Introduction
Pancreatic cancer (PC) is one of the most malignant
tumors in humans, and its 5-year survival rate is <5% (1). Additionally, the development and
metastasis of PC frequently goes undetected, and in China ~80% of
patients with PC are inoperable (2). Unfortunately, the incidence of PC is
increasing, therefore, effective new medicines and therapeutic
targets are required.
Green tea is a popular drink consumed worldwide, and
a number of epidemiological studies have indicated an association
between tea consumption and reduced incidence of cancer (3–5).
(−)-Epigallocatechin-3-gallate (EGCG) is the most abundant catechin
in green tea, and has been shown to inhibit inflammation,
oxidation, viruses and oncogenesis (6–9).
EGCG acts on numerous molecular targets, and has been demonstrated
to have inhibitory ability during the initiation and progression
stages of carcinogenesis (10–12).
It can inhibit the growth and metastasis of a number of types of
cancer (13–15) through a variety of mechanisms
(16,17), including modulation of the
phosphatidylinositide 3-kinase (PI3K)/protein kinase B
(Akt)/mechanistic target of rapamycin (mTOR) pathway (18).
PTEN is a potent tumor-suppressor gene and a
significant negative regulator of the PI3K/Akt/mTOR pathway. The
PI3K/Akt/mTOR pathway modulates cellular functions, including
proliferation, differentiation and migration (19). The dysregulation of this pathway
has been associated with many types of cancer (20), including PC. PI3K/Akt/mTOR pathway
activity promotes cancer cell proliferation, invasion and
metastasis and inhibits apoptosis (21).
In a previous study, EGCG was demonstrated to
upregulate the expression of PTEN and downregulate the expression
of phosphorylated (p)-Akt and p-mTOR in human PC cells (22). In the present study, PC cells with
or without PTEN knockdown were treated with EGCG, and the
alterations in apoptosis and protein expression of PI3K/Akt/mTOR
pathway targets were examined to investigate the therapeutic
mechanisms of EGCG in human PC.
Materials and methods
Lentiviral-based RNA interference
knockdown of PTEN in PC cells
The human PC cell lines, PANC-1 and BxPC-3, were
purchased from the Shanghai Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences (Shanghai, China). PTEN
knockdown in PANC-1 and BxPC-3 cells was performed as previously
described (23). Briefly,
lentiviral transduction was used to steadily express short hairpin
RNAs (shRNAs) that target PTEN. shRNA constructs were obtained from
Sigma-Aldrich (St. Louis, MO, USA). Both the PTEN shRNA construct
(TRCN0000219043, including the shRNA for human PTEN) and the
luciferase shRNA construct (TRCN0000072247, including the shRNA as
a control) were used to produce recombinant lentiviral particles.
The PC cells were transfected with the viral particles containing
PTEN or luciferase shRNAs for 24 h using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
after which the cells were placed into fresh RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.). The supernatants were
collected at 36, 48, 60 and 72 h following transduction, and the
supernatants were filtered using 0.45 µm low protein-binding
filters (EMD Millipore, Billerica, MA, USA). Subsequently, the
viral particles were centrifuged at 20,000 × g at 4°C for 2 h and
then resuspended in fresh RPMI-1640 medium. The lentiviral
particles (shPTEN and shLuc) were then introduced to PANC-1 and
BxPC-3 cells at a multiplicity of infection of 40. The PTEN
knockdown was examined by Western blotting in triplicate
experiments.
Cell culture and treatment
PC cells were incubated at 37°C in a 95% air and 5%
CO2 atmosphere in Roswell Park Memorial Institute 1640
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (GE Healthcare Life Sciences, Logan, UT, USA). The
cells with or without PTEN knockdown were treated with 40
µg/ml EGCG (Sigma-Aldrich) for 48 h, with control cells
treated with deionized water. Subsequently, cell proliferation was
examined using a Cell Counting kit-8 (CCK-8) assay. Apoptosis was
detected by flow cytometry. The expression of genes and proteins in
the PI3K/Akt/mTOR signaling pathway were analyzed by reverse
transcription-polymerase chain reaction (RT-PCR) and western
blotting.
Cell proliferation assays
PC cell proliferation was measured by CCK-8 assays
as previously described (24).
Briefly, the cells (with/without PTEN knockdown) were plated in
96-well plates of 5,000 cells/well. Following culture at 37°C for
24 h, the cells were treated with 40 µg/ml EGCG for 24, 48
and 72 h. The cells were incubated with CCK-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) solution (10 µl/well)
for 2 h, and then the absorbance was measured at 450 nm using a
microplate reader (Model 680; Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The alterations in cell growth were calculated as the
Inhibition Ratio (%) = (1-treated group OD values/control group OD
values) × 100. The experiments were performed in triplicate.
Apoptosis assays
The apoptosis rates of PANC-1 and BxPC-3 cells were
examined using an annexin V-fluorescein isothiocyante (FITC)
apoptosis detection kit (BioVision, Inc., Milpitas, CA, USA) as
described previously (22).
Briefly, the cells were dissociated using trypsin, and 10 µl
annexin V-FITC and 10 µl propidium iodide were then added to
the cells in the dark for 10 min. Stained cells were analysed by
flow cytometry using a FACSCalibur flow cytometer (BD Biosciences,
Franklin, NJ, USA). Cells in the lower right quadrant of the dot
plot were considered to be in early apoptosis, and those in the
upper right quadrant were in late apoptosis. The experiments were
performed in triplicate.
RT-PCR analysis
PI3K, Akt, PTEN, and mTOR mRNA expression was
analyzed by RT-PCR as previously described (22). Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as the internal control. Total RNA
was extracted from cells using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) and was treated with DNase (Promega
Corporation, Madison, WI, USA), prior to reverse transcription into
cDNA using the RETROscript™ kit (cat. no. AM1710; Thermo Fisher
Scientific, Inc.), which contained dNTPs, a RNase inhibitor, M-MLV
reverse transcriptase and RT buffer (Tris-HCl, KCl,
MgCl2 and DTT). RT-PCR was conducted using an
AccessQuick RT-PCR System (Promega Corporation). A total of 30
cycles of amplification were performed using the following
conditions: Denaturation at 94°C for 30 sec, annealing at 58°C for
30 sec, and extension at 72°C for 1 min. The primer sequences were
as follows: PI3K forward, 5′-AGGAGCGGTACAGCAAAGAA-3′ and reverse,
5′-GCCGAACACCTTTTTGAGTC-3′; AKT forward, 5′-TGAAAACCTTCTGTG
GGACC-3′ and reverse, 5′-TGGTCCTGGTTGTAGAAG GG-3′; PTEN forward,
5′-CAGAAAGACTTGAAGGCGTAT-3′ and reverse,
5′-CGTCGTGTGGGTCCTGAGTGA-3′; mTOR forward, 5′-CTG
GGACTCAAATGTGTGCAGTTC-3′ and reverse, 5′-GAACAATAGGGT
GAATGATCCGGG-3′; and GAPDH forward, 5′-GGAAGGTGAAGGTCGGAGT-3′ and
reverse, 5′-CCTGGAAGATGGTGATGGG-3′. The PCR products were separated
by 1% agarose gel electrophoresis and stained with ethidium
bromide, and the results analyzed using NIH Image 1.60 software
(National Institutes of Health, Bethesda, MD, USA). The experiments
were performed in triplicate.
Western blotting
Protein extraction and western blotting were
conducted as previously described (22). In brief, the cells were rinsed with
phosphate-buffered saline, and lysed with lysis buffer for 30 min.
Subsequently, the lysates were centrifuged at 12,000 × g for 10
min, and the protein concentrations were measured using a
bicinchoninic acid protein assay kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Following this, the proteins were separated by
10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 80
V for 1.5 h, and then transferred onto polyvinylidene fluoride
membranes (EMD Millipore) at 100 V for 2.5 h. Following incubation
in bovine serum albumin (Gibco; Thermo Fisher Scientific, Inc.) at
4°C for 1 h, the membranes were incubated with the following
primary antibodies: Rabbit anti-PTEN (1:1,000; cat. no. 9188),
rabbit anti-PI3K (1:1,000; cat. no. 4249), rabbit anti-Akt
(1:1,000; cat. no. 4685), mouse anti-p-Akt (1:500; cat. no. 12694),
rabbit anti-mTOR (1:1,000; cat. no. 2983), rabbit anti-p-mTOR
(1:1,000; cat. no. 5536) and mouse anti-β-actin (1:500; cat. no.
3700) monoclonal antibodies (Cell Signaling Technology, Inc.,
Danvers, MA, USA) in Tris-buffered saline-Tween-20; (Sigma-Aldrich)
overnight. Membranes were then incubated with horseradish
peroxidase-conjugated goat anti-rabbit (1:1,000; cat. no. 7074) and
goat anti-mouse (1:2,500; cat. no. 7076) secondary antibodies (Cell
Signaling Technology, Inc.). Following rinsing, the bands were
detected using an enhanced chemiluminescence detection system (GE
Healthcare Life Sciences, Chalfont, UK). Relative protein levels
were normalized to β-actin as the internal control. The experiments
were performed in triplicate.
Statistical analysis
Statistical analysis was performed using SPSS
software, version 13.0 (SPSS, Inc., Chicago, IL, USA). The data are
presented as the mean ± standard deviation. Differences between
groups were examined using one-way analysis of variance followed by
Fisher's least significant difference test. P<0.05 was
considered to indicate a statistically significant difference.
Results
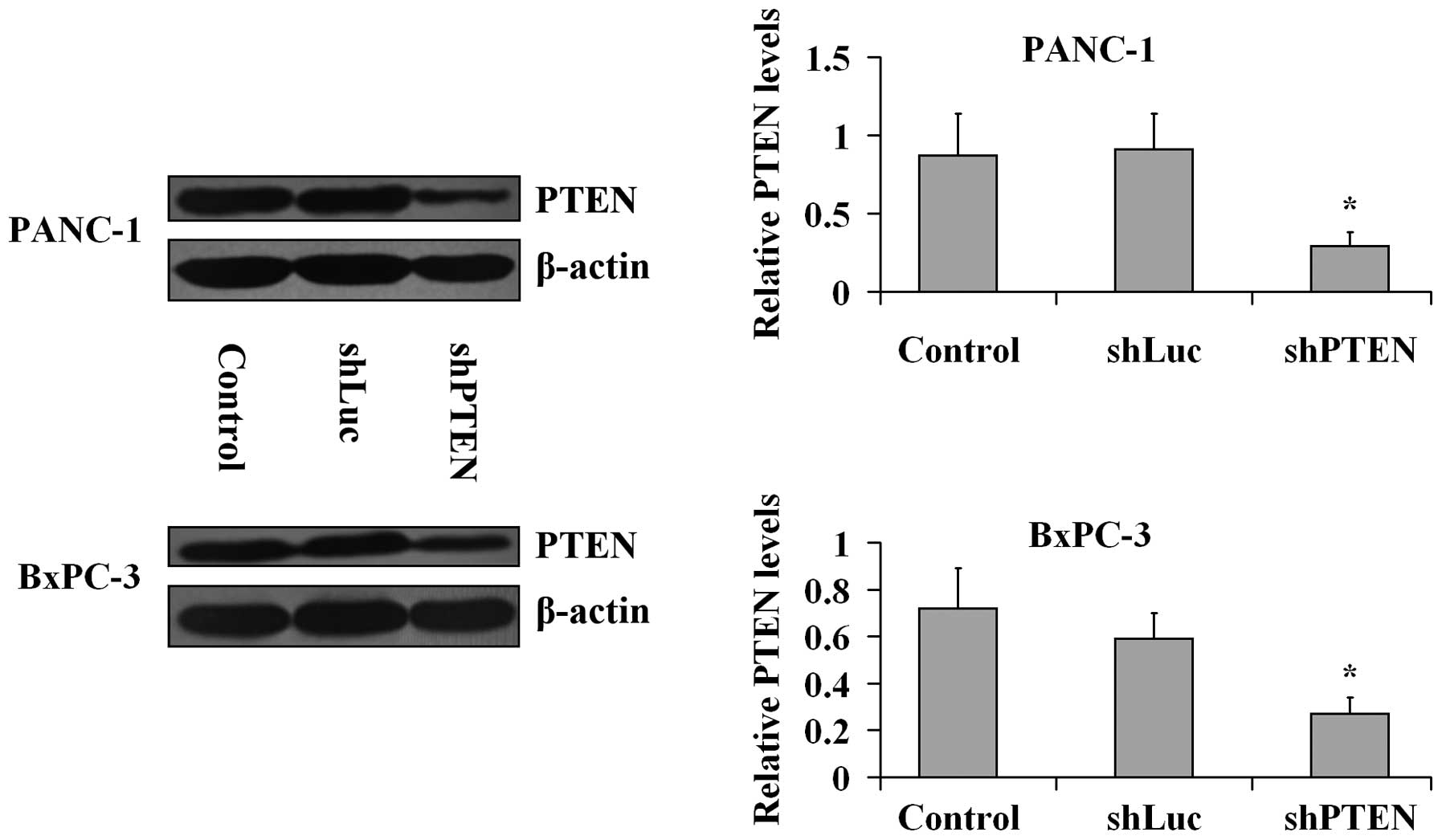
PTEN knockdown by RNA interference in PC
cells
PANC-1 and BxPC-3 cells were transfected with the
viral particles including PTEN or luciferase shRNAs for 24 h and
the knockdown of PTEN was confirmed by western blotting analysis
(Fig. 1). The β-actin expression
levels in the treated group did not differ with the levels in the
control (untreated) group (P>0.05). PTEN expression levels were
significantly lower in shPTEN groups compared with the control
group (P<0.05), Furthermore, the expression levels of PTEN in
the shLuc groups were not different compared with the controls
(P>0.05).
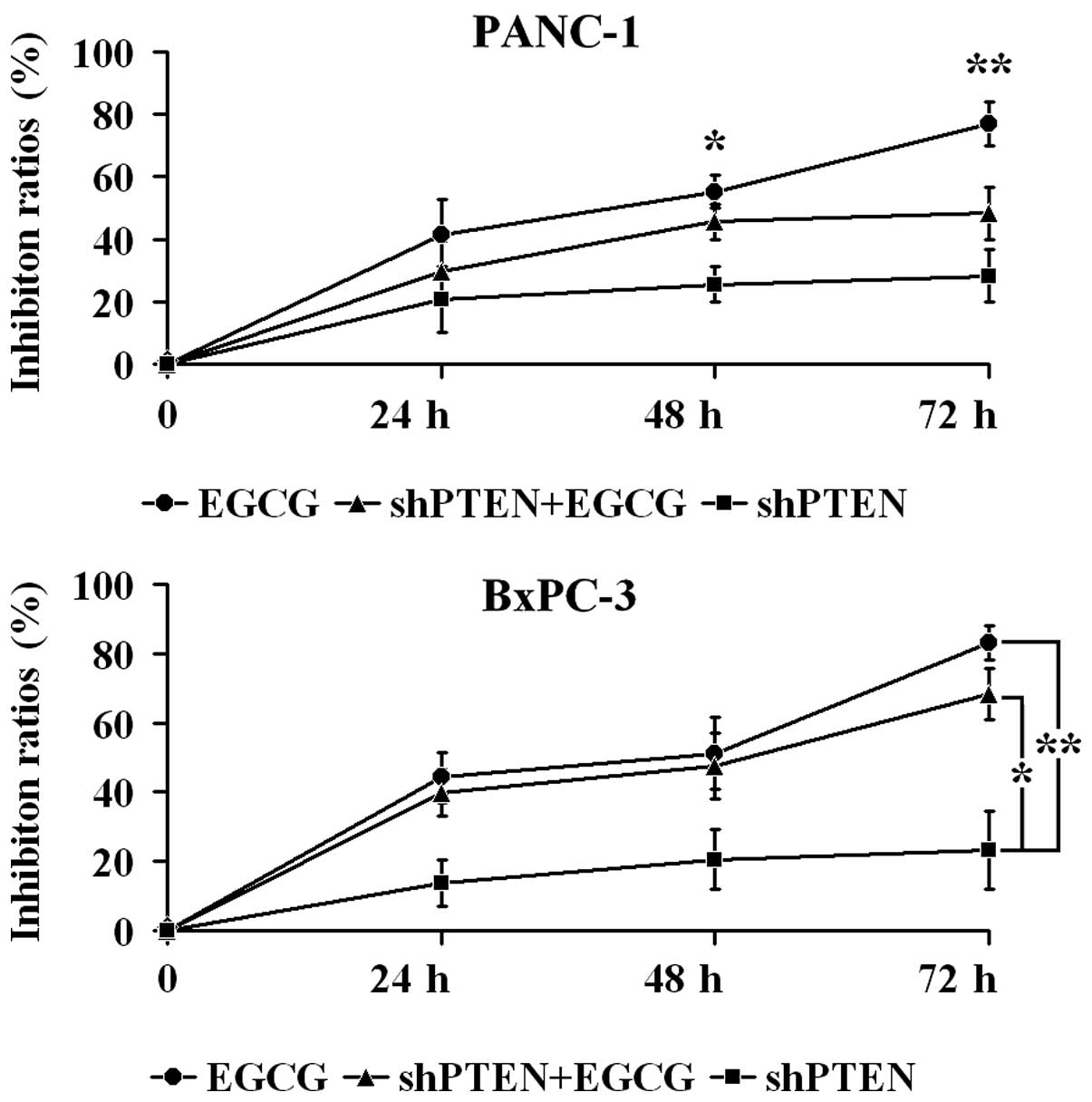
EGCG inhibits PC cell proliferation via
PTEN
PANC-1 and BxPC-3 cells with or without PTEN
knockdown were cultured in medium with or without 40 µg/ml
EGCG for 24, 48 and 72 h, and proliferation was examined by CCK-8
assays (Fig. 2). In PANC-1 cells,
the inhibition ratio in the EGCG group at 48 and 72 h was
significantly higher compared with the shPTEN group (P<0.05 and
P<0.01, respectively). Furthermore, the inhibition ratio in the
shPTEN+EGCG group was not significantly different compared with
shPTEN group (P>0.05). In BxPC-3 cells, the inhibition ratios in
the EGCG group were significantly greater compared with the shPTEN
group (P<0.01). Additionally, the inhibition ratios in the
shPTEN+EGCG group was significantly higher than the shPTEN group
(P<0.05).
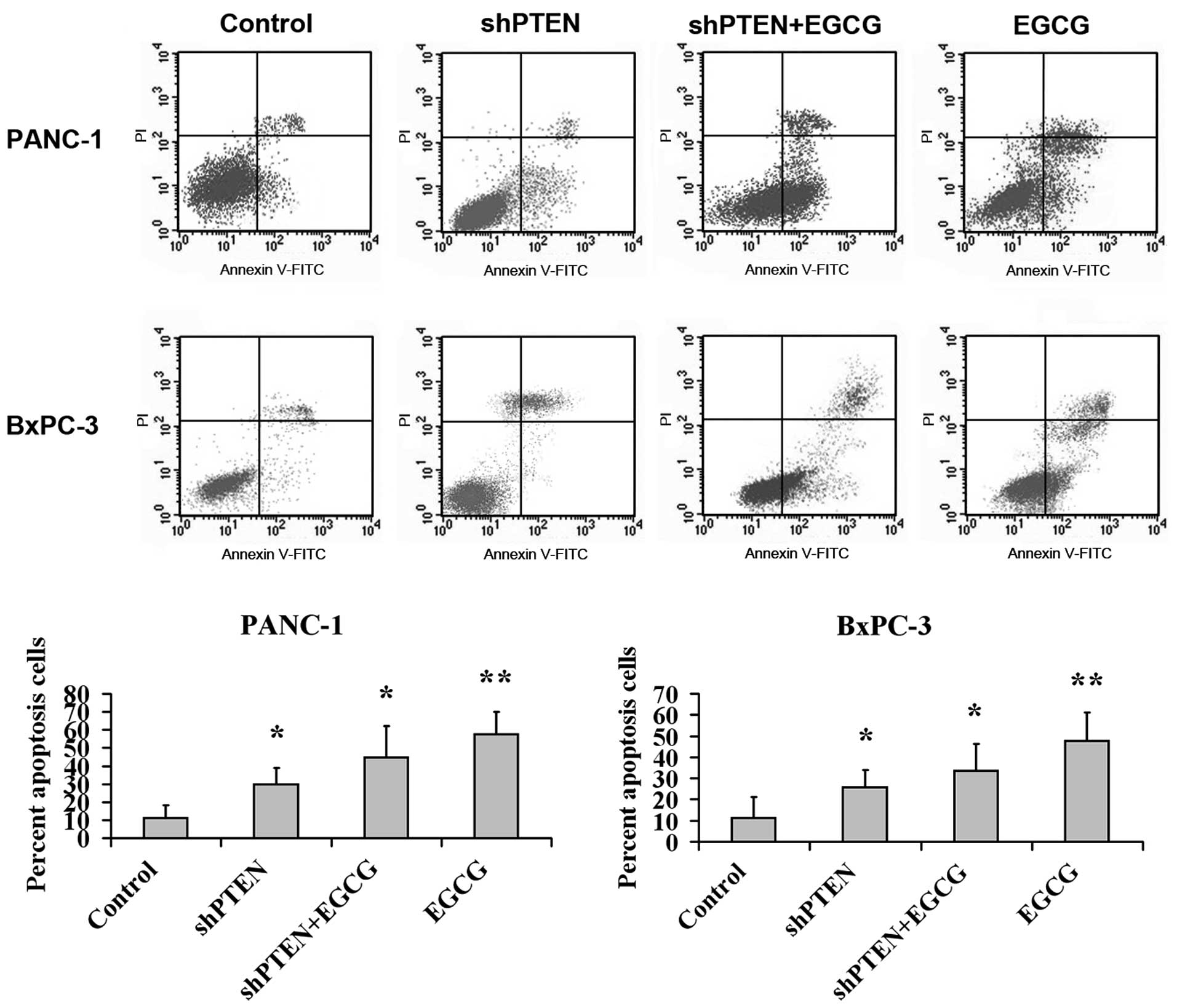
Effect of EGCG on PC cell apoptosis via
PTEN
PANC-1 and BxPC-3 cells with or without PTEN
knockdown were cultured in medium with or without 40 µg/ml
EGCG for 48 h, and the apoptotic rate was analyzed by flow
cytometry (Fig. 3). This indicated
that the apoptotic ratios in the EGCG group were substantially
higher compared with the control (untreated) group (P<0.01).
Additionally, the apoptotic rates in the shPTEN and shPTEN+EGCG
groups were substantially higher than the control group
(P<0.05).
EGCG regulates the expression of genes
and proteins in the PI3K/Akt/mTOR pathway in PC cells via PTEN
PANC-1 and BxPC-3 cells with or without PTEN
knockdown were cultured in medium with or without 40 µg/ml
EGCG for 48 h, and the mRNA expression of PI3K, PTEN, Akt and mTOR
is presented in Fig. 4. The mRNA
expression levels of PTEN in the shPTEN group were significantly
lower compared with the control (untreated) group (P<0.05).
Furthermore, the mRNA expression of PTEN in the EGCG group was
significantly higher compared with the control group (P<0.01).
However, the mRNA expression levels of PTEN in the shPTEN+EGCG
group did not differ compared with the control group
(P>0.05).
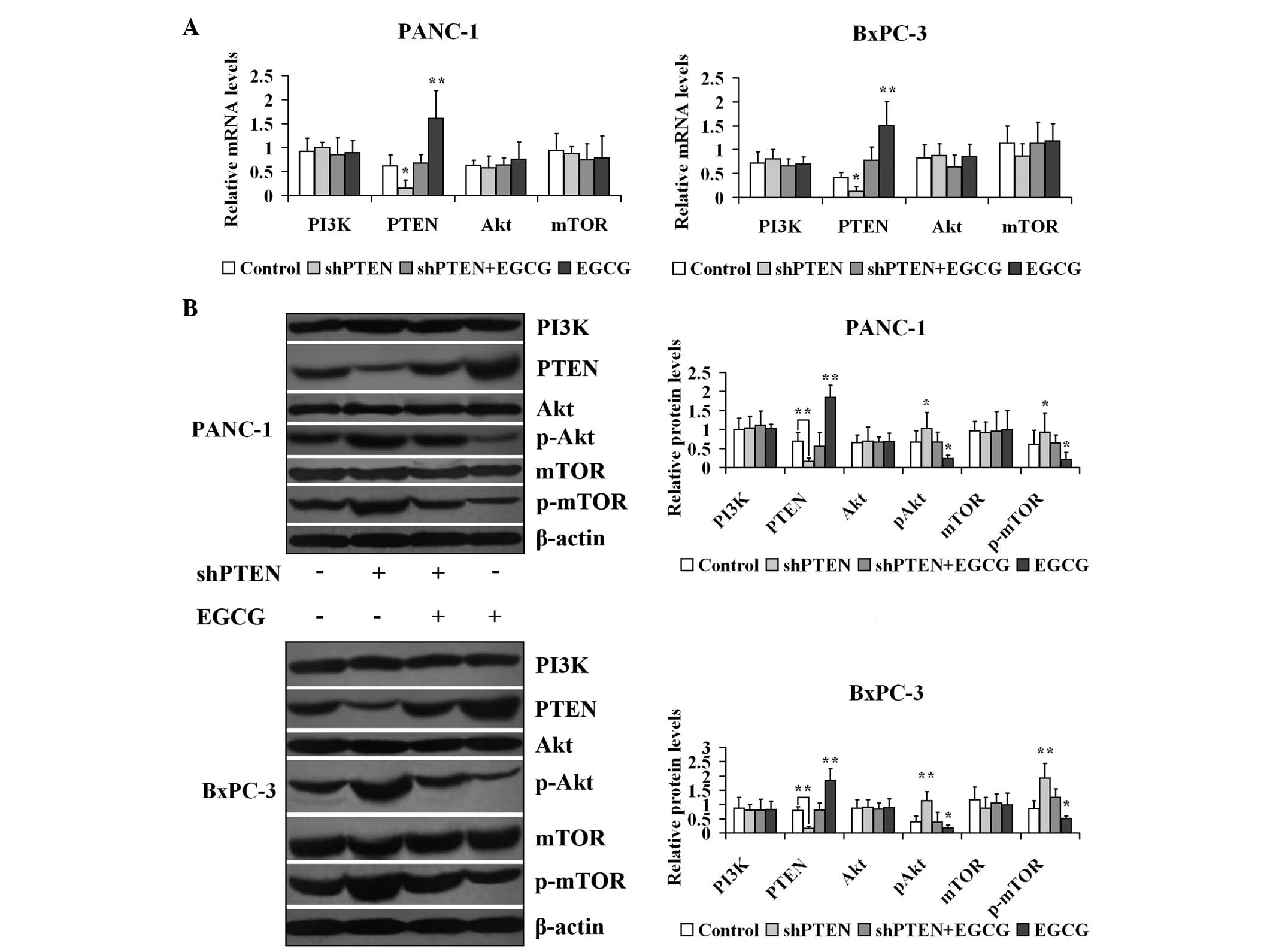 | Figure 4Effect of EGCG on expression of genes
and proteins involved in the PI3K/Akt/mTOR pathway in prostate
cancer cells via PTEN. (A) PANC-1 and BxPC-3 cells with or without
PTEN knockdown were treated with 40 µg/ml EGCG for 48 h.
PI3K, PTEN, Akt and mTOR mRNA expression levels were measured by
reverse transcription-polymerase chain reaction. (B) PANC-1 and
BxPC-3 cells with or without PTEN knockdown were treated with 40
µg/ml EGCG for 48 h. PI3K, PTEN, Akt, pAkt, mTOR and p-mTOR
expression levels were measured by western blotting.
*P<0.05, **P<0.01 vs. control group.
EGCG, (−)-epigallocatechin-3-gallate; PI3K, phosphoinositide
3-kinase; Akt, protein kinase B; mTOR, mechanistic target of
rapamycin; PTEN, phosphatase and tensin homolog deleted on
chromosome 10; sh, short hairpin; p-, phosphorylated. |
Subsequently, the effect of EGCG on the protein
expression of PI3K, PTEN, Akt, p-Akt, mTOR and p-mTOR in PANC-1 and
BxPC-3 cells was investigated with or without PTEN knockdown
(Fig. 4). The β-actin expression
levels in the treated groups were unaltered compared with the
control groups (P>0.05). The protein expression levels of PTEN
in the shPTEN group were significantly lower compared with the
control (untreated) group (P<0.01). Furthermore, the expression
of PTEN in the EGCG group was markedly higher compared with the
control group (P<0.01). However, the expression of PTEN in the
shPTEN+EGCG group did not differ compared with the control group
(P>0.05). The expression levels of p-Akt and p-mTOR in the
shPTEN group were significantly greater compared with the control
(untreated) group (P<0.05). Additionally, the p-Akt and p-mTOR
expression levels in the EGCG groups were significantly lower
compared with the control group (P<0.05). However, the p-Akt and
p-mTOR expression levels in the shPTEN+EGCG group were unaltered
compared with the control group (P>0.05).
Discussion
In the present study, using PANC-1 cells the
inhibition ratio in normal cells following EGCG treatment at 48 and
72 h was observed to be significantly higher compared with shPTEN
cells (P<0.05 and P<0.01, respectively), with the inhibition
ratio in shPTEN cells following EGCG treatment unaltered compared
with shPTEN cells (P>0.05). In BxPC-3 cells, the inhibition
ratios in normal cells following EGCG treatment were significantly
higher compared with shPTEN cells (P<0.01), with the inhibition
ratios in shPTEN cells following EGCG treatment substantially
higher than in the shPTEN cells (P<0.05). These data indicate
that PTEN was involved in EGCG inhibiting PC cell proliferation,
with the knockdown of PTEN reducing the inhibitory effect of EGCG
on PC cell proliferation. Furthermore, the previous findings
support the present study. In a previous study, the proliferation
of PANC-1 cells was inhibited following treatment with 40
µg/ml EGCG for 24, 48 and 72 h (22). In addition, Zhang et al
(25) reported that loss of PTEN
promoted proliferation and invasion in PC cells, and Ma et
al (26) demonstrated that
knockdown of PTEN was able to upregulate cell invasiveness and
proliferation in PC cells. Furthermore, Lyn-Cook et al
(27) demonstrated that EGCG
suppressed pancreatic cell growth by approximately 90%. Differences
in the methods or cell lines used in these studies may explain the
discrepancies between these studies and the present study.
The current study indicated that the apoptotic rates
in normal cells following EGCG treatment were significantly higher
compared with the control group (P<0.01), and the apoptotic
rates in the shPTEN cells with or without EGCG treatment were
significantly higher compared with the control group (P<0.05).
These results suggested that PTEN was involved in EGCG promoting PC
cell apoptosis, and that the absence of PTEN may attenuate the
apoptosis-promoting ability of EGCG in PC cells. These results are
supported by previous studies. In a previous study, the apoptosis
ratio in PANC-1 cells following 40 µg/ml EGCG treatment over
24 h was 28.56±1.56% (22).
Qanungo et al (28)
reported that EGCG induced the apoptosis of human PC Mia Paca-2
cells and that the apoptotic rate was ~2.5–25% following treatment
with 0.025–0.2 mM EGCG for 24 h. The differences in the cell types
and EGCG concentrations used in these previous studies may account
for the variation in these rates.
PTEN is an important negative modulator of the
PI3K/Akt/mTOR pathway, as it can weaken upstream signals.
Deactivation of PTEN leads to activated PI3K/Akt/mTOR signaling. In
the present study, the mRNA and protein expression of PTEN in
normal cells following treatment with EGCG were significantly
higher compared with the controls (P<0.01). The mRNA and protein
expression levels of PTEN in shPTEN cells following EGCG treatment
were comparable with the control cells (P>0.05). The expression
levels of p-Akt and p-mTOR in shPTEN cells were significantly
higher compared with the controls (P<0.05), whilst the p-Akt and
p-mTOR expression levels in normal cells treated with EGCG alone
were significantly lower compared with the control cells
(P<0.05). The expression levels of p-Akt and p-mTOR in shPTEN
cells treated with EGCG were comparable with the control cells
(P>0.05). Previous studies have demonstrated that EGCG is able
suppress the PI3K/Akt/mTOR pathway by downregulating p-Akt and
p-mTOR expression based on the presence of PTEN, instead of
regulating Akt and mTOR (22,29).
These data indicate that EGCG-induced upregulation of PTEN
expression is a prohibitive mechanism on the PI3K/Akt/mTOR pathway
and that the loss of PTEN may attenuate the inhibitory effect of
EGCG on the PI3K/Akt/mTOR pathway in human PC cells. However,
previous studies support the results of the present study. In a
previous study, EGCG upregulated the expression levels of PTEN and
downregulated the expressions of p-Akt and p-mTOR in PANC-1 cells
(22). Additionally, Zhang et
al (25) reported that loss of
PTEN resulted in increased expression of p-Akt and p-mTOR in PC
cells and Shankar et al (30) reported that EGCG inhibited the
phosphorylation of PI3K and p-Akt in PC tissues and promoted PTEN
expression, however with no influence on Akt. Nevertheless, certain
studies have indicated that EGCG is able to modulate the expression
levels of PI3K, mTOR or Akt in certain types of cancer. Shen et
al (31) reported that EGCG
treatment resulted in a reduction in the mRNA and protein
expression levels of PI3K and Akt in hepatoma, and Li et al
(32) found that loss of PTEN
resulted in increased expression levels of Akt, p-Akt, and p-mTOR
in endometrial cancer cells. Additionally, Van Aller et al
(33) indicated that EGCG was able
to inhibit the expression of PI3K, mTOR and p-Akt in MDA-MB-231 and
A549 cells, and Shimizu et al (34) demonstrated that EGCG can inhibit
the expression levels of Akt and p-Akt in colorectal cancer
xenograft tumors. Furthermore, Shirakami et al (35) reported that EGCG can repress Akt
expression in human hepatoma HuH7 cell xenografts, and Ichimatsu
et al (36) reported that
EGCG can repress the activation of PI3K in JB6Cl41 cells. However,
alterations in the expression levels of PI3K, Akt and mTOR were not
observed in the current study, therefore further study is
required.
In conclusion, EGCG was able to inhibit
proliferation and induce apoptosis in PC cells via PTEN, with the
loss of PTEN reducing the ability of EGCG to inhibit proliferation
and promote apoptosis in PC cells. In addition, EGCG is able to
downregulate the expression levels of p-Akt and p-mTOR to regulate
then PI3K/Akt/mTOR pathway via PTEN. Furthermore, this regulatory
effect may contribute to the apoptosis-inducing and
anti-proliferative properties of EGCG. However, further study is
required to fully elucidate the regulatory effect of EGCG on
components downstream of the PI3K/Akt/mTOR signal pathway.
Acknowledgments
The current study was supported by the Science and
Technology Research Grant of Education Department of Heilongjiang
Province, China (grant no. 12541921).
References
|
1
|
Ghaneh P, Costello E and Neoptolemos JP:
Biology and management of pancreatic cancer. Gut. 56:1134–1152.
2007.PubMed/NCBI
|
|
2
|
Zhang Y and Shi X: Progress in the
chemotherapy of pancreatic carcinoma. World Chin J Digestology.
17:1422–1426. 2009.
|
|
3
|
Suganuma M, Okabe S, Sueoka N, Sueoka E,
Matsuyama S, Imai K, Nakachi K and Fujiki H: Green tea and cancer
chemo-prevention. Mutat Res. 428:339–344. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shankar S, Ganapathy S and Srivastava RK:
Green tea poly-phenols: Biology and therapeutic implications in
cancer. Front Biosci. 12:4881–4899. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang CS, Ju J, Lu G, Xiao H, Hao X, Sang S
and Lambert JD: Cancer prevention by tea and tea polyphenols. Asia
Pac J Clin Nutr. 17(Suppl 1): S245–S248. 2008.
|
|
6
|
Hastak K, Gupta S, Ahmad N, Agarwal MK,
Agarwal ML and Mukhtar H: Role of P53 and NF-kappaB
inepigal-locatechin3-gallate-induced apoptosis of LNCaP cells.
Oncogene. 22:4851–4859. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maeda-Yamamoto M, Suzuki N, Sawai Y,
Miyase T, Sano M, Hashimoto-Ohta A and Isemura M: Association of
suppression of extracellular signal-regulated kinase
phosphorylation by epigallocatechin gallate with the reduction of
matrix metalloproteinase activities in human fibrosarcoma HT1080
cells. J Agic Food Chem. 51:1858–1863. 2003. View Article : Google Scholar
|
|
8
|
Roy M, Chakrabarty S, Sinha D,
Bhattacharya RK and Siddiqi M: Anticlastogenic, antigenotoxic and
apoptoic antivity of epigallocatechin gallate: A green tea
polyphenol. Mutat Res. 523–524:33–41. 2003. View Article : Google Scholar
|
|
9
|
Mittal A, Pate MS, Wylie RC, Tollefsbol TO
and Katiyar SK: EGCG downregulates telomerase in human breast
carcinoma MCF-7 cells, leading to suppression of cell viability and
induction of apotosis. Int J Oncol. 24:703–710. 2004.PubMed/NCBI
|
|
10
|
Mukhtar H and Ahmad N: Tea polyphenols:
Prevention of cancer and optimizing health. Am J Clin Nutr.
71(Suppl 6): S1698–S1702; discussion S1703–S1704. 2000.
|
|
11
|
Lambert JD and Yang CS: Cancer
chemopreventive activity and bioavailability of tea and tea
polyphenols. Mutat Res. 523–524:201–208. 2003. View Article : Google Scholar
|
|
12
|
Bode AM and Dong Z: Targeting signal
transduction pathways by chemopreventive agents. Mutat Res.
555:33–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang CS, Chung JY, Yang G, Chhabra SK and
Lee MJ: Tea and tea polyphenols in cancer prevention. J Nutr.
130(2S Suppl): S472–S478. 2000.
|
|
14
|
Leone M, Zhai D, Sareth S, Kitada S, Reed
JC and Pellecchia M: Cancer prevention by tea polyphenols is linked
to their direct inhibition of antiapoptotic Bcl-2-family proteins.
Cancer Res. 63:8118–8121. 2003.PubMed/NCBI
|
|
15
|
Pellecchia M and Reed JC: Inhibition of
anti-apoptotic Bcl-2 family proteins by natural polyphenols: New
avenues for cancer chemoprevention and chemotherapy. Curr Pharm
Des. 10:1387–1398. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang CS, Lambert JD, Hou Z, Ju J, Lu G and
Hao X: Molecular targets for the cancer preventive activity of tea
polyphenols. Mol Carcinog. 45:431–435. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanwar J, Taskeen M, Mohammad I, Huo C,
Chan TH and Dou QP: Recent advances on tea polyphenols. Front
Biosci (Elite Ed). 4:111–131. 2012. View
Article : Google Scholar
|
|
19
|
Morgan TM, Koreckij TD and Corey E:
Targeted therapy for advanced prostate cancer: Inhibition of the
PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 9:237–249. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Downward J: PI3-kinase, Akt and cell
survival. Semin Cell Dev Biol. 15:177–182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu G, Zhang W, Bertram P, Zheng XF and
McLeod H: Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR
pathway in common human tumors. Int J Oncol. 24:893–900.
2004.PubMed/NCBI
|
|
22
|
Liu S, Wang XJ, Liu Y and Cui YF:
PI3K/AKT/mTOR signaling is involved in
(−)-epigallocatechin-3-gallate-induced apoptosis of human
pancreatic carcinoma cells. Am J Chin Med. 41:629–642. 2013.
View Article : Google Scholar
|
|
23
|
Lin CF, Young KC, Bai CH, Yu BC, Ma CT,
Chien YC, Chiang CL, Liao CS, Lai HW and Tsao CW: Rosiglitazone
regulates anti-inflammation and growth inhibition via PTEN. Biomed
Res Int. 2014:7879242014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie F, Su M, Qiu W, Zhang M, Guo Z, Su B,
Liu J, Li X and Zhou L: Kaempferol promotes apoptosis in human
bladder cancer cells by inducing the tumor suppressor, PTEN. Int J
Mol Sci. 14:21215–21226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Zhang J, Xu K, Xiao Z, Sun J, Xu
J, Wang J and Tang Q: PTEN/PI3K/mTOR/B7-H1 signaling pathway
regulates cell progression and immuno-resistance in pancreatic
cancer. Hepatogastroenterology. 60:1766–1772. 2013.
|
|
26
|
Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A,
Takahashi H, Wakasugi T, Funahashi H, Sato M and Takeyama H: IGF-1
mediates PTEN suppression and enhances cell invasion and
proliferation via activation of the IGF-1/PI3K/Akt signaling
pathway in pancreatic cancer cells. J Surg Res. 160:90–101. 2010.
View Article : Google Scholar
|
|
27
|
Lyn-Cook BD, Rogers T, Yan Y, Blann EB,
Kadlubar FF and Hammons GJ: Chemopreventive effects of tea extracts
and various components on human pancreatic and prostate tumor cells
in vitro. Nutr Cancer. 35:80–86. 1999. View Article : Google Scholar
|
|
28
|
Qanungo S, Das M, Haldar S and Basu A:
Epigallocatechin-3-gallate induces mitochondrial membrane
depolarization and caspase-dependent apoptosis in pancreatic cancer
cells. Carcinogenesis. 26:958–967. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang LY, Li X and Han YZ: Neuroprotection
by epigallocatechin gallate against bupivacaine anesthesia induced
toxicity involves modulation of PI3/Akt/PTEN signalling in N2a and
SH-SY5Y cells. Int J Clin Exp Med. 8:15065–15075. 2015.
|
|
30
|
Shankar S, Marsh L and Srivastava RK: EGCG
inhibits growth of human pancreatic tumors orthotopically implanted
in Balb C nude mice through modulation of FKHRL1/FOXO3a and
neuropilin. Mol Cell Biochem. 372:83–94. 2013. View Article : Google Scholar
|
|
31
|
Shen X, Zhang Y, Feng Y, Zhang L, Li J,
Xie YA and Luo X: Epigallocatechin-3-gallate inhibits cell growth,
induces apoptosis and causes S phase arrest in hepatocellular
carcinoma by suppressing the AKT pathway. Int J Oncol. 44:791–796.
2014.PubMed/NCBI
|
|
32
|
Li T, Yang Y, Li X, Xu C and Meng L: EGFR-
and AKT-mediated reduction in PTEN expression contributes to
tyrphostin resistance and is reversed by mTOR inhibition in
endometrial cancer cells. Mol Cell Biochem. 361:19–29. 2012.
View Article : Google Scholar
|
|
33
|
Van Aller GS, Carson JD, Tang W, Peng H,
Zhao L, Copeland RA, Tummino PJ and Luo L: Epigallocatechin gallate
(EGCG), a major component of green tea, is a dual
phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res
Commun. 406:194–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shimizu M, Shirakami Y, Sakai H, Yasuda Y,
Kubota M, Adachi S, Tsurumi H, Hara Y and Moriwaki H:
(−)-Epigallocatechin gallate inhibits growth and activation of the
VEGF/VEGFR axis in human colorectal cancer cells. Chem Biol
Interact. 185:247–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shirakami Y, Shimizu M, Adachi S, Sakai H,
Nakagawa T, Yasuda Y, Tsurumi H, Hara Y and Moriwaki H:
(−)-Epigallocatechin gallate suppresses the growth of human
hepatocellular carcinoma cells by inhibiting activation of the
vascular endothelial growth factor-vascular endothelial growth
factor receptor axis. Cancer Sci. 100:1957–1962. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ichimatsu D, Nomura M, Nakamura S,
Moritani S, Yokogawa K, Kobayashi S, Nishioka T and Miyamoto K:
Structure-activity relationship of flavonoids for inhibition of
epidermal growth factor-induced transformation of JB6 Cl 41 cells.
Mol Carcinog. 46:436–445. 2007. View
Article : Google Scholar : PubMed/NCBI
|