Introduction
Reactive oxygen species (ROS) are a class of
oxygen-derived molecules, which include hydrogen peroxide
(H2O2), super-oxide anion
(O2•−) and hydroxyl radical (•OH).
These elemental molecules are considered to be deleterious or
harmful to cells and tissues; however, it has been reported that
ROS may regulate several cellular events, including gene
expression, differentiation and cell proliferation (1,2). In
addition, ROS may act as secondary messengers to manipulate
distinct signal transduction pathways in the cardiovascular and
pulmonary systems (3,4). ROS are usually generated as
by-products of mitochondrial respiration; however, they may also be
specifically produced by various oxidases (5). The major metabolic pathways contain
superoxide dismutases, which metabolize O2•−
to H2O2 (6).
Further metabolism by catalase or glutathione (GSH) peroxidase,
produces O2 and H2O (7). Cells possess diverse antioxidant
systems to control their redox state, which is important for the
balance between cell survival and death. Oxidative stress may be
the result of overproduction of ROS or downregulation of
antioxidants, which induces irreversible alterations to proteins,
lipids and DNA, resulting in cell death and tissue damage (8–10).
Vascular smooth muscle cells (VSMCs) in the medial
layer of blood vessels are a dynamic component of the vascular
system. When these cells are cultured in normal media, they exhibit
a contractile phenotype for the regulation of blood pressure. In
response to pathological stimuli, VSMCs may undergo hypertrophy or
proliferation, which leads to various vascular diseases, including
hypertension, restenosis and atherosclerosis (3,4).
VSMCs contain several sources of ROS, including NADPH oxidase and
mitochondrial respiration. In VSMCs, ROS mediate several
pathophysiological processes, including growth, migration,
apoptosis and secretion of inflammatory cytokines, and
physiological processes at numerous signaling levels (3,4).
Particularly relevant to the pulmonary vascular system is
modulation of ROS levels by tissue oxygen concentration (4). ROS induce an increase in
intracellular calcium concentration and contraction in human
pulmonary artery smooth muscle cells (HPASMCs), consequently
contributing to the cellular response induced by various
vasoconstrictor stimuli, including hypoxia (4). ROS are involved in the development of
pulmonary hypertension, ultimately inducing right ventricular
failure, which may result in fatality (4). Therefore, it is critical to
understand the various functions of ROS in the physiology and
pathophysiology of VSMCs. In particular, an improved understanding
of how ROS regulate proliferation and apoptosis of VSMCs may allow
for the development of novel strategies to treat or prevent
vascular diseases.
Pyrogallol (PG; benzene-1,2,3-triol) is derived from
hardwood plants. Due to its capability to generate free radicals,
PG is frequently used to investigate the function of
O2•− in several biological systems (11–13).
For example, PG induces O2•−-mediated cell
death in various types of cancer, including lung, gastric and
cervical cancer (13–16). However, to the best of our
knowledge, the effects of PG on normal VSMCs have not yet been
elucidated. PG-induced cytotoxicity in VSMCs in vitro may be
of interest for toxicological research, considering the toxic
potential of PG on VSMCs. In the present study, the effects of
exogenous H2O2 and PG on the cell growth and
death of HPASMCs were investigated, with regards to changes in
intracellular ROS and GSH levels. In addition, the effects of
N-acetyl cysteine (NAC; an established antioxidant) and
L-buthionine sulfoximine (BSO; an inhibitor of GSH synthesis) were
examined on H2O2 or PG-induced HPASMC
death.
Materials and methods
Cell culture
The primary HPASMCs were obtained from PromoCell
GmbH (Heidelberg, Germany) and were maintained in a humidified
incubator containing 5% CO2 at 37°C. HPASMCs were
cultured in Complete Smooth Muscle Cell Growth Medium 2 (PromoCell
GmbH). The cells were grown in 100-mm plastic tissue culture dishes
(Nunc; Sigma-Aldrich, St. Louis, MO, USA), and were washed and
detached with 30 mM Hepes buffered saline solution, trypsin-EDTA
and trypsin neutralization solution (PromoCell GmbH). HPASMCs
between passages four and six were used for subsequent
experiments.
Reagents
H2O2 and PG were purchased
from Sigma-Aldrich. PG was dissolved in water. The Pan-caspase
inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
(Z-VAD-FMK) was obtained from R&D Systems, Inc. (Minneapolis,
MN, USA) and was dissolved in dimethyl sulfoxide (Sigma-Aldrich).
NAC and BSO were obtained from Sigma-Aldrich. NAC was dissolved in
buffer [20 mM Hepes (pH 7.0)] and BSO was dissolved in water. Based
on previous studies (14,17), cells were pretreated with or
without 15 μM Z-VAD-FMK, 2 mM NAC or 10 μM BSO for 1
h at 37°C prior to treatment with H2O2 or
PG.
Cell growth and cell number assays
The growth rate of HPASMCs treated with
H2O2 or PG was indirectly determined
according to 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Sigma-Aldrich) dye absorbance, as previously
described (18). Changes in viable
and dead cell counts were determined by trypan blue cell counting.
Briefly, 5.0×103 cells/well were seeded in 96-well
microtiter plates (Nunc; Sigma-Aldrich) for the MTT assays, and
2×105 cells/well were seeded in 24-well plates (Nunc;
Sigma-Aldrich) for cell counting. Following exposure to the
indicated concentrations of H2O2 or PG (0,
100, 250, 500, 750 and 1,000 μM) for 24 h at 37°C, cells in
the 96-well plates were used for MTT assays, and cells in the
24-well plates were collected with trypsin for trypan blue cell
counting.
Annexin V-fluorescein isothiocyanate
(FITC) staining for cell death detection
Apoptosis was determined by staining cells with
Annexin V-FITC (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA; excitation/emission=488/519 nm) as described
previously (19). Cells were
incubated with the indicated concentrations of
H2O2 or PG (0, 100, 250, 500, 750 and 1,000
μM) for 24 h at 37°C in the presence or absence of
Z-VAD-FMK, NAC or BSO. Annexin V-FITC staining was analyzed using a
FACStar flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA).
Detection of intracellular ROS
levels
Intracellular ROS levels were detected using the
oxidation-sensitive fluorescent probe,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
excitation/emission=495/529 nm; Invitrogen; Thermo Fisher
Scientific, Inc.) as described previously (19). Briefly, 1.0×106 cells/ml
were aliquoted in a flow cytometer tube (BD Biosciences) and were
treated with 500 μM H2O2 or PG in the
presence of 20 μM H2DCFDA at 37°C. The level of
dichlorofluorescein (DCF) fluorescence was evaluated using a
FACStar flow cytometer at 0, 10, 30, 60, 120 and 180 min. DCF (ROS)
levels were expressed as mean fluorescence intensity (MFI). The
levels of mitochondrial O2•− were
specifically detected using MitoSOX Red mitochondrial
O2•− indicator (excitation/emission=510/580
nm; Invitrogen; Thermo Fisher Scientific, Inc.) as previously
described (20). Briefly,
1.0×106 cells in a 60-mm culture dish were incubated
with the indicated concentrations of H2O2 or
PG (0, 100, 250, 500, 750 and 1,000 μM) at 37°C for 24 h in
the presence or absence of Z-VAD-FMK or NAC. Cells were incubated
with 5 μM MitoSOX Red at 37°C for 30 min. MitoSOX Red
fluorescence was assessed using a FACStar flow cytometer and the
levels were expressed as MFI.
Detection of intracellular GSH
levels
GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA;
excitation/emission=522 nm/595 nm; Invitrogen; Thermo Fisher
Scientific, Inc.) as previously described (18,19).
Briefly, 1.0×106 cells/ml were aliquoted in a flow
cytometer tube (BD Biosciences) and were treated at 37°C with 500
μM H2O2 or PG in the presence of 5
μM CMFDA. The level of 5-chloromethyl-fluorescein (CMF)
fluorescence was evaluated using a FACStar flow cytometer at the
indicated times (0, 10, 30, 60, 120 and 180 min). CMF (GSH) levels
were expressed as MFI. In addition, 1.0×106 cells in a
60-mm culture dish were incubated with the indicated amounts of
H2O2 or PG (0, 100, 250, 500, 750 and 1,000
μM) for 24 h at 37°C in the presence or absence of
Z-VAD-FMK, NAC or BSO. Following the treatment, cells were
incubated with 5 μM CMFDA at 37°C for 30 min. CMF
fluorescence was assessed using a FACStar flow cytometer. Negative
CMF staining (GSH depletion) of cells is expressed as the
percentage of (−) CMF cells.
Statistical analysis
Data are presented as the mean ± standard deviation
of three independent experiments. Data were analyzed using Instat
software, version 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Student's t-test, or one-way analysis of variance with Tukey's
honest significant difference test as post-hoc analysis, was used
to determine if there was a significant difference between the
means of various treatment groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of H2O2 and
PG on cell growth and death of HPASMCs
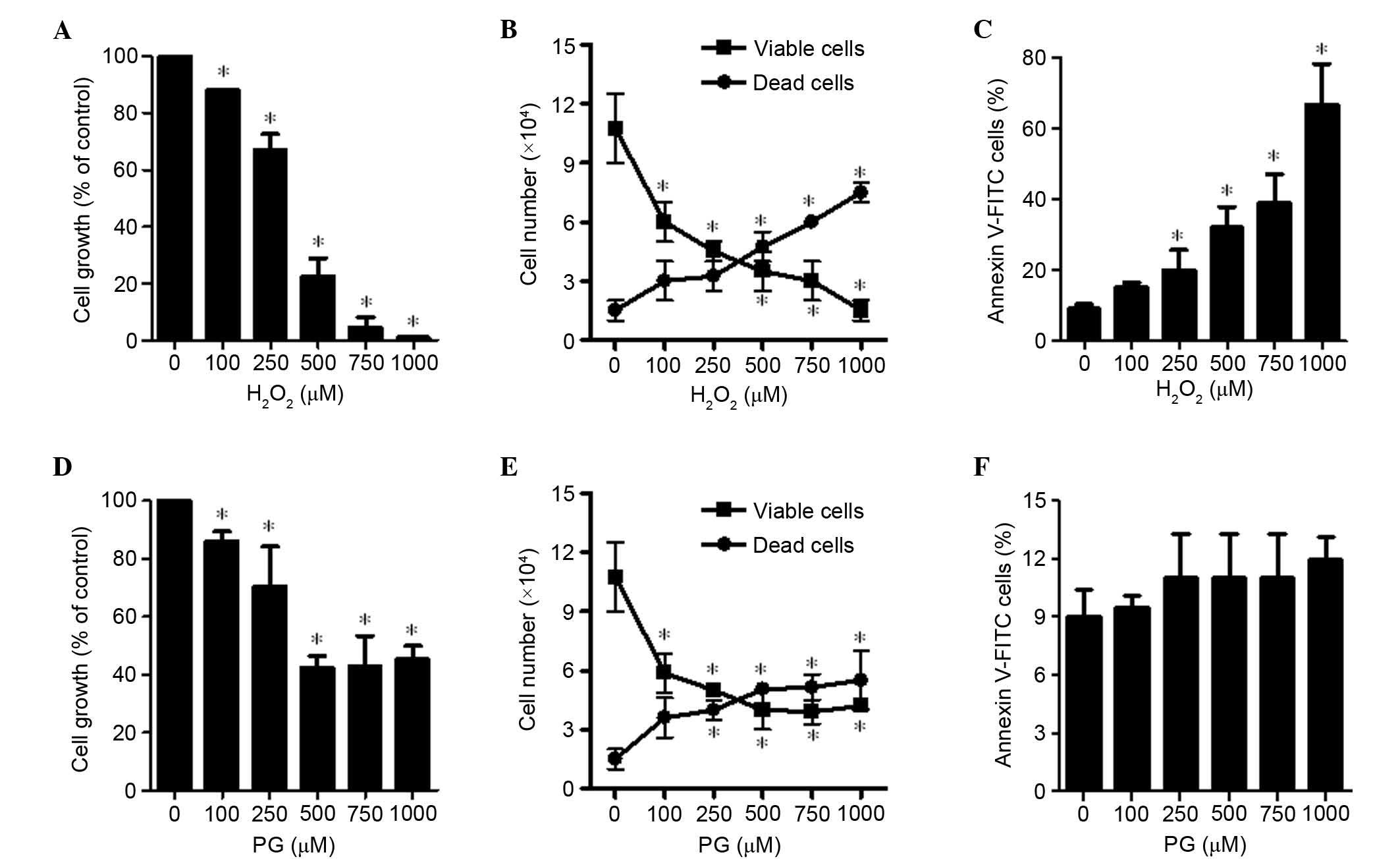
The effects of H2O2 and PG
were examined on HPASMCs 24 h after treatment. Treatment with
H2O2 led to a dose-dependent inhibition of
HPASMCs with a half maximal inhibitory concentration
(IC50) of 250–500 μM (P=0.006; Fig. 1A). In addition, as the
concentration of H2O2 increased from 100 to
1,000 μM the population of viable (trypan blue-negative)
HPASMCs was significantly reduced, whereas the number of dead
(trypan blue-positive) cells increased in a dose-dependent manner
(P<0.001; Fig. 1B). The ratio
of dead cells to viable cells was increased by
H2O2 treatment. Furthermore, the number of
Annexin V-stained cells was increased in a dose-dependent manner
(Fig. 1C). When HPASMCs were
exposed to 500 μM PG, their growth was decreased by ~50%
(P<0.001; Fig. 1D). However,
this effect was not dose-dependent, since 750 and 1,000 μM
PG did not inhibit cell proliferation to the same extent as 500
μM PG (Fig. 1D). In
addition, PG increased the ratio of dead to viable cells; however,
higher doses of PG did not additionally increase the ratio
(Fig. 1E). The doses of PG used
did not significantly increase the proportion of Annexin V-stained
cells (Fig. 1F).
Effects of H2O2 and
PG on ROS levels in HPASMCs
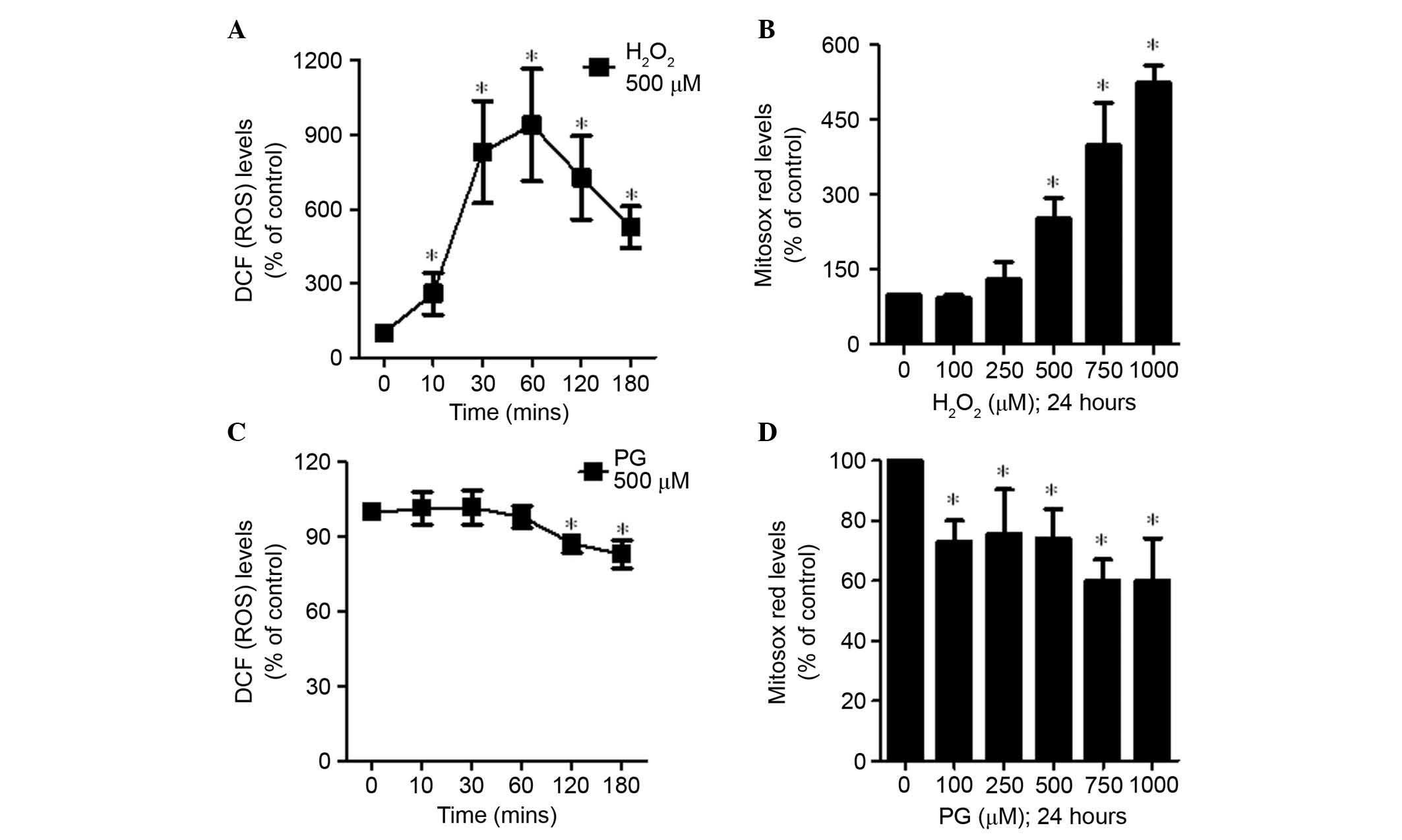
To assess intracellular ROS levels in
H2O2 and PG-treated HPASMCs,
H2DCFDA and MitoSOX Red dyes were used. Treatment with
500 μM H2O2 increased ROS (DCF) levels
gradually from 10 until 60 min, followed by a reduction (Fig. 2A). In addition,
H2O2 treatment led to significantly increased
levels of mitochondrial O2•−, as detected by
MitoSOX Red dye, in a dose-dependent manner (P=0.031; Fig. 2B). Conversely, 500 μM PG did
not increase ROS (DCF) levels in HPASMCs at the 10, 30 or 60 min
time points, and ROS levels were significantly decreased at 120 and
180 min (P=0.047; Fig. 2C). In
addition, the levels of mitochondrial O2•−
were significantly decreased following treatment with PG (P=0.013;
Fig. 2D).
Effects of Z-VAD-FMK, NAC or BSO on cell
death and ROS levels in H2O2 or PG-treated
HPASMCs
For this experiment, 500 μM
H2O2 and PG was selected as a suitable dose
to differentiate the levels of cell death in the presence or
absence of Z-VAD-FMK, NAC or BSO. Application of Z-VAD-FMK and NAC
led to a decrease in apoptotic cell death in
H2O2-treated HPASMCs (Fig. 3A). In addition, these agents
attenuated ROS (DCF) levels in H2O2-treated
HPASMCs from 10 min, with NAC having a strong effect (Fig. 3B). NAC also significantly
attenuated the levels of mitochondrial O2•−
in H2O2-treated HPASMCs (P=0.049; Fig. 3C). In PG-treated HPASMCs, Z-VAD-FMK
did not alter the proportion of Annexin V-stained cells, whereas
NAC and BSO significantly increased the proportion (P=0.010;
Fig. 3D). Z-VAD-FMK and NAC
decreased ROS (DCF) levels in PG-treated HPASMCs at the earlier
time points of 10, 30 and 60 min (Fig.
3E). However, NAC increased the ROS (DCF) levels at 120 and 180
min (Fig. 3E). In addition,
Z-VAD-FMK did not affect the levels of mitochondrial
O2•− in PG-treated HPASMCs, whereas NAC
significantly increased the O2•− levels in
these cells (P=0.004; Fig.
3F).
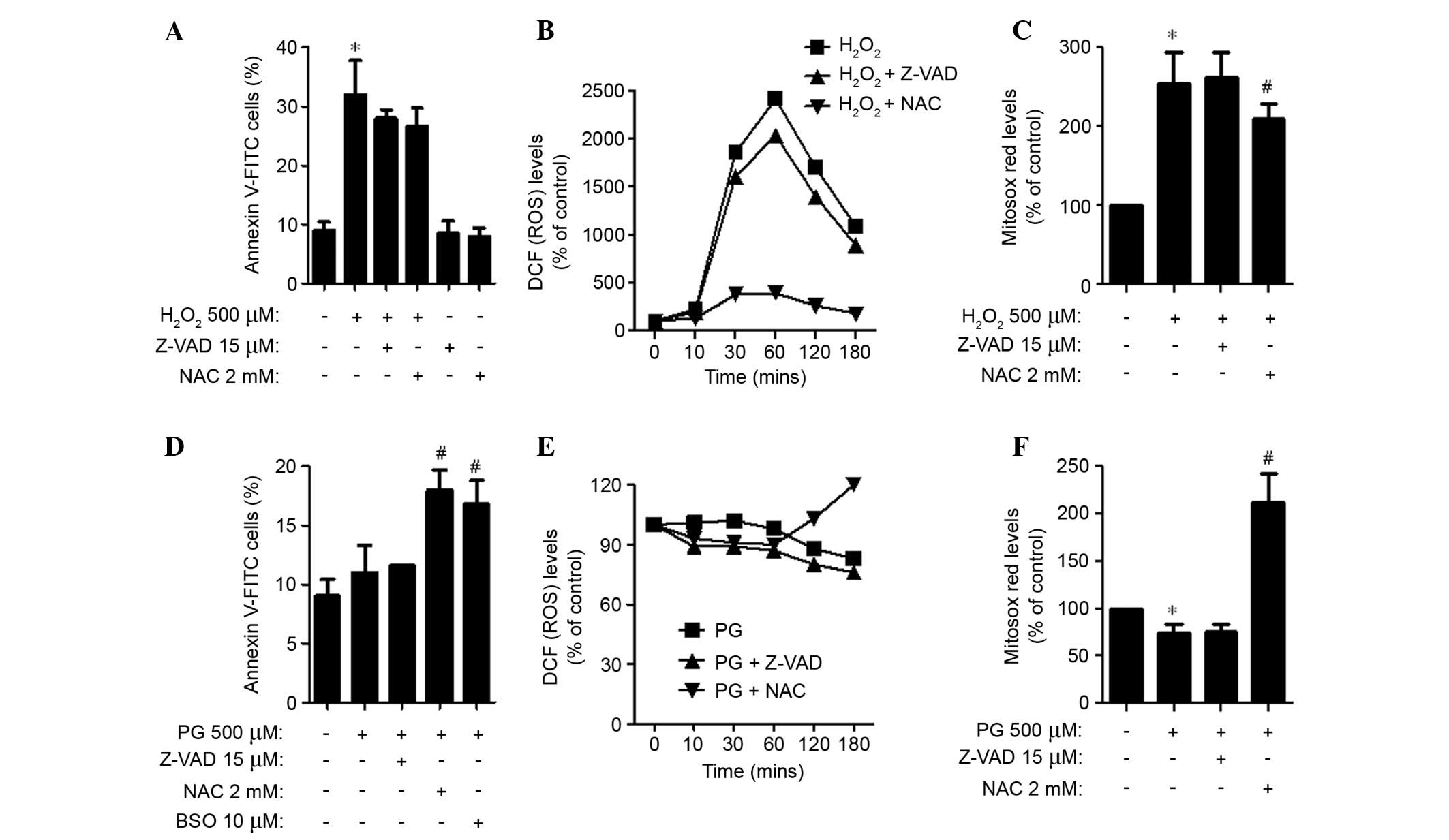 | Figure 3Effects of Z-VAD -FMK, NAC or BSO on
cell death and ROS levels in (A–C) H2O2 or
(D–F) PG-treated human pulmonary artery smooth muscle cells. (A and
D) Percentages of Annexin V-FITC positive cells, as measured using
a FACStar flow cytometer. (B and E) DCF (ROS) levels (% of control)
at the indicated time points. (C and F) Levels of mitochondrial
O2•− (% of control), as detected by MitoSOX
Red dye at 24 h. Data are presented as the mean ± standard
deviation. *P<0.05 vs. the control group (three
independent experiments). #P<0.05 vs. 500 μM
H2O2 or PG groups.
H2O2, hydrogen peroxide; PG, pyrogallol; NAC,
N-acetyl cysteine; BSO, L-buthionine sulfoximine, Z-VAD-FMK,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone; FITC, fluorescein
isothiocyanate; ROS, reactive oxygen species; DCF,
dichlorofluorescein; O2•−, superoxide
anion. |
Effects of H2O2 and
PG on GSH levels in HPASMCs
CMFDA dye was used to evaluate intracellular GSH
levels in H2O2 and PG-treated HPASMCs.
Treatment with 500 μM H2O2 decreased
GSH (CMF) levels at 10 min compared with 0 min (P=0.022); however,
the level was gradually recovered for the remainder of the
treatment time (Fig. 4A).
H2O2 dose-dependently increased the number of
GSH-depleted cells in HPASMCs (Fig.
4B). Conversely, Z-VAD-FMK and NAC reduced GSH depletion in
H2O2-treated HPASMCs (Fig. 4C). When cells were exposed to PG,
GSH (CMF) levels were transiently decreased in HPASMCs at 10 min
(P=0.005; Fig. 4D). The decreased
level was gradually and partially recovered from 30 min onwards;
however, the GSH level was significantly reduced compared with the
control group (P=0.013; Fig. 4D).
PG (500 μM) also significantly increased the number of
GSH-depleted cells (P=0.014; Fig.
4E). However, relatively high doses of PG (750 and 1,000
μM) did not strongly increase the number of GSH-depleted
cells (Fig. 4E). Z-VAD-FMK did not
affect GSH depletion in PG-treated HPASMCs; however, NAC and BSO
significantly intensified GSH depletion in these cells (P=0.042;
Fig. 4F).
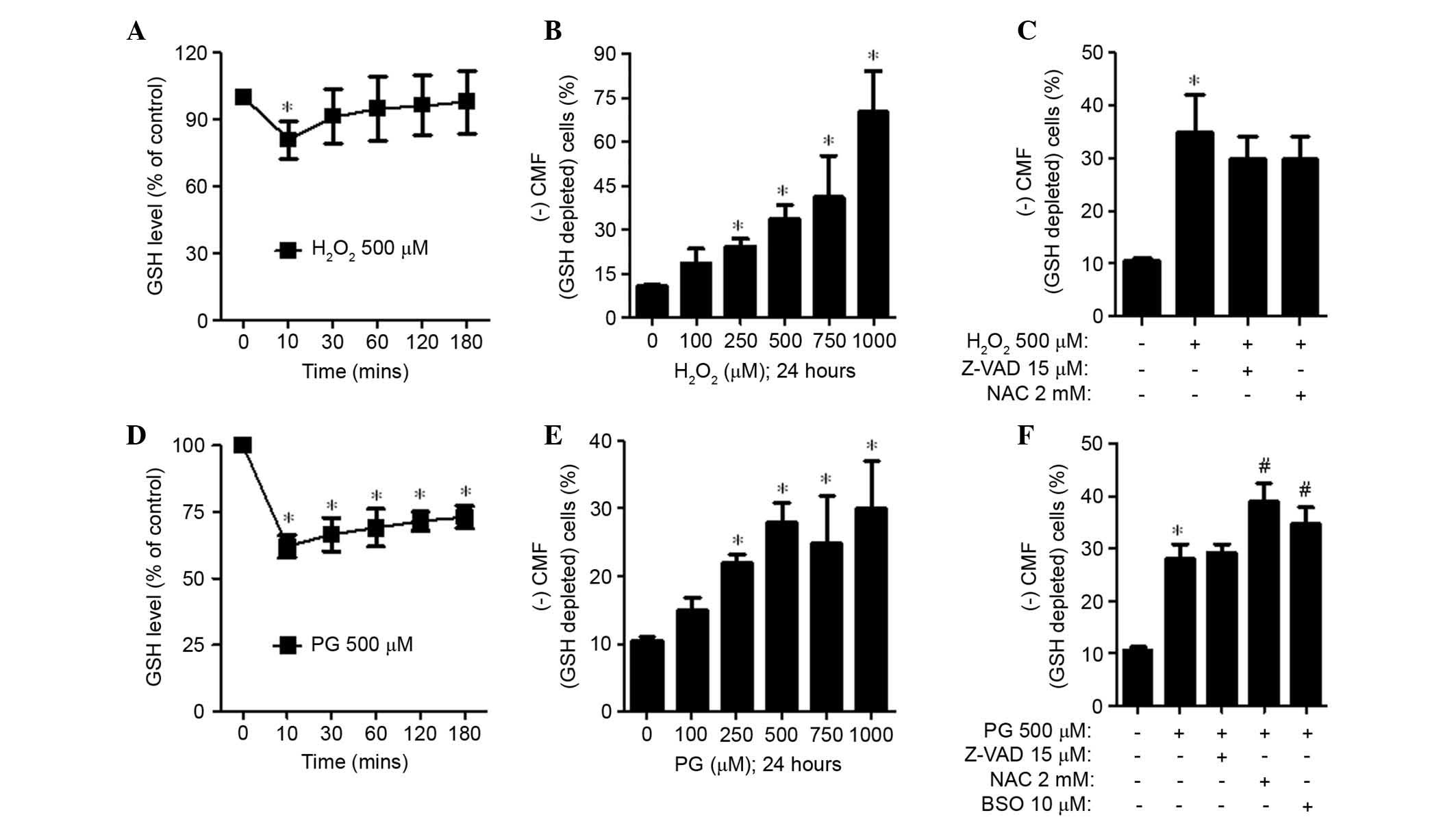 | Figure 4Effects of Z-VAD-FMK, NAC or BSO on
GSH levels in (A–C) H2O2 or (D–F) PG-treated
HPASMCs. GSH levels in HPASMCs were measured using a FACStar flow
cytometer. (A and D) GSH (CMF) levels (% of control) at the
indicated time points. (B, C, E and F) (−)CMF (GSH-depleted) cells
(%) at 24 h. Data are presented as the mean ± standard deviation.
*P<0.05 vs. the control group (three independent
experiments). #P<0.05 vs. 500 μM PG groups.
H2O2, hydrogen peroxide; PG, pyrogalllol;
HPASMCs, human pulmonary artery smooth muscle cells; NAC, N-acetyl
cysteine; BSO, L-buthionine sulfoximine, Z-VAD-FMK,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone; GSH, glutathione;
CMF, 5-chloromethyl-fluorescein. |
Discussion
ROS are involved in various physiological and
pathophysiological processes of VSM systems via the manipulation of
cell proliferation, cell hypertrophy, migration, inflammation,
contraction, and death of VSMCs (3,4). The
present study aimed to elucidate the cytotoxic effects of exogenous
H2O2 and PG on HPASMCs with respect to
changes in intracellular ROS and GSH levels. Previous studies have
reported that exogenous ROS generators lead to VSMC death (21,22).
However, they may also be associated with proliferation of these
cells (23,24), and intracellular ROS are essential
for the survival of VSMCs (25).
The present study demonstrated that H2O2
decreased the proliferation of HPASMCs, with an IC50 of
250–500 μM at 24 h. In addition, these doses were determined
to partially induce apoptosis, as exhibited by the Annexin
V-staining of cells and Z-VAD-FMK treatment. However, the precise
exposure times and concentrations of exogenous oxidants have not
been precisely defined in order to determine their effects on cell
growth, cell survival and death of VSMCs. Notably, the relatively
higher doses of PG (750 and 1,000 μM) did not induce
apoptosis or growth inhibition of HPASMCs. Previous studies have
stated that the IC50 values of PG range between 20 and
50 mM in lung, gastric and cervical cancer cells (13–16);
however, the susceptibility of HPASMCs to PG is low compared with
that of cancer cells. In addition, the susceptibility of HPASMCs is
lower compared with normal endothelial cells (26). Therefore, HPASMCs appeared to be
cytotoxically resistant to PG compared with other cell types,
including cancer cells.
H2O2 and
O2•− are among the primary ROS involved in
various cell signaling pathways. The toxicity of ROS is usually
mediated by •OH (4). In
the present study, ROS levels (as determined by DCF) were increased
in HPASMCs treated with 500 μM H2O2
for 10 min. In addition, H2O2 increased the
levels of mitochondrial O2•−. These results
indicated that exogenous H2O2 may damage the
mitochondria and induce cell death via the generation of
O2•−. Exogenous H2O2
may lead to a greater production of ROS by a self-amplifying
mechanism (ROS-induced ROS generation), which may be converted into
the toxic ROS •OH via the Fenton reaction, leading to
death of HPASMCs. As expected, NAC attenuated cell death in
H2O2-treated HPASMCs and markedly decreased
ROS (DCF) levels at the earlier time points; the levels of
mitochondrial O2•− were also decreased. In
addition, Z-VAD-FMK decreased apoptotic cell death and ROS (DCF)
levels in H2O2-treated HPASMCs. Notably,
although PG has been established to generate
O2•− in biological systems (11–13),
it did not increase ROS (DCF) levels in HPASMCs at the earlier time
points in the present study. In addition, PG significantly
decreased the levels of mitochondrial O2•−.
It is possible that a failure in the generation of
O2•− in PG-treated HPASMCs led to a cytotoxic
resistance to PG compared with other cell types. Notably, NAC
decreased ROS (DCF) levels in PG-treated HPASMCs at 10, 30 and 60
min; however, it increased the levels at 120 and 180 min. NAC
significantly increased the levels of mitochondrial
O2•− and cell death of PG-treated HPASMCs. In
the present study, NAC acted as a pro-oxidant in PG-treated cells,
which led to an increase in O2•− levels, as
opposed to an antioxidant in PG-treated HPASMCs, consequently
intensifying cell death. In addition, a previous study reported
that NAC enhances growth inhibition and death in gallic
acid-treated lung cancer, which was accompanied by an increase in
O2•− levels (17). Therefore, NAC may act as an
antioxidant or a pro-oxidant depending on co-incubated agents and
should be used with caution since it may induce vascular toxicity
via the increased generation of O2•−.
The GSH content of cells is inversely proportionate
to the induction of apoptosis (27). In the present study,
H2O2 treatment led to a dose-dependent
increase in the number of GSH-depleted cells. Conversely, Z-VAD-FMK
and NAC prevented H2O2-induced GSH depletion.
In addition, PG increased the number of GSH-depleted cells.
Treatment with NAC led to the PG-induced death of HPASMCs and
significantly intensified GSH depletion. Furthermore, BSO increased
GSH depletion in PG-treated HPASMCs and enhanced cell death. These
results supported the conclusions of previous studies, which stated
that the intracellular GSH content has an important effect on cell
death (20,28–30).
H2O2 decreased GSH levels at 10 min; however,
the levels were recovered from 30 min onwards. Conversely, the
transient decrease of GSH levels in PG-treated HPASMCs was
significant and the levels did not fully recover. Since PG did not
strongly affect ROS (DCF) levels at the earlier time points, it may
reduce GSH levels as opposed to generating ROS in HPASMCs.
In conclusion, H2O2 and PG
induced growth inhibition and death of HPASMCs via GSH depletion.
However, when exposed to PG, HPASMCs were not significantly
affected compared with other cell types investigated in previous
studies. NAC attenuated cell death and GSH depletion in
H2O2-treated HPASMCs, whereas it intensified
cell death and GSH depletion in PG-treated HPASMCs. The results of
the present study indicated that exogenous oxidants may disturb the
various physiological properties of VSMCs through altering the
balance between cell survival and death. Therefore, it is
imperative that future research efforts aim to define precise
signaling pathways and mechanisms involved in vascular toxicity
triggered by endogenous and exogenous ROS. Future research efforts
may allow for the elucidation of more effective prevention and
therapeutic strategies for vascular diseases in response to
oxidative stress.
Acknowledgments
The present study was supported by a grant from the
National Research Foundation of Korea (NRF) funded by the Korean
government (MSIP; grant no. 2008-0062279).
Abbreviations:
|
HPASM
|
human pulmonary artery smooth
muscle
|
|
PG
|
pyrogallol
|
|
ROS
|
reactive oxygen species
|
|
FITC
|
fluorescein isothiocyanate
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
NAC
|
N-acetyl cysteine
|
|
BSO
|
L-buthionine sulfoximine
|
|
GSH
|
glutathione
|
References
|
1
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Niño A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: A review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perez-Vizcaino F, Cogolludo A and Moreno
L: Reactive oxygen species signaling in pulmonary vascular smooth
muscle. Respir Physiol Neurobiol. 174:212–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: A comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilcox CS: Reactive oxygen species: Roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen TJ, Jeng JY, Lin CW, Wu CY and Chen
YC: Quercetin inhibition of ROS-dependent and -independent
apoptosis in rat glioma C6 cells. Toxicology. 223:113–126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dasmahapatra G, Rahmani M, Dent P and
Grant S: The tyrphostin adaphostin interacts synergistically with
proteasome inhibitors to induce apoptosis in human leukemia cells
through a reactive oxygen species (ROS)-dependent mechanism. Blood.
107:232–240. 2006. View Article : Google Scholar
|
|
10
|
Wallach-Dayan SB, Izbicki G, Cohen PY,
Gerstl-Golan R, Fine A and Breuer R: Bleomycin initiates apoptosis
of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J
Physiol Lung Cell Mol Physiol. 290:L790–L796. 2006. View Article : Google Scholar
|
|
11
|
Saeki K, Hayakawa S, Isemura M and Miyase
T: Importance of a pyrogallol-type structure in catechin compounds
for apoptosis-inducing activity. Phytochemistry. 53:391–394. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yamada J, Yoshimura S, Yamakawa H, Sawada
M, Nakagawa M, Hara S, Kaku Y, Iwama T, Naganawa T, Banno Y, et al:
Cell permeable ROS scavengers, Tiron and Tempol, rescue PC12 cell
death caused by pyrogallol or hypoxia/reoxygenation. Neurosci Res.
45:1–8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim SW, Han YW, Lee ST, Jeong HJ, Kim SH,
Kim IH, Lee SO, Kim DG, Kim SH, Kim SZ and Park WH: A superoxide
anion generator, pyrogallol, inhibits the growth of HeLa cells via
cell cycle arrest and apoptosis. Mol Carcinog. 47:114–125. 2008.
View Article : Google Scholar
|
|
14
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar
|
|
15
|
Han YH, Kim SH, Kim SZ and Park WH:
Pyrogallol inhibits the growth of human pulmonary adenocarcinoma
A549 cells by arresting cell cycle and triggering apoptosis. J
Biochem Mol Toxicol. 23:36–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park WH, Park MN, Han YH and Kim SW:
Pyrogallol inhibits the growth of gastric cancer SNU-484 cells via
induction of apoptosis. Int J Mol Med. 22:263–268. 2008.PubMed/NCBI
|
|
17
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
You BR, Kim SH and Park WH: Reactive
oxygen species, glutathione, and thioredoxin influence suberoyl
bishydroxamic acid-induced apoptosis in A549 lung cancer cells.
Tumour Biol. 36:3429–3439. 2015. View Article : Google Scholar
|
|
19
|
You BR, Shin HR, Han BR and Park WH: PX-12
induces apoptosis in Calu-6 cells in an oxidative stress-dependent
manner. Tumour Biol. 36:2087–2095. 2015. View Article : Google Scholar
|
|
20
|
You BR and Park WH: Arsenic trioxide
induces human pulmonary fibroblast cell death via increasing ROS
levels and GSH depletion. Oncol Rep. 28:749–757. 2012.PubMed/NCBI
|
|
21
|
Li PF, Dietz R and von Harsdorf R:
Reactive oxygen species induce apoptosis of vascular smooth muscle
cell. FEBS Lett. 404:249–252. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnson TM, Yu ZX, Ferrans VJ, Lowenstein
RA and Finkel T: Reactive oxygen species are downstream mediators
of p53-dependent apoptosis. Proc Natl Acad Sci USA. 93:11848–11852.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rao GN and Berk BC: Active oxygen species
stimulate vascular smooth muscle cell growth and proto-oncogene
expression. Circ Res. 70:593–599. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rao GN, Lassègue B, Griendling KK and
Alexander RW: Hydrogen peroxide stimulates transcription of c-jun
in vascular smooth muscle cells: Role of arachidonic acid.
Oncogene. 8:2759–2764. 1993.PubMed/NCBI
|
|
25
|
Brown MR, Miller FJ Jr, Li WG, Ellingson
AN, Mozena JD, Chatterjee P, Engelhardt JF, Zwacka RM, Oberley LW,
Fang X, et al: Overexpression of human catalase inhibits
proliferation and promotes apoptosis in vascular smooth muscle
cells. Circ Res. 85:524–533. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Pyrogallol-induced endothelial cell death is related
to GSH depletion rather than ROS level changes. Oncol Rep.
23:287–292. 2010.
|
|
27
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Kim SZ, Kim SH and Park WH:
Induction of apoptosis in arsenic trioxide-treated lung cancer A549
cells by buthionine sulfoximine. Mol Cells. 26:158–164.
2008.PubMed/NCBI
|
|
29
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of arsenic trioxide-induced apoptosis in HeLa cells by
diethyldithiocarbamate or buthionine sulfoximine. Int J Oncol.
33:205–213. 2008.PubMed/NCBI
|
|
30
|
Wu XX, Ogawa O and Kakehi Y: Enhancement
of arsenic trioxide-induced apoptosis in renal cell carcinoma cells
by L-buthionine sulfoximine. Int J Oncol. 24:1489–1497.
2004.PubMed/NCBI
|