is a large,
extracellular matrix glycoprotein that not only serves as an
important calcium-binding microfibrillar structural molecule, but
also serves as a regulator of TGF-β signaling (6). TGFBR2 encodes a transmembrane,
serine-threonine kinase domain-containing protein, TGF-β receptor
2. Mutations affecting the intracellular kinase domain of this
protein are able to disturb TGF-β signaling, which subsequently
leads to similar features of MFS-1 (7).
The causal mutation types of the two genes include
missense mutations affecting conserved residues, nonsense
mutations, deletions, in-frame and splice-site mutations, and gene
disruptions (1,7). However, how different mutations of
the genes lead to the various phenotypes of MFS has yet to be fully
elucidated (8); therefore, a rapid
determination of the genetic basis of MFS is vital to an improved
understanding, and optimal management, of this lethal disease.
Current molecular analytical methods for MFS are
single strand conformation polymorphism analysis, direct capillary
sequencing and denaturing high-performance liquid chromatography.
For the detection of large insertions/deletions, fluorescence in
situ hybridization and multiplex ligation-dependent probe
amplification are employed (4,7).
Comparatively long turnaround times and/or high cost are the major
disadvantages of the above-mentioned assays, with respect to their
clinical use (9). Next-generation
sequencing (NGS) technologies have the potential to solve these
problems by rapidly dissecting large regions at low cost (10). The technique of semiconductor
sequencing, which is rapidly gaining in popularity, is notable for
having the lowest price, shortest running time, minimum start DNA
amount and flexible sequencing-chip reagents (11).
The current study presents the most rapid,
comprehensive, cost-efficient and reliable assay for the genetic
diagnosis of MFS in routine clinical practice. By using this assay,
pathogenic loss-of-function mutations in two unrelated families
were identified.
Materials and methods
Ethics statement and participant
recruitment
The present study was approved by the Ethics
Committee of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology (Wuhan, China), and written
informed consent was obtained from each participant. The two MFS
pedigrees were recruited at the Tongji Hospital between February
and August 2014. A total of 400 unrelated healthy controls were
randomly recruited from healthy individuals undergoing routine
health examinations in the Tongji Hospital during the corresponding
period. This cohort was determined to be free of MFS by physical
examination, medical history inquiry and definitive imaging
examination (echocardiography or computer tomography).
Case reports
Concerning family 1
The proband (patient 2) was a 26-year-old woman who
complained of an acute exacerbation of a deep, oppressive
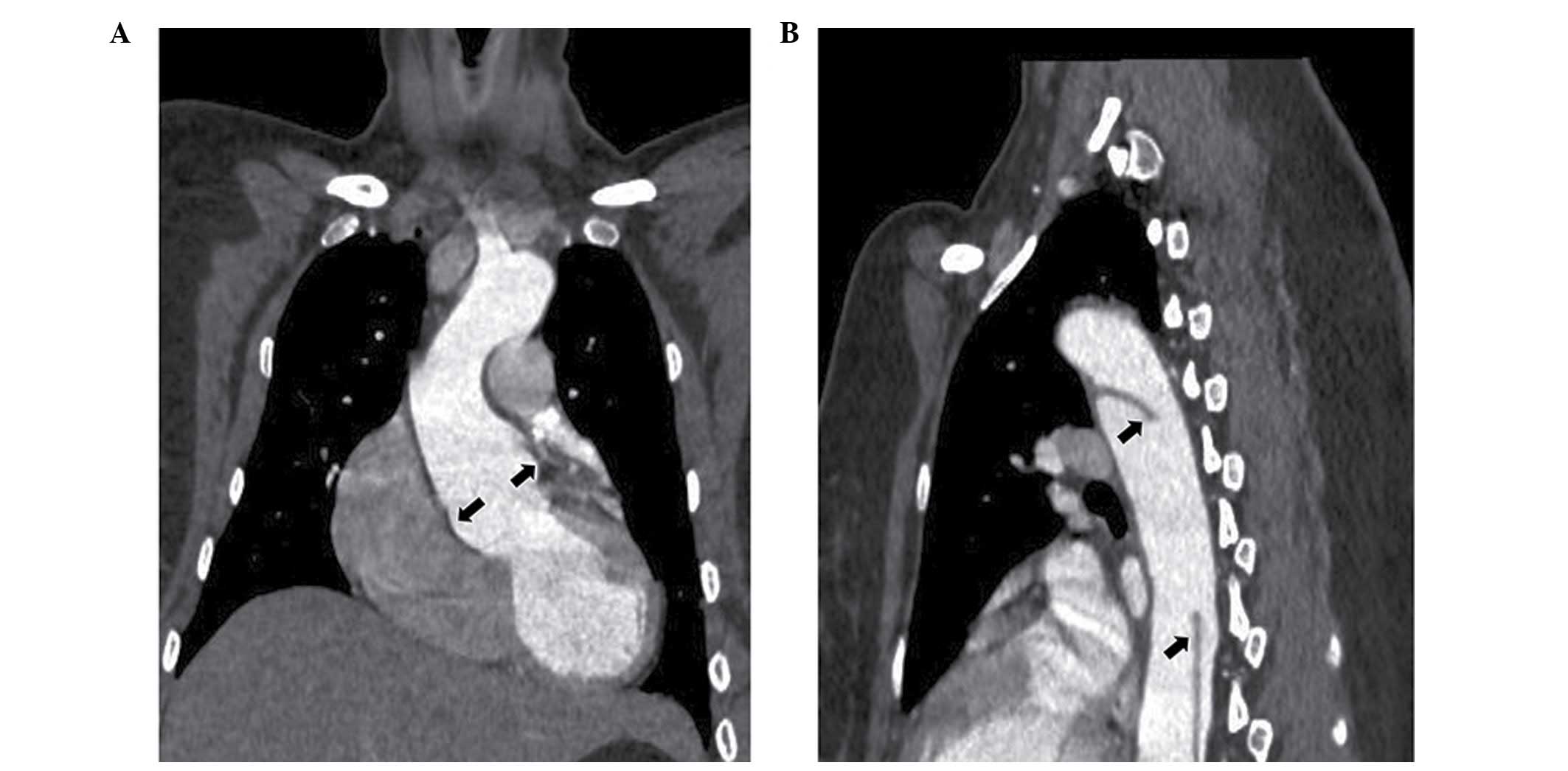
thoracodorsal pain at 31 weeks of pregnancy (Table I). After computed tomography
angiography had been performed, the patient was diagnosed with a
type B aortic dissection (Fig. 1).
Her physical examination presented bilateral ptosis, down-slanting
palpebral fissures, malar flattening and a reduced ratio of upper
segment to the lower segment. The patient had mild myopia (three
diopters bilateral). Her bilateral 'thumb signs' and 'wrist signs'
were positive. The skin striae were bilateral on the shoulders,
lumbar and knee regions, excluding the striae gravidarum.
Echocardiography detected a dilatation of the aortic sinus to 37 mm
without marked aortic valve regurgitation. Following a cesarean
section for the delivery of an infant girl, the patient underwent
replacement of the thoracic and abdominal aortas.
 | Table IClinical manifestations of the two
families. |
Table I
Clinical manifestations of the two
families.
| General feature | Family 1
| Family 2
|
|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|
| Age (years) | 29 | 26 | 5 | 33 |
| Gender | Female | Female | Female | Male |
| Height (cm) | 175 | 175 | 125 | 178 |
| Weight (kg) | 63 | 67.5 | 19 | 70.5 |
| Cardiovascular
manifestations | | | | |
| Aortic
dilatation | − | + | − | + |
| Aortic
dissection | − | + | − | + |
| Z-score not <2
points | − | + | − | + |
| Mitral valve
prolapse | − | − | − | + |
| Ocular
manifestations | | | | |
| Myopia >3
diopters | − | + | − | − |
| Ectopia lentis | − | − | − | − |
| Systemic
features | | | | |
| Thumb sign | + | + | + | + |
| Wrist sign | + | + | + | + |
| Pectus carinatum
deformity | − | − | − | − |
| Pectus excavatum or
chest asymmetry | − | − | − | − |
| Hindfoot
deformity | − | − | − | − |
| Pes planus | + | + | + | − |
| Pneumothorax | − | − | − | − |
| Dural ectasia | − | − | − | − |
| Protrusio
acetabuli | − | − | − | − |
| Decreased upper
body length to lower length | + | + | − | + |
| Scoliosis or
thoracolumbar kyphosis | − | − | − | − |
| Reduced extension
at elbows <170° | − | − | − | − |
| Craniofacial
features | + | + | + | + |
| Skin striae | + | + | − | − |
| Total score based
on revised Ghent criteria | 7 | 8 | 5 | 6 |
The elder sister (patient 1) with her 5-year-old
daughter (patient 3) of the proband presented similar clinical
features (Table I). The mother of
the proband suffered from sudden death at the age of 45. On the
basis of the oral description and photographs, the patient (height
175 cm, weight 67.5 kg) had atypical marfanoid facial features and
arachnodactyly. Other healthy relatives lacked positive
phenotypical findings of MFS in family 1.
Concerning family 2
The proband (patient 4) was a 33-year-old man who
presented typical marfanoid craniofacial features without ectopia
lentis. The patient exhibited skeletal features such as
arachnodactyly, bilateral positive wrist signs and thumb signs, and
excessive spinal curvature (Table
I). The patient had undergone thoracic aorta replacement due to
type B aortic dissection 2 years previously. Echocardiography on a
Vivid E9 cardiovascular ultrasound system (GE Healthcare Life
Sciences, Shanghai, China) detected a dilative aortic root, with a
diameter of 47 cm. Since the dilatation of the aortic root was
causing aggravation and the aortic valve developed severe
regurgitation, the patient underwent a secondary operation for the
aortic valve and ascending aorta replacement 1 year subsequently.
His father was described to present similar clinical
characteristics, and the patient suffered from sudden death at the
age of 35. His mother, however, was healthy and presented with a
normal phenotype.
MFS resequencing panel design and
next-generation sequencing
To recruit a maximum coverage of the mutation
spectrum of MFS, a specific targeted resequencing panel was
designed, including two genes predisposing to MFS, FBN1 and
TGFBR2 (Table II). Ion
torrent adapter-ligated libraries were prepared using the Ion
AmpliSeq Library kit 2.0 (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) following the manufacturer's protocol. The libraries of
two patients with MFS were pooled together. Subsequent emulsion PCR
and enrichment of the sequencing beads of the pooled libraries were
performed using the Ion OneTouch 2 system (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol, as
previously described (12).
Finally, 500 flows (125 cycles) of sequencing were performed on the
Ion 318 Chip Kit v2 using an Ion PGM Sequencing 200 Kit v2 on the
Ion PGM System (Thermo Fisher Scientific, Inc.). There were 90-bp,
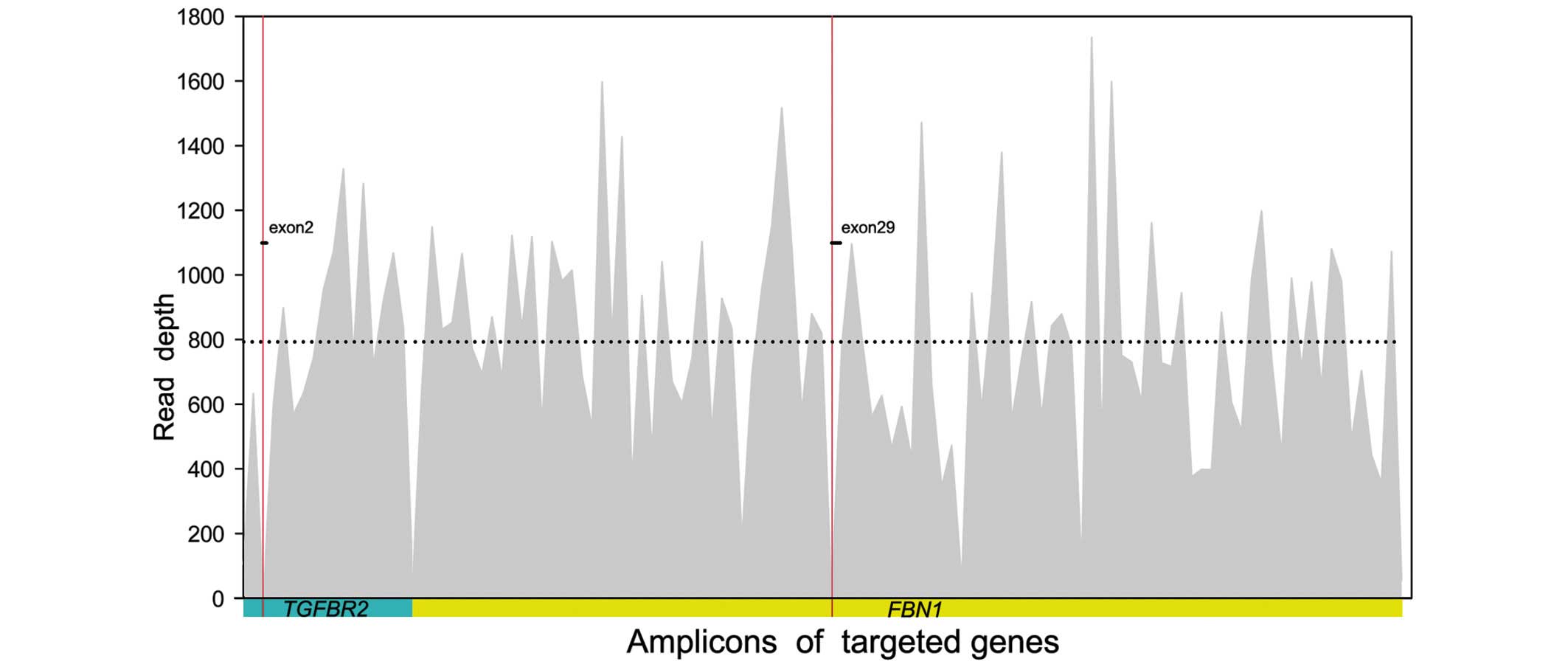
technically uncovered regions of the two targeted genes (Fig. 2), which were carefully sequenced
directly by Sanger sequencing (the primer details are shown in
Table III). Prior to mutation
screening, the panel was validated with four positive controls (MFS
patients whose pathogenic mutations had been identified previously
by Sanger sequencing).
| Table IIGenes selected for Marfan
syndrome-specific semiconductor sequencing. |
Table II
Genes selected for Marfan
syndrome-specific semiconductor sequencing.
| Gene | Chromosome | Ensembl | Target (bp) | Misseda (bp) | Coverageb (%) | Exon (n) | Amplicon (n) |
|---|
| FBN1 | 15 |
ENSG00000166147 | 8,588 | 28 | 99.70 | 65 | 98 |
| TGFBR2 | 3 |
ENSG00000163513 | 1,717 | 62 | 96.50 | 8 | 16 |
| Table IIIPrimers for uncovered region for
Sanger sequencing |
Table III
Primers for uncovered region for
Sanger sequencing
| Gene | Position | Forward primer | Reverse primer |
|---|
| FBN1 |
chr15:48777666-48777693 |
5′-AGGAACCTACTGAGAGATTCAACAT-3′ |
5′-ATCCCATTGAAGAAAGCACG-3′ |
| TGFBR2 |
chr3:30664691-30664752 |
5′-TACCAGGAAAACAGAAAAAAGAAGTG-3′ |
5′-GTGGACAAAACCCTCAAAGAAGA-3′ |
Bioinformatics analysis
Raw data were initially processed with the Ion
Torrent platform-specific software Torrent Suite™ v. 4.2.1, to
perform an alignment with the hg19 human reference genome, analyze
coverage and call variants. Subsequently, all variants were
annotated with an online-software Variant Effect Predictor
(13). Pathogenic MFS mutations
ought to be locatable in the UMD-FBN1 database (14) or in the Human Gene Mutation
Database (HGMD®) (15).
To determinate likely pathogenic novel mutations, all missense
substitutions were predicted and scored using the Sorting
Intolerant from Tolerant (SIFT) (16) and Polymorphism Phenotyping v2
(PolyPhen-2) (17) prediction
algorithms. The putative pathogenic novel mutations were further
confirmed that were not in the University of California Santa Cruz
(UCSC) common single nucleotide polymorphism (SNP) database
(18). Conservation evaluation was
performed using the online software COBALT algorithm (19).
Sanger sequencing validation
All mutations detected with NGS were validated by
Sanger sequencing using the BigDye Terminator v3.1 Cycle Sequencing
kit (Applied Biosystems; Thermo Fisher Scientific, Inc.), followed
by capillary electrophoresis on an 3500XL Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The putative causal
mutations were then targeted for Sanger sequencing (cascade
testing) in 11 family members in family 1, and in the mother of
proband in family 2, respectively. According to the revised Ghent
nosology (1), the de novo
missense mutation of FBN1 was also validated by direct
Sanger sequencing in the 400 matched healthy controls to exclude
pathogenic mutations in the FBN1 gene.
Results
Sequencing output and coverage
By semiconductor sequencing of selected regions of
the two MFS genes in the probands of the two pedigrees, an average
output of 589,714 mapped reads were achieved, with 89.87% on target
per sample. In total, 99.11% of the amplicons were covered at least
once, 97.17% of the amplicons were covered at least 20 times, and
92.14% of the amplicons were covered at least 100 times. The mean
uniformity of base coverage was 91.55% in this panel. The average
read-depth in the target region was 794 fold (Fig. 2).
Mutation detection and Sanger sequencing
validation
In the two probands, 19 (16 in patient 2; 15 in
patient 4) known or novel variants were detected by semiconductor
sequencing. Following Sanger sequencing validation, all variants
were determined. Of these 19 variants, 12 (63.16%) were annotated
as non-coding and 2 (10.53%) were annotated as synonymous, whereas
5 (26.32%) were non-synonymous, including three missense mutations,
one nonsense mutation and one frame-shift insertion, resulting in
the replacements of amino acids (Table IV).
| Table IVMutations detected by semiconductor
sequencing. |
Table IV
Mutations detected by semiconductor
sequencing.
| Patient | Position | Type | Genotype | Gene | Function | Exon | Protein | Coding | dbSNPID |
|---|
| 2 | Chrl5:48729541 | INS | -/CA | FBN1 | Ins/fs | 52 | p.Gly2120fs | c.6355_56
insTG | Novel |
| 2,4 | Chrl5:48779530 | SNV | G/C | FBN1 | mis | 28 | p.Proll48Ala | c.3442C>G | rs140598 |
| 2,4 | Chrl5:48807637 | SNV | T/T | FBN1 | mis | 12 | p.Cys472Tyr | C.1415G>A | rs4775765 |
| 2 | Chrl5:48818402 | SNV | T/C | FBN1 | mis | 9 | p.Thr305Ala | c.913A>G | Novel |
| 4 | Chrl5:48805749 | SNV | G/A | FBN1 | nons | 13 | p.Arg529X | C.1585C>T | rs137854476a |
| 2,4 | Chrl5:48797307 | SNV | A/G | FBN1 | syno | 16 | WT | C.1875T>C | rs25458 |
| 2,4 | Chrl5:48720526 | SNV | G/C | FBN1 | intr | – | – | – | rs363832 |
| 2,4 | Chrl5:48755168 | SNV | T/C | FBN1 | intr | – | – | – | rs9806595 |
| 2,4 | Chrl5:48759994 | DEL | T/- | FBN1 | intr | – | – | – | rs3214935 |
| 2,4 | Chrl5:48779201 | DEL | ATAAC/- | FBN1 | intr | – | – | – | rs72132658 |
| 2,4 | Chrl5:48779232 | DEL | TAAAA/- | FBN1 | intr | – | – | – | rs72158035 |
| 2,4 | Chrl5:48779402 | SNV | C/T | FBN1 | intr | – | – | – | rs11853943 |
| 2,4 | Chrl5:48789634 | SNV | T/C | FBN1 | intr | – | – | – | rs140605 |
| 2 | Chrl5:48826422 | DEL | AG/- | FBN1 | intr | – | – | – | rs72041020 |
| 4 | Chrl5:48826427 | SNV | G/A | FBN1 | intr | – | – | – | rs3837725 |
| 2,4 | Chrl5:48826455 | SNV | T/C | FBN1 | intr | – | – | – | rs57512865 |
| 2 | Chr3:30713842 | SNV | C/T | TGFBR2 | syno | 4 | WT | c.H67C>T | rs2228048 |
| 2,4 | Chr3:30686414 | SNV | G/G | TGFBR2 | intr | – | – | – | rs1155705 |
| 4 | Chr3:30713126 | SNV | A/A | TGFBR2 | intr | – | – | – | rsll466512 |
Pathogenic mutations identification
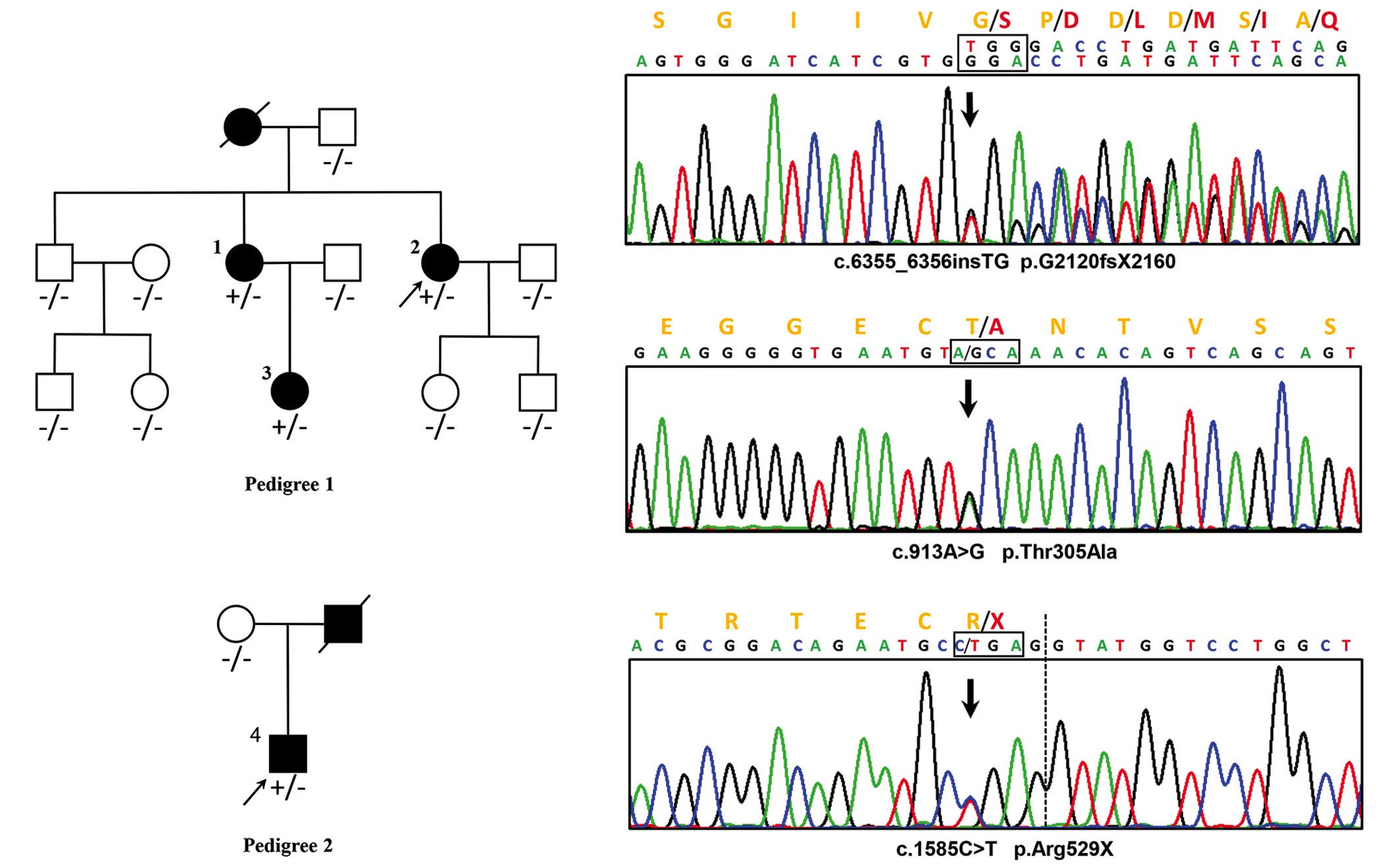
A reported MFS pathogenic nonsense mutation,
c.1585C>T, p.Arg529X (rs137854476), was identified in
FBN1 in patient 4 by filtering the HGMD and ClinVar
databases (15,20). Two novel FBN1 mutations were
identified in patient 2, including a frameshift insertion,
c.6355_6356insTG (p.G2120fsX2160), and a missense mutation,
c.913A>G (p.Thr305Ala). Neither of the mutations existed in
known databases (UMD-FBN1, HGMD, ClinVar, UCSC common SNP, dbSNP
and the 1000-genome-project) or in published articles (Fig. 3).
Via cascade testing, the insertion c.6355_6356insTG
was detected in patients 1 and 3, and the remaining nine
asymptomatic family members, as well as 400 healthy matched
controls, consistently with the phenotype, exhibited no mutation at
the identical position. The missense mutation, c.913A>G, was
detected in the healthy brother of the proband (patient 2) and in
two of the 400 healthy controls. Arg529X was not detected in the
asymptomatic mother of patient 4 in family 2.
Bioinformatics analysis
The de novo missense mutation c.913A>G
underwent in silico functional analysis and a conservation
test. The score of online predictive tools revealed no tendency of
protein damaging (SIFT=0.45 and Polyphen-2=0) (21,22).
In the conservation test, the affected residue in patients and 13
other species was revealed to be evolutionarily non-conserved,
which implied that the alternation of amino acids in this position
is less likely to damage protein function. Therefore, this missense
mutation was identified as being a rare, probably benign,
Chinese-specific polymorphism.
Discussion
Currently, the diagnosis of MFS remains
predominantly based on clinical manifestation (1). The genetic dissection of this lethal
disease helps to establish the diagnosis of familial FS and to
predict neonatal patients and young children, even when the
clinical manifestations are not yet evident (23). In the present study, a rapid and
convenient measure for routine MFS genetic diagnosis has been
described. With this panel and only 20 ng input genomic DNA,
physicians are able to go from blood samples to variants in a
single day. Compared with the Sanger method, which takes 2 months
to screen the whole FBN1 gene for 10 samples, the turnaround
time of NGS is sharply reduced. The split cost of the NGS reagents
was one-tenth that of the reagents required for the Sanger
procedure. In addition, this MFS special panel, covering all the
FBN1 and TGFBR2 coding exons and their flanking
regions, simplified the data analysis procedure and was more
practical for clinical use. It is the most rapid and convenient
method among similar panels reported previously (10,11,24).
Using this assay, genotypes of two unrelated MFS pedigrees were
screened. A novel pathogenic frameshift mutation, c.6355_6356insTG
(p.G2120fsX2160), a reported pathogenic nonsense mutation,
c.1585C>T (p.Arg529X, rs137854476), and a rare, probably benign,
Chinese-specific polymorphism, c.913A>G (p.Thr305Ala) in the
FBN1 gene, were demonstrated.
In family 1, three patients with MFS (patients 1, 2
and 3) were demonstrated to harbor a novel FBN1 heterozygous
insertion, c.6355_6356insTG, which resulted in a frame-shift
mutation and truncated the original 2,871 amino-acid full-length
protein to a 2,160-residue protein. This mutation is in exon 52,
located between TGF-like domain 8 and the epidermal growth factor
(EGF)-like domain 36, according to UniProtKB (25). Since a string of amino acids was
replaced and the EGF-like domains 37–47 of FBN1 were lost,
our hypothesis was that there could be a severe functional defect
in the mutated translation product. According to the revised Ghent
criteria of causal FBN1 mutation, this insertion was able to
be identified as a novel pathogenic mutation. In addition, a novel
missense mutation, c.913A>G (p.Thr305Ala) in FBN1, was
also detected in the asymptomatic brother of the proband (patient
2) and two of the 400 negative controls. The pathogenic effect was
also excluded by in silico functional analysis (SIFT=0.45,
Polyphen-2=0) and a conservation test. On the basis of this
evidence, it is reasonable to surmise that the novel missense
mutation c.913A>G is a benign SNP in the Chinese population.
In family 2, the proband (patient 4) was
demonstrated to harbor a FBN1 heterozygous nonsense
mutation, c.1585C>T, which caused translation of the protein to
stop at the 529th amino acid residue. This mutation, p.Arg529X
located in exon 13, is a reported MFS pathogenic mutation, also
known as rs137854476. The affected proband (patient 4) in the
present study presented phenotypes which were similar to those of
previous cases (26).
All patients (patients 1,2,3 and 4) in the present
study fulfilled the MFS diagnostic criteria, in accordance with the
genetic diagnosis results. However, since all the patients with MFS
in this study were young, particularly patient 3, the typical
marfanoid symptoms may not have been fully manifested. These should
be monitored, since the phenotypes are variable and
time-dependent.
To date, the technically uncovered regions and
sequencing bias are inevitable (9). The results of NGS need to be
validated using Sanger method. This panel recruited only
MFS-associated genes, which limited the usefulness with respect to
other inherited aortic disease diagnoses. Globally, NGS methods
have achieved a comparable, or even more optimal, sequencing
quality compared with standard assays.
In conclusion, the present study has provided what
is, at present, the most rapid, cost-effective and reliable
semiconductor MFS-specific resequencing assay for clinical routine
use. A novel pathogenic insertion and a rare, probably benign
Chinese-specific polymorphism in FBN1 were revealed. This
study enriches the mutation spectrum of FBN1, and will
facilitate the molecular diagnosis of MFS.
Acknowledgments
This study was funded by the National Natural
Science Foundation of China (no. 81370201). We would like to thank
the patients for consenting to participate in this study and
publication.
References
|
1
|
Loeys BL, Dietz HC, Braverman AC,
Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y,
Jondeau G, Faivre L, Milewicz DM, et al: The revised Ghent nosology
for the Marfan syndrome. J Med Genet. 47:476–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Drolsum L, Rand-Hendriksen S, Paus B,
Geiran OR and Semb SO: Ocular findings in 87 adults with Ghent-1
verified Marfan syndrome. Acta Ophthalmol. 93:46–53. 2015.
View Article : Google Scholar
|
|
3
|
Cañadas V, Vilacosta I, Bruna I and Fuster
V: Marfan syndrome. Part 1: Pathophysiology and diagnosis. Nat Rev
Cardiol. 7:256–265. 2010.PubMed/NCBI
|
|
4
|
Loeys B, De Backer J, Van Acker P,
Wettinck K, Pals G, Nuytinck L, Coucke P and De Paepe A:
Comprehensive molecular screening of the FBN1 gene favors locus
homogeneity of classical Marfan syndrome. Hum Mutat. 24:140–146.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Waldmuller S, Muller M, Warnecke H, Rees
W, Schöls W, Walterbusch G, Ennker J and Scheffold T: Genetic
testing in patients with aortic aneurysms/dissections: A novel
genotype/phenotype correlation? Eur J Cardiothorac Surg.
31:970–975. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Robinson PN, Arteaga-Solis E, Baldock C,
Collod-Béroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA,
Judge DP, et al: The molecular genetics of Marfan syndrome and
related disorders. J Med Genet. 43:769–787. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mizuguchi T, Collod-Beroud G, Akiyama T,
Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M,
Morisaki H, et al: Heterozygous TGFBR2 mutations in Marfan
syndrome. Nat Genet. 36:855–860. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Faivre L, Collod-Beroud G, Loeys BL, Child
A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K,
Arslan-Kirchner M, et al: Effect of mutation type and location on
clinical outcome in 1,013 probands with Marfan syndrome or related
phenotypes and FBN1 mutations: An international study. Am J Hum
Genet. 81:454–466. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Churko JM, Mantalas GL, Snyder MP and Wu
JC: Overview of high throughput sequencing technologies to
elucidate molecular pathways in cardiovascular diseases. Circ Res.
112:1613–1623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baetens M, Van Laer L, De Leeneer K,
Hellemans J, De Schrijver J, Van De Voorde H, Renard M, Dietz H,
Lacro RV, Menten B, et al: Applying massive parallel sequencing to
molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat.
32:1053–1062. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiao Y, Liu Y, Yang K, Yang Y, Zhou X, Lu
C, Xiao J, Liu F and Zhang X: Next generation sequencing as a rapid
molecular diagnosis for Marfan syndrome in a Chinese family with
mutations in the fibrillin-1 gene. Clin Chim Acta. 439:58–60. 2015.
View Article : Google Scholar
|
|
12
|
Li Z, Huang J, Zhao J, Chen C, Wang H,
Ding H and Wang DW and Wang DW: Rapid molecular genetic diagnosis
of hypertrophic cardiomyopathy by semiconductor sequencing. J
Transl Med. 12:1732014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ensembl Variant Effect Predictor.
Available online: http://asia.ensembl.org/info/docs/tools/vep/index.html.
15–March. 2015
|
|
14
|
UMD-FBN1 database. Available online:
http://www.umd.be/FBN1/.
15–March. 2015
|
|
15
|
The Human Gene Mutation Database.
Available online: http://www.hgmd.cf.ac.uk.
15–March. 2015
|
|
16
|
SIFT. Available online: http://sift.jcvi.org/.
15–March. 2015
|
|
17
|
PolyPhen-2. Available online: http://genetics.bwh.harvard.edu/pph2/.
15–March. 2015
|
|
18
|
UCSC common SNP database. Available
online: https://genome.ucsc.edu/cgi-bin/hgVai.
15–March. 2015
|
|
19
|
COBALT Cobalt Constraint-based Multiple
Protein Alignment Tool. Available online: http://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi.
15–March. 2015
|
|
20
|
ClinVar database. Available online:
http://www.ncbi.nlm.nih.gov/clinvar/variation/16452/.
15–March. 2015
|
|
21
|
Sequence SIFT. Available online:
http://sift.jcvi.org/sift-bin/catfile.csh?/opt/www/sift/tmp/2996.siftresults.predictions+Predictions+PREsift-bin/catfile.csh?/opt/www/sift/tmp/2996.siftresults.predictions+Predictions+PRE.
15–June. 2015
|
|
22
|
PolyPhen-2 report for P35555 T305A.
Available online: http://genetics.bwh.harvard.edu/ggi/pph2/a914c296bdbd9ce-b1e8afb68bef4854ebc5f1c2b/2976072.html.
15–June. 2015
|
|
23
|
Potter KJ, Creighton S, Armstrong L,
Eydoux P, Duncan W, Penny DJ, Fan Y and Gibson WT: The c.7409G>A
(p.Cys2470Tyr) Variant of FBN1: Phenotypic Variability across Three
Generations. Mol Syndromol. 4:125–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Proost D, Vandeweyer G, Meester JAN,
Salemink S, Kempers M, Ingram C, Peeters N, Saenen J, Vrints C,
Lacro RV, et al: Performant mutation identification using targeted
next generation sequencing of fourteen thoracic aortic aneurysm
genes. Hum Mutat. 36:808–814. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
UniProtKB. Available online: http://www.uniprot.org/uniprot/P35555.
15–March. 2015
|
|
26
|
Rand-Hendriksen S, Tjeldhorn L, Lundby R,
Semb SO, Offstad J, Andersen K, Geiran O and Paus B: Search for
correlations between FBN1 genotype and complete Ghent phenotype in
44 unrelated Norwegian patients with Marfan syndrome. Am J Med
Genet A. 143A. pp. 1968–1977. 2007, View Article : Google Scholar
|