Introduction
Hypoxic pulmonary hypertension (HPH) is a severe
disease detrimental to human health. The pathophysiology of HPH is
contraction and proliferation of human pulmonary artery smooth
muscle cells (HPASMCs) as well as small pulmonary arterial
remodeling (1). The pathogenesis
mechanism of HPH is not well characterized and no effective therapy
for HPH is currently available. Although calcium channel
antagonists, prostacyclins, endothelin receptor antagonists and
phosphodiesterase type 5 (PDE5) inhibitors may treat pulmonary
hypertension, these drugs did not inhibit or reverse pulmonary
vascular remodeling due to hypoxia (2,3).
Therefore, clinical use of these expensive drugs is limited.
Krick et al reported that the K+
channels on the HPASMC membrane were closely associated with the
vasomotor tone (4). The activity
of K+ channels on the HPASMC membrane decreased in
hypoxia, leading to increased intracellular K+
concentration, decreased activities of caspases and nuclease, and
decreased apoptosis of HPASMCs. On the other hand, decreased
activity of K+ channels may depolarize the cell
membrane, open voltage-dependent Ca2+ channels and
increase Ca2+ influx (5). Furthermore, the release of
Ca2+ from sarcoplasmic reticulum increased free
intracellular Ca2+ ([Ca2+]cyt) and
promoted vascular smooth muscle contraction (2,3).
Increase of [Ca2+]cyt is a second messenger
for many proliferation factors, and may induce the transition from
synthesis phase to mitosis phase, promoting cell proliferation and
aggravating pulmonary vascular remodeling (6–8).
Currently, ATP-sensitive K+
(KATP) is the only known compensatory open K+
channel in ischemia and hypoxia, representing an important
compensatory mechanism (9,10). The KATP channels are a
group of widely distributed inward rectifier K+
channels, which are heterologous octamers
[(SUR/Kir6.x)4] consisting of the inward rectifier
K+ channel Kir6.x family and sulfonylurea receptor (SUR)
family (11).
Pulmonary artery endothelial cells may be injured in
chronic hypoxia, creating an imbalance of vasoactive substances
secreted by endothelial cells, and may affect the pulmonary artery
(12). The expression of
vasoconstrictors increased ET-1, angiotensin II (Ang II) and
5-hydroxytryptamine (5-HT), while the expression of vasodilators
decreased prostaglandin I2 (PGI2), calcitonin
gene-related peptide, adenosine and nitric oxide (NO). These
vasoconstrictors act on KATP channels to promote
pulmonary artery smooth muscle contraction and increase pulmonary
vascular resistance, thereby promoting HPASMC proliferation and
pulmonary vascular remodeling leading to pulmonary hypertension.
KATP channel is an important pathway of pulmonary
hypertension. Therefore, KATP channel opener is a
promising novel drug for HPH.
Iptakalim (IPT) is a novel KATP channel
opener and glibenclamide (GLI) is an antagonist to KATP
channel. Wang reported that IPT could increase outward potassium
current in rat pulmonary artery smooth muscle cell membrane
(13). This could not only prevent
rat pulmonary hypertension induced by hypoxia or ET-1, but also
reverse pulmonary vascular remodeling and right ventricular
hypertrophy in hypoxic rats, thus preventing rat pulmonary
hypertension effectively (14). In
addition, IPT could open KATP channels on rabbit
pulmonary artery smooth muscle cell membrane, which inhibits
Ca2+ influx and decrease cytoplasmic Ca2+
concentration, thus inhibiting ET-1-induced rabbit pulmonary artery
smooth muscle cell contraction and proliferation (15).
Cell proliferation and apoptosis maintain
homeostasis in normal cells. In B-cell lymphoma 2 (Bcl-2) gene
family, Bcl-2 is the first gene found to be associated with cell
proliferation and apoptosis. Bcl-2 proteins are located on inner
mitochondrial membrane, endoplasmic reticulum and nuclear membrane.
The main biological function of Bcl-2 is to prolong cell life,
enhancing cell resistance to various apoptosis-inducing factors.
Since the finding by Oltvai et al (16) that Bcl-2-associated X protein (Bax)
could accelerate apoptosis, the regulation of apoptosis by Bax has
been under investigation. Bax formed heterodimers with
anti-apoptosis Bcl-2 to inhibit Bcl-2 and induce cell apoptosis.
Higher Bax/Bcl-2 ratio promoted cell apoptosis and vice versa
(17).
In the present study, a cell proliferation/toxicity
detection kit [Cell Counting Kit-8 (CCK-8)] and
5-ethynyl-2′-deoxyuri-dine (EdU) incorporation assay were used to
determine the effect on HPASMC proliferation by ET-1, western
blotting to evaluate the expression of Bcl-2 and Bax to detect
apoptosis. The potential therapeutic effect of IPT on HPH was
clarified on the cellular level.
Materials and methods
Cell culture
HPASMCs were purchased from Sciencell Research
Laboratories (Carlsbad, CA, USA). Cells were routinely maintained
in the medium containing 2% fetal bovine serum, 1% smooth muscle
cell growth supplement (SMCGS) and 1% penicillin/streptomycin, and
incubated in a humidified incubator at 37°C with 5% CO2.
The medium was replaced every other day. Cells were passaged every
2 days, and the cells of the 2nd-6th passage were used in the
study.
Reagents
ET-1 (Sigma, St. Louis, MO, USA), IPT (Institute of
Pharmacology and Toxicology, Academy of Military Medical Sciences),
GLI (Sigma), CCK-8 kit (Beyotime Institute of Biotechnology,
Shanghai, China), rabbit anti-Bax polyclonal antibody and rabbit
anti-Bcl-2 polyclonal antibody (both from Cell Signaling
Technology, Inc., Danvers, MA, USA) were used.
HPASMC viability by CCK-8 assay
Adherent HPASMCs in the logarithmic phase were
digested and inoculated in 96-well plates, 100 μl/well in
triplicate. Drugs were added after cell adherence for 24 h. CCK-8
solution was prepared in serum-free SMCM medium and 100 μl
CCK-8 medium was added into wells instead of medium followed by 2-h
incubation. Subsequently, the absorbance values were measured at
450 and 620 nm with a microplate reader (Thermo Fisher, Shanghai,
China).
Cell viability was calculated as (OD450 -
OD620 in treatment group)/(OD450 -
OD620 in control group) × 100%. Experiments were
performed in triplicate independently.
HPASMC proliferation by EdU incorporation
assay
EdU incorporation assay was performed following the
manufacturer's instructions. Briefly, HPASMCs in the logarithmic
phase were digested and inoculated in a 24-well plate with
coverslip. Drugs of a pre-determined concentration were added after
cell adherence, 50 μmol/l EdU medium were added for 2-h
incubation. EdU medium was then discarded, the cells were washed
with phosphate-buffered saline (PBS) for 5 min × 2, fixed with 4%
paraformaldehyde at room temperature (RT) for 30 min, decolorized
in 2 mg/ml glycine for 5 min and washed with PBS for 5 min. Cell
permeation was performed by incubating the cells with 0.5% Triton
X-100 for 10 min followed by washing. Staining buffer (1X Apollo)
was added and incubated in the dark at RT for 30 min. The staining
buffer was discarded, permeation reagent (PBS containing 0.5%
Triton X-100) was added and incubated for 10 min × 2–3 times.
Permeation reagent was discarded, the cells were washed with
methanol for 5 min, 1–2 times and then washed with PBS for 5 min.
Staining solution (1X Hoechst 33342) was freshly prepared and added
to wells for 30-min incubation in the dark at RT. The cells were
washed 1–3 times and observed under a fluorescence microscope
(EUROIMMUN, Lubeck, Germany). Cell images were taken randomly and
the percentage of positive cells was calculated. The assay was
performed in triplicate.
Western blotting for Bax and Bcl-3 in
HPASMCs to assay apoptosis
Cells were collected for protein extraction and
loaded on SDS-PAGE gel for electrophoresis under constant voltage
70 V, and 120 V after protein migration into the separating gel.
The proteins were transferred to PVDF membrane (Runwelltac,
Shanghai, China) under a constant current 300 mA for 90 min. The
membrane was blocked in 5% skimmed milk for 2 h. Primary antibody
(Bax, Bcl-2, 1:1,000; β-actin, 1:5,000) was added for incubation
overnight in 4°C. The following day, the membrane was washed with
TBST for 10 min × 3 and then secondary antibody (horseradish
peroxidase (HRP)-labelled goat anti-rabbit antibody, 1:10,000) was
added and incubated at RT for 1 h. The membrane was washed with
TBST for 10 min × 3 and subjected to color development.
Statistical analysis
SPSS 13.0 software (IBM Corporation, Armonk, NY,
USA) was used for statistical analysis. Quantitative data are
presented as mean ± SD. One-way analysis of variance (ANOVA) was
used for comparison between multiple groups, and the LSD method was
used for comparison between two groups. Independent sample t-test
was used in comparison between two groups. P<0.05 was considered
of statistical significance.
Results
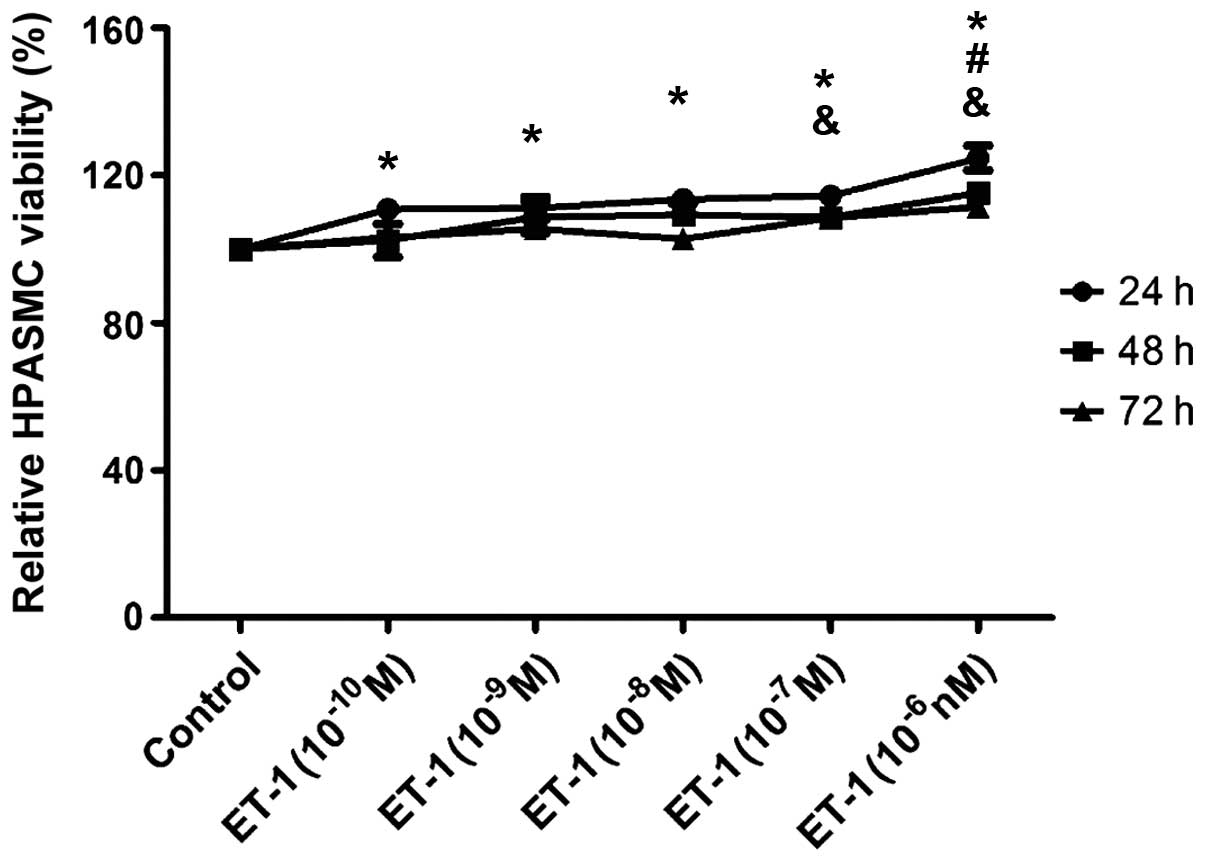
Effect on HPASMC viability by ET-1
HPASMCs were incubated with ET-1 10−10,
10−9, 10−8, 10−7 and
10−6 M for 24, 48 and 72 h. As shown in Fig. 1, ET-1 increases HPASMC viability
and the effect was enhanced with increasing ET-1 concentration. In
addition, with different ET-1 concentrations, the HPASMC viability
at 48 and 72 h was similar to that at 24 h and the effect on
HPASMCs by ET-1 was not significantly time-dependent. Therefore, we
selected ET-1 at 10−6 M concentration for 24 h for
further investigatation.
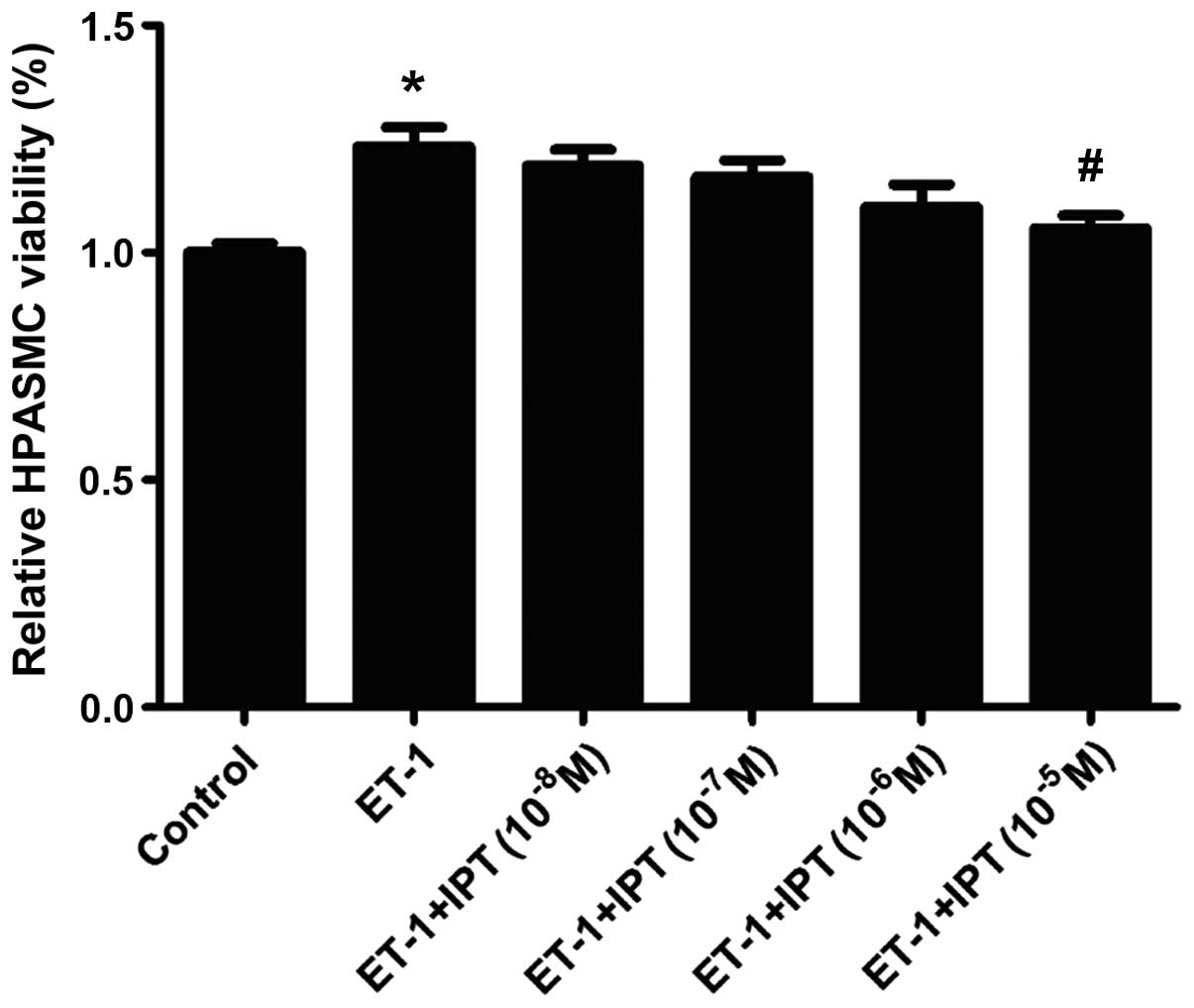
Effect on ET-1-induced HPASMC
proliferation by IPT
HPASMCs were incubated with IPT 10−8,
10−7, 10−6 and 10−5 M, and ET-1
10−6 M for 24 h. CCK-8 assay was used to evaluate the
change of HPASMC viability after incubation with varying
concentrations of IPT. As shown in Fig. 2, IPT inhibited ET-1 induced
increasing HPASMC viability and this effect was enhanced with
increasing IPT concentration. In comparison with ET-1 alone, IPT
(10−5 M)+ET-1 treatment decreased HPASMC viability
significantly (P<0.05).
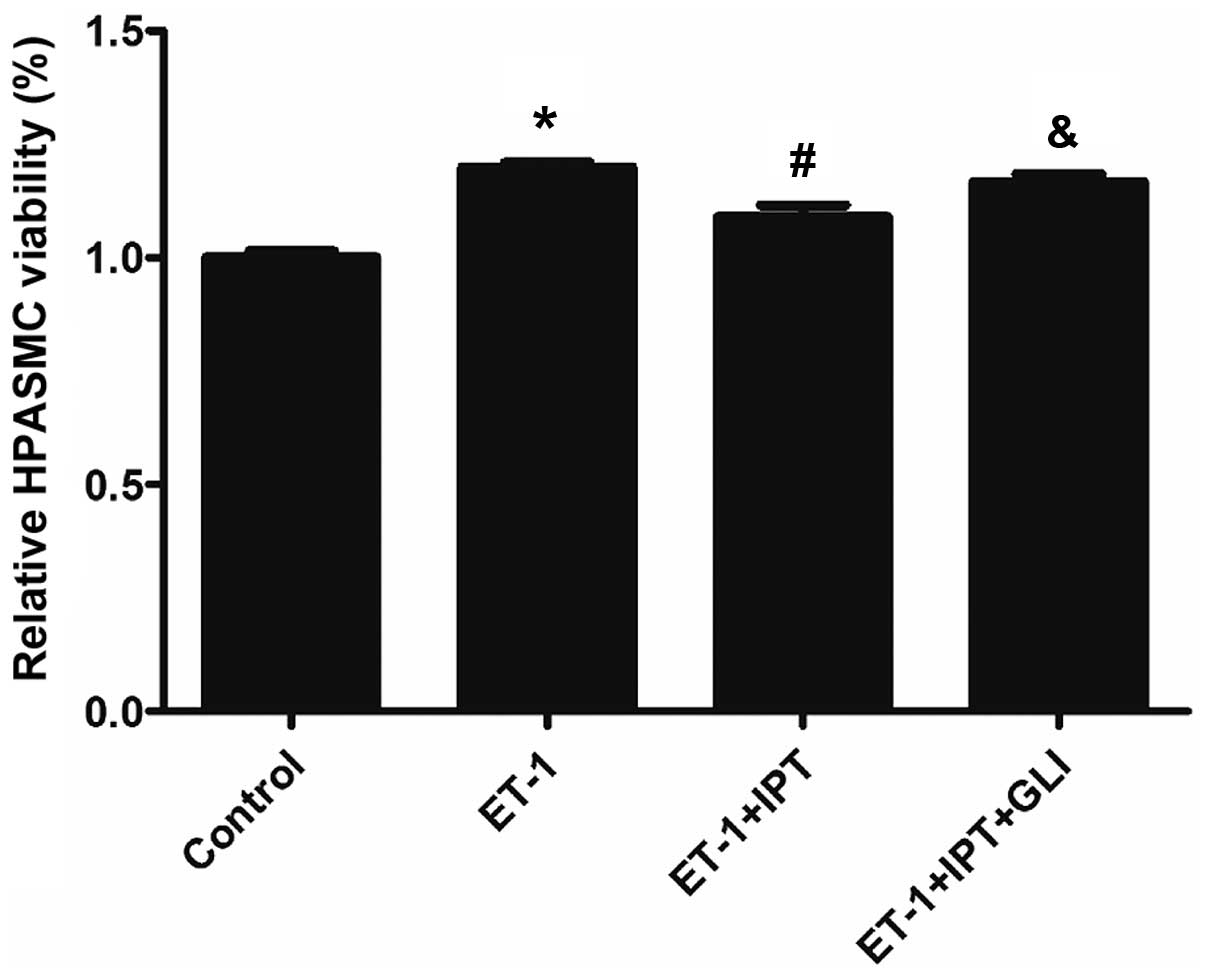
Effect on the ET-1-induced increase of
HPASMC viability by IPT after blocking K+ channel
K+ channel blocker GLI was used to block
IPT-induced K+ channel opening, to determine whether IPT
was able to inhibit the ET-1-induced increase of HPASMC viability.
Four groups were included in this analysis, i.e., control, ET-1,
ET-1+IPT and ET-1+IPT+GLI groups, with ET-1 at 10−6 M,
IPT at 10−5 M, GLI at 10−5 M concentration.
In the ET-1+IPT+GLI group, GLI was added 30 min prior to ET-1 and
IPT, and the incubation time was 24 h. As shown in Fig. 3, after blocking K+
channel by GLI, HPASMC viability in the ET-1+IPT+GLI group was
significantly higher than that in the ET-1+IPT group (P<0.05).
This result demonstrated that GLI partially reversed the effect on
ET-1-induced HPASMC viability by IPT, indicating that the
inhibition of the ET-1-induced increase of HPASMC viability by IPT
may occur through the opening K+ channels.s
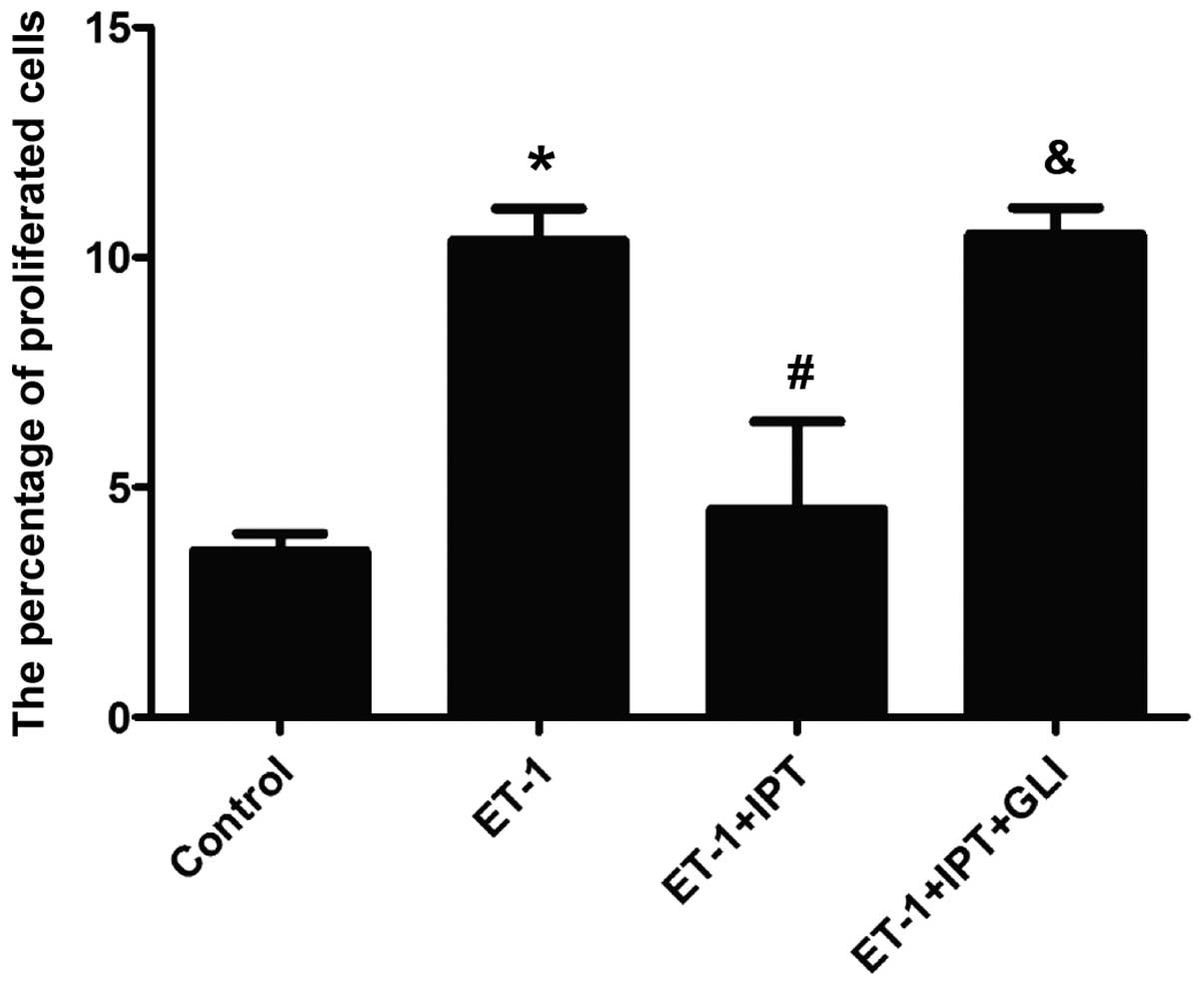
Twenty-four hours after incubation, EdU staining was
performed for HPASMCs in all 4 groups. The percentage of
EdU-stained cells in total cells was calculated on the basis of 5
fields (20X) randomly selected in each group. As shown in Figs. 4 and 5, the percentage of proliferated cells in
the control, ET-1, ET-1+IPT and ET-1+IPT+GLI groups was 3.61±0.38,
10.35±0.71, 4.51±1.92 and 10.50±0.58%, respectively. After the
addition of GLI, the percentage of proliferated cells in the
ET-1+IPT+GLI group was significantly higher than that in the
ET-1+IPT group (P<0.05).
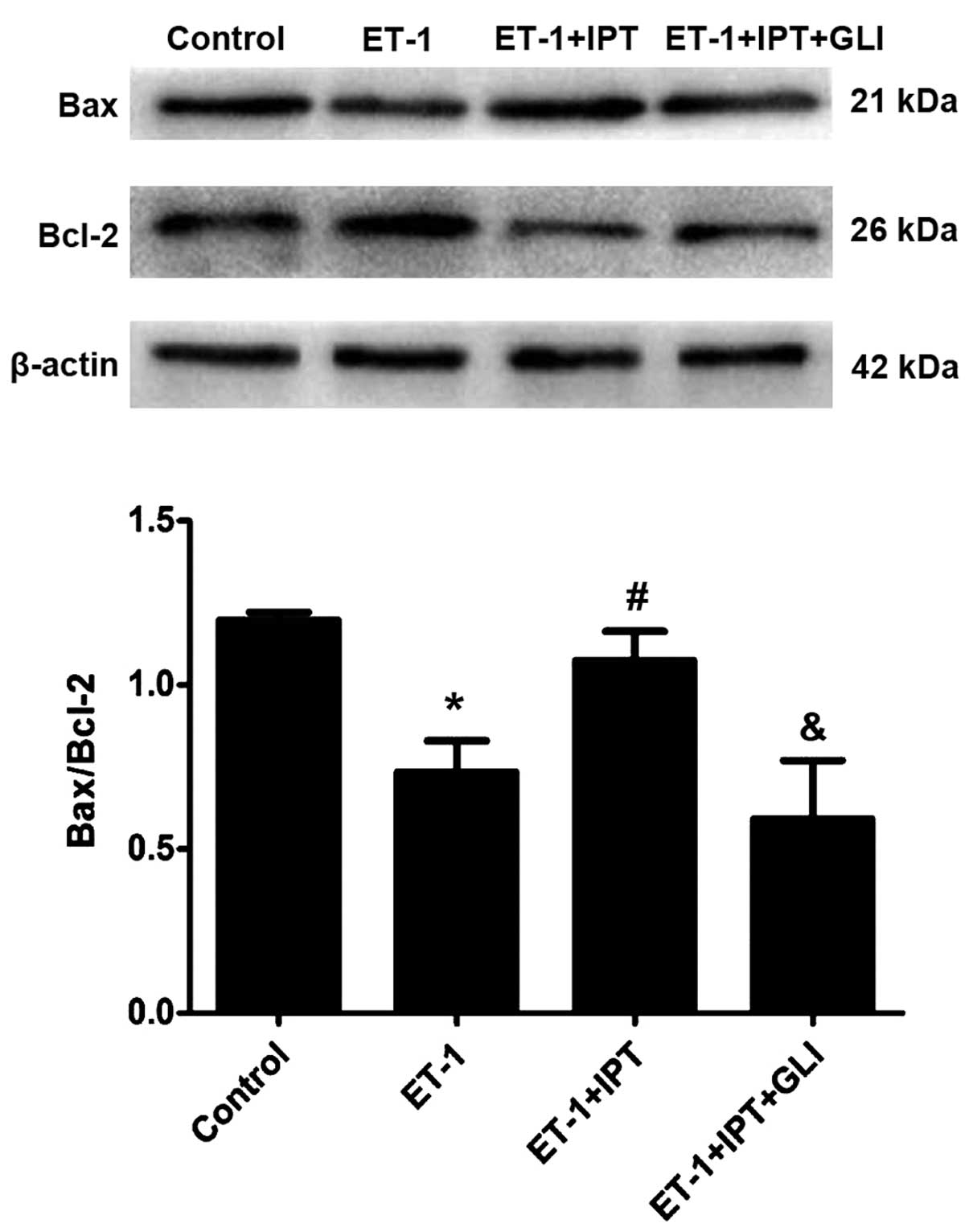
Effect on ET-1-induced change of
apoptotic protein expression in HPASMCs by IPT after blocking
K+ channel
After incubation with drugs for 24 h, the expression
of apoptosis-related Bax and Bcl-2 in HPASMCs was evaluated by
western blotting. As shown in Fig.
6, ET-1 downregulated Bax and upregulated Bcl-2, led to lower
Bax/Bcl-2 ratio (P<0.05) vs. control group. IPT upregulated Bax
and downregulated Bcl-2, leading to higher Bax/Bcl-2 ratio
(p<0.05) vs. ET-1 group. GLI could block K+ channels,
downregulated Bax and upregulated Bcl-2, leading to lower Bax/Bcl-2
ratio (P<0.05) vs. ET-1+IPT group.
Discussion
Pulmonary hypertension is a disease detrimental to
human health, its incidence is only second to hypertension and
coronary heart disease in cardiovascular diseases (18–20).
The pathophysiological characteristics are predominantly pulmonary
vasoconstriction and pulmonary vascular remodeling. Pulmonary
hypertension potentially causes right-sided heart failure and death
(21). In China, the incidence of
pulmonary hypertension due to hypoxia and respiratory diseases is
on the increase, and the reasons include: i) smoking, chronic
bronchitis and chronic obstructive pulmonary disease (COPD) due to
cigarette smoking, common pulmonary diseases that lead to HPH; ii)
the incidence of respiratory disorder during sleep is increasing
due to an increase in obese population, with pulmonary hypertension
due to higher pulmonary vascular resistance; and iii) high altitude
pulmonary hypertension, a common chronic hypoxic pulmonary disease
in China as compared to other countries (22). Therefore, the investigation of
pathogenesis mechanism underlying HPH and development of new
therapeutic drugs are of important clinical significance.
The pathogenesis mechanism of HPH is not well
established. Currently, the main mechanism underlying higher
pulmonary vascular resistance due to chronic hypoxia is pulmonary
vascular proliferation and remodeling (23). On the one hand, hypoxia induces the
production of reactive oxygen species in mitochondria and inhibits
the activity of voltage-gated K+ channels on the HPASMC
membrane leading to depolarization of the cell membrane and influx
of extracellular Ca2+, which may cause vascular smooth
muscle cell contraction. On the other hand, hypoxia may cause
dysfunction in pulmonary artery endothelial cells, leading to the
decreased production of vasodilator factors (NO and prostacyclin)
and overexpression of vasoconstrictive and pro-proliferation
factors (thromboxane A2 and ET-1). An imbalance between
vasodilators and vasoconstrictors may cause increased small
pulmonary arterial tone. The proliferation of endothelial cells,
smooth muscle cells and fibroblasts may lead to small pulmonary
artery remodeling and increased synthesis of extracellular matrix,
such as collagen, elastin and fibronectin (24).
ET-1 is a vasoconstrictor synthetized and secreted
by pulmonary vascular endothelial cells. Excess ET-1 may cause
pulmonary vascular smooth muscle cell contraction and proliferation
(25,26). ET-1 binds to the endothelin-A
(ETA) receptor, inhibit G protein (Gi), adenylate
cyclase or phospholipase C (PLC), and KATP channels
(27), leading to a decreased
outflow of intracellular K+, cell membrane
depolarization, voltage-dependent Ca2+ channel opening,
increased influx of extracellular Ca2+, release of
Ca2+ in sarcoplasmic reticulum, which would increase
free intracellular Ca2+, promoting the expression of the
genes related to cell proliferation (c-Myc and c-Fos), and inducing
cell proliferation (28).
In the present study, we stimulated HPASMCs with
ET-1 to develop the model of HPASMC proliferation. We found that
IPT inhibited ET-1 and induced HPASMC proliferation to promote
HPASMC apoptosis. To verify whether the inhibition of ET-1-induced
HPASMC proliferation by IPT was mediated through KATP
channels, we blocked KATP channel with KATP
channel blocker GLI and evaluated the effect on IPT. As shown in
Figs. 4 and 5, in ET-1+IPT+GLI group, GLI could block
IPT, thus IPT could not inhibit ET-1-induced HPASMC proliferation.
This result confirms that IPT could inhibit ET-1 induced HPASMC
proliferation through opening KATP channels.
It was reported that mitochondria was sensitive to
hypoxia, mitochondrial dysfunction was closely associated with cell
apoptosis and played important roles in the pathogenesis of HPH.
Hypoxia could inhibit the release of cytochrome c
(cyt-c) from mitochondria into cytoplasm, thus inhibiting
the mitochondrial apoptotic pathway, leading to HPASMC
proliferation and pulmonary hypertension (29–32).
The release of mitochondrial cyt-c and other pro-apoptotic
components was regulated by Bcl-2 family, upstream of the
mitochondrial apoptosis pathway. Bcl-2 family proteins were
classified into anti-apoptosis proteins (Bcl-2, Bcl-xL and Bcl-W)
and pro-apoptosis proteins (Bax, Bcl-Xs and Bak). Among others,
Bcl-2 and Bax were similar in structure and antagonistic to each
other (33). Bcl-2 and Bax played
important regulatory roles in the mitochondrial apoptosis pathway.
The present findings demonstrate that ET-1 downregulated Bax and
upregulated Bcl-2 in HPASMCs to inhibit cell apoptosis while IPT
reversed the effect of ET-1, indicating that IPT regulated the
proliferation and apoptosis of HPASMCs by increasing the Bax/Bcl-2
ratio. KATP channel inhibitor GLI could partially
reverse the effect of IPT, indicating that IPT could increase
Bax/Bcl-2 ratio through opening the KATP channel, and
inhibited ET-1- induced HPASMC proliferation.
In conclusion, IPT inhibited ET-1-induced HPASMC
proliferation and promoted apotosis through opening KATP
channels, therefore, it may be a promising novel drug for HPH.
References
|
1
|
Mandegar M and Yuan JX: Role of
K+ channels in pulmonary hypertension. Vascul Pharmacol.
38:25–33. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saji T, Myoishi M, Sugimura K, Tahara N,
Takeda Y, Fukuda K, Olschewski H, Matsuda Y, Nikkho S and Satoh T:
Efficacy and safety of inhaled iloprost in japanese patients with
pulmonary arterial hypertension- insights from the IBUKI and AIR
studies. Circ J. 80:835–842. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Provencher S and Granton JT: Current
treatment approaches to pulmonary arterial hypertension. Can J
Cardiol. 31:460–477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krick S, Platoshyn O, Sweeney M, Kim H and
Yuan JX: Activation of K+ channels induces apoptosis in
vascular smooth muscle cells. Am J Physiol Cell Physiol.
280:C970–C979. 2001.PubMed/NCBI
|
|
5
|
Shimoda LA, Wang J and Sylvester JT:
Ca2+ channels and chronic hypoxia. Microcirculation.
13:657–670. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonnet S and Archer SL: Potassium channel
diversity in the pulmonary arteries and pulmonary veins:
implications for regulation of the pulmonary vasculature in health
and during pulmonary hypertension. Pharmacol Ther. 115:56–69. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang J, Juhaszova M, Rubin LJ and Yuan XJ:
Hypoxia inhibits gene expression of voltage-gated K+
channel α subunits in pulmonary artery smooth muscle cells. J Clin
Invest. 100:2347–2353. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moudgil R, Michelakis ED and Archer SL:
The role of K+ channels in determining pulmonary
vascular tone, oxygen sensing, cell proliferation, and apoptosis:
implications in hypoxic pulmonary vasoconstriction and pulmonary
arterial hypertension. Microcirculation. 13:615–632. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wiener CM, Banta MR, Dowless MS, Flavahan
NA and Sylvester JT: Mechanisms of hypoxic vasodilation in ferret
pulmonary arteries. Am J Physiol. 269:L351–L357. 1995.PubMed/NCBI
|
|
10
|
Standen NB and Quayle JM: K+
channel modulation in arterial smooth muscle. Acta Physiol Scand.
164:549–557. 1998. View Article : Google Scholar
|
|
11
|
Tang Y, Long CL, Wang RH, Cui W and Wang
H: Activation of SUR2B/Kir6.1 subtype of adenosine
triphosphate-sensitive potassium channel improves pressure
overload-induced cardiac remodeling via protecting endothelial
function. J Cardiovasc Pharmacol. 56:345–353. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hampl V, Bíbová J, Banasová A, Uhlík J,
Miková D, Hnilicková O, Lachmanová V and Herget J: Pulmonary
vascular iNOS induction participates in the onset of chronic
hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol.
290:L11–L20. 2006. View Article : Google Scholar
|
|
13
|
Wang H: Pharmacological characteristics of
the novel antihypertensive drug, iptakalim hydrochloride, and its
molecular mechanisms. Drug Dev Res. 58:65–68. 2003. View Article : Google Scholar
|
|
14
|
Xie W, Wang H, Wang H and Hu G: Effects of
iptakalim hydrochloride, a novel KATP channel opener, on
pulmonary vascular remodeling in hypoxic rats. Life Sci.
75:2065–2076. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie W, Wang H, Ding J, Wang H and Hu G:
Anti-proliferating effect of iptakalim, a novel KATP
channel opener, in cultured rabbit pulmonary arterial smooth muscle
cells. Eur J Pharmacol. 511:81–87. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin XM, Oltvai ZN and Korsmeyer SJ: BH1
and BH2 domains of Bcl-2 are required for inhibition of apoptosis
and heterodimerization with Bax. Nature. 369:321–323. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Humbert M, Sitbon O, Chaouat A, Bertocchi
M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot
F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M,
Hachulla E and Simonneau G: Pulmonary arterial hypertension in
France: Results from a national registry. Am J Respir Crit Care
Med. 173:1023–1030. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peacock AJ, Murphy NF, McMurray JJ,
Caballero L and Stewart S: An epidemiological study of pulmonary
arterial hypertension. Eur Respir J. 30:104–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duffels MG, Engelfriet PM, Berger RM, van
Loon RL, Hoendermis E, Vriend JW, van der Velde ET, Bresser P and
Mulder BJ: Pulmonary arterial hypertension in congenital heart
disease: An epidemiologic perspective from a Dutch registry. Int J
Cardiol. 120:198–204. 2007. View Article : Google Scholar
|
|
21
|
van de Veerdonk MC, Marcus JT, Westerhof
N, de Man FS, Boonstra A, Heymans MW, Bogaard HJ and Vonk NA: Signs
of right ventricular deterioration in clinically stable patients
with pulmonary arterial hypertension. Chest. 147:1063–1071. 2015.
View Article : Google Scholar
|
|
22
|
Zhang S, Li G, Tian L, Guo Q and Pan X:
Short-term exposure to air pollution and morbidity of COPD and
asthma in East Asian area: A systematic review and meta-analysis.
Environ Res. 148:15–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hassoun PM, Mouthon L, Barberà JA,
Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML,
Michelakis ED, Morrell NW, et al: Inflammation, growth factors, and
pulmonary vascular remodeling. J Am Coll Cardiol. 54(Suppl):
S10–S19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stenmark KR, Tuder RM and El KK: Metabolic
reprogramming and inflammation act in concert to control vascular
remodeling in hypoxic pulmonary hypertension. J Appl Physiol
(1985). 119:1164–1172. 2015. View Article : Google Scholar
|
|
25
|
Li P, Oparil S, Sun JZ, Thompson JA and
Chen YF: Fibroblast growth factor mediates hypoxia-induced
endothelin - a receptor expression in lung artery smooth muscle
cells. J Appl Physiol (1985). 95:643–651; discussion 863. 2003.
View Article : Google Scholar
|
|
26
|
Davie N, Haleen SJ, Upton PD, Polak JM,
Yacoub MH, Morrell NW and Wharton J: ET(A) and ET(B) receptors
modulate the proliferation of human pulmonary artery smooth muscle
cells. Am J Respir Crit Care Med. 165:398–405. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sato K, Morio Y, Morris KG, Rodman DM and
McMurtry IF: Mechanism of hypoxic pulmonary vasoconstriction
involves ET(A) receptor-mediated inhibition of K(ATP) channel. Am J
Physiol Lung Cell Mol Physiol. 278:L434–L442. 2000.PubMed/NCBI
|
|
28
|
Sweeney M, Yu Y, Platoshyn O, Zhang S,
McDaniel SS and Yuan JX: Inhibition of endogenous TRP1 decreases
capacitative Ca2+ entry and attenuates pulmonary artery
smooth muscle cell proliferation. Am J Physiol Lung Cell Mol
Physiol. 283:L144–L155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu HL, Zhang ZX, Zhao JP, Wang T and Xu
YJ: Effect of opening of mitochondrial ATP-sensitive K+
channel on the distribution of cytochrome C and on proliferation of
human pulmonary arterial smooth muscle cells in hypoxia. Sheng Li
Xue Bao. 58:262–268. 2006.In Chinese. PubMed/NCBI
|
|
30
|
Hu HL, Wang T and Zhang ZX: The effect on
the change of oxygen free radicals and cell proliferation in
hypoxic human pulmonary artery smooth muscle cells by mitochondrial
membrane potential. Chin J Tubercul Respiratory Diseases.
11:727–730. 2000.In Chinese.
|
|
31
|
Hu HL, Wang T, Zhang ZX, Zhao JP and Xu
YJ: Effect of diazoxide on change of H2O2 in
rat pulmonary artery smooth muscle cells and proliferation of
hypoxic rat pulmonary artery smooth muscle cells. Chin J
Pathophysiol. 23:2002–2006. 2007.In Chinese.
|
|
32
|
Hu HL, Wang T and Zhang ZX: The effect on
the distribution of cytochrome C and cell proliferation in hypoxic
rat pulmonary artery smooth muscle cells by mitochondrial membrane
potential. Proceedings of Huazhong University of Science and
Technology (Medicine). 166–169. 2802008.In Chinese.
|
|
33
|
Zeng H, Kong X, Peng H, Chen Y, Cai S, Luo
H and Chen P: Apoptosis and Bcl-2 family proteins, taken to chronic
obstructive pulmonary disease. Eur Rev Med Pharmacol Sci.
16:711–727. 2012.PubMed/NCBI
|