Introduction
The meninges, the membranes located between the
skull and brain, consist of three layers: Dura mater, arachnoid
mater and pia mater. The latter two layers are often collectively
termed leptomeninges, as they have a similar embryonic origin and
are closely associated (1). It has
been demonstrated that leptomeningeal fibrosis is important in the
pathogenesis of communicating hydrocephalus following subarachnoid
hemorrhage (2,3). The predominant pathological features
of leptomeningeal fibrosis include highly increased proliferation
of leptomeningeal mesothelial cells and the accumulation of
extracellular matrix (ECM), however the specific mechanisms are not
clear (4–7). A previous study regarding meningeal
fibrosis primarily focused on imaging and histology (7). However, investigations of its
pathogenesis at the molecular level are lacking. Establishing an
in vitro culture system of meningeal mesothelial cells
(MMCs) could provide an improved experimental model for mechanistic
studies of meningeal fibrosis.
Transforming growth factor-β1 (TGF-β1) is a critical
fibrosis-inducing cytokine. It promotes cell proliferation, and
stimulates overexpression and deposition of ECM proteins, such as
fibronectin and collagen (8). The
occurrence of many fibrotic diseases, including pulmonary fibrosis
and renal fibrosis, has been demonstrated to be associated with the
overexpression of TGF (9).
Connective tissue growth factor (CTGF), an important downstream
factor, could mediate the pro-fibrotic effects of TGF-β1, by
promoting mitosis and fibroblast proliferation, inducing collagen
synthesis, and mediating cell adhesion and chemotaxis (10). A previous study indicated that
TGF-β1 is an important factor for the induction of meningeal
fibrosis (11). TGF-β1 promotes
fibrosis by inducing mRNA and protein expression of CTGF in MMCs,
suggesting the possibility of blocking TGF-β1 signaling pathway to
delay the progression of meningeal fibrosis. However, TGF-β1 exerts
many important physiological effects, including immune modulation
and the anti-inflammatory response, suggesting that blocking TGF-β1
function in a direct manner would be clinically unfeasible.
Therefore, the identification of novel downstream targets of TGF-β1
is required to specifically inhibit the pro-fibrotic effect of
TGF-β1. Previous studies have revealed that the p38 signaling
pathway mediates TGF-β1-induced fibrosis in various types of cells,
such as lung fibroblasts, mesangial cells and renal fibroblasts
(12). However, whether the p38
signaling pathway is involved in TGF-β1-induced meningeal fibrosis
remains unclear.
The present study aimed to establish a cellular
model of TGF-β1-induced meningeal fibrosis with primarily cultured
MMCs. The specific role of the p38 mitogen-activated protein kinase
(MAPK) signaling pathway in TGF-β1-induced meningeal fibrosis of
mesothelial cells was investigated to identify novel therapeutic
targets for the prevention and treatment of communicating
hydrocephalus.
Materials and methods
Materials
High-glucose Dulbecco's modified Eagle's medium
(DMEM) and fetal calf serum (FCS) were purchased from Hyclone (GE
Healthcare Life Sciences, Logan, UT, USA). The following rabbit
anti-mouse IgG antibodies were purchased from Wuhan Boster
Biological Technology, Ltd. (Wuhan, China): Keratin (cat. no.
BA2266-1), vimentin (cat. no. BS-0756R) and factor VIII (cat. no.
BS-0434R). TGF-β1 was obtained from R&D Systems China Co., Ltd.
(Shanghai, China). TRIzol reagent was purchased from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Rabbit
anti-mouse CTGF antibody was obtained from Abcam (Shanghai, China).
Rabbit anti-mouse p38 MAPK antibody, rabbit anti-mouse
phosphorylated-p38 MAPK antibody and horseradish peroxidase (HRP)
-conjugated goat anti-rabbit IgG were all purchased from Cell
Signaling Technologies (Shanghai, China).
Methods
Cell culture and characterization
Ten Sprague Dawley rats, aged 3–5 days, were
obtained from the Laboratory Animal Center of Xinxiang Medical
University (Xinxiang, China). The rats were immediately sacrificed
by overdose of pentobarbital sodium (200 mg/kg i.p.; Sangon Biotech
Co., Ltd., Shanghai, China) and whole brains were removed under
sterile conditions, and washed with pre-cooled phosphate-buffered
saline (PBS) (13). Leptomeninges
were carefully peeled off in ice-cold DMEM (with 10% FCS) using
ophthalmic tweezers and blood vessels were removed. Tissues were
minced and triturated three times, and allowed to settle by gravity
for 5 min. The supernatant was discarded and 5 ml complete medium
(DMEM with 10% FCS, 100 U/ml penicillin and 100 U/ml streptomycin)
was added. The tissues were pipetted up and down repeatedly before
being transferred to 6-well plates and cultured in an incubator at
37°C with 95% humidity. After 24 h, one-half of the culture medium
was replaced with fresh medium and impurities were removed by
pipetting. Three days later, tissue fragments were aspirated and
discarded. The culture medium was subsequently refreshed every
three days. Cells were passaged routinely at 80% confluency. After
three passages, the expression levels of markers of MMCs were
investigated using immunofluorescence staining. Briefly, cells were
fixed with 4% paraformaldehyde for 15 min and permeabilized with
Triton X-100/PBS for 30 min. After two washes with PBS, cells were
blocked with 4% bovine serum albumin (BSA; Sangon Biotech Co.,
Ltd.) for 1 h, followed by incubation with primary antibodies
(keratin, vimentin and factor VIII) diluted 1:100 in Triton
X-100/PBS/1% BSA at 4°C overnight. Following removal of the primary
antibodies, cells were washed with PBS twice and the secondary
antibody, Invitrogen Alexa Fluor 488 goat anti-rabbit (cat. no.
A11008; Thermo Fisher Scientific, Inc) with Triton X-100/PBS
(dilution, 1:100) was added and incubated for 3 h at room
temperature. For imaging, an Olympus IX71 (Nikon Corporation,
Kanagawa, Japan) fluorescent microscope was used.
Cell processing and testing
MMCs of passage 3–8 were plated in a 60-ml flask at
a density of 1×105/ml. At 70–80% confluency, the cells
were treated with different concentrations of TGF-β1 (0, 1, 2 and 4
ng/ml), or pretreated with SB203580 (0, 1, 5 and 10 µM) for
1 h followed by treatment with 2 ng/ml TGF-β1. Treated cells were
cultured in an incubator at 37°C with 5% CO2 and 95%
humidity for a further 48 h before being harvested for total RNA
and protein extractions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
according to the manufacturer's instructions, and reverse
transcribed to cDNA using SuperScript II Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.). mRNA levels were
measured using an ABI Prism 7500 (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and SYBR Green qPCR Master mix (Kapa Biosystems,
Inc., Wilmington, MA, USA). Gene-specific primers used for qPCR
were as follows: Forward, 5′-TTGCCAAGCCTGTCAAGTTTG-3′ and reverse,
5′-AATGGCAGGCACAGGTCTTG-3′ for CTGF; forward,
5′-GGTCGGTGTGAACGGATTTG-3′ and reverse, 5′-GCTTCCCATTCTCAGCCTTGA-3′
for GAPDH. The target mRNA level of control cells normalized to the
level of GAPDH mRNA was set at 1 (14).
Western blotting
Cells were washed with PBS, and protein was
extracted with lysis buffer [20 mM Tris-HCl (pH 7.4), 150 mM NaCl,
1 mM EDTA, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride and
Roche's complete protease inhibitors (Roche Diagnostics, Shanghai,
China)] and centrifuged at 15,000 × g for 15 min at 4°C (15). The protein concentration of the
supernatants was determined using a Protein Assay kit II (Bio-Rad,
Hercules, CA, USA). For western blotting, samples were separated by
electrophoresis (150 V for 1.5 h), on a 12–15% SDS-PAGE gel and
transferred onto polyvinylidene fluoride membranes. After blocking
with 0.1% Tween-20 in PBS containing 5% skimmed milk, the membranes
were incubated with the above-mentioned primary antibodies,
keratin, vimentin and factor VIII. They were further incubated with
a HRP-conjugated donkey anti-rabbit IgG (1:1,000; cat. no. D110056;
Sangon Biotech Co., Ltd.) for 1 h. After three washes, the
membranes were developed using chemiluminescence substrate.
Immunoblot signals were quantified by measuring the immunoreactive
protein band density with ImageJ 1.48 software (National Institutes
of Health, Bethesda, MA, USA). For all immunoblot assays, β-actin
served as a loading control.
Statistical analysis
Data were expressed as the mean ± standard error of
the mean. The group means were compared by two-way analysis of
variance, and the significance of differences was determined by
post hoc testing using Bonferroni's method. P<0.05 was
considered to indicate a statistically significant difference.
Results
Characterization of primary cultured rat
MMCs
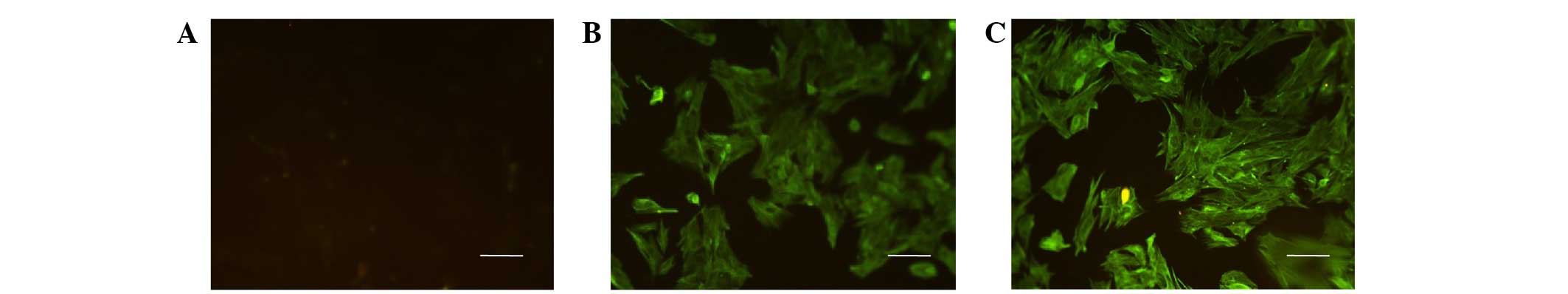
After 24 h incubation of explant cultures, small
quantities of spindle-shaped cells were observed. Cells then
gradually expanded and exponentially growing cells exhibited a
reticular-like growth pattern, while cell cultures displayed a
typical 'cobblestone' pattern upon reaching confluence. After three
passages, cells exhibited similar morphology to the primarily
isolated cells. The cells were positive for keratin and vimentin,
but negative for factor VIII following immunofluorescence staining
(Fig. 1). Keratin and vimentin are
commonly used markers for identifying mesothelial cells (16,17).
Keratin and vimentin were predominantly localized in the cytoplasm,
which is consistent with the characteristics of mesothelial cells.
These results suggested that MMCs had been successfully
isolated.
TGF-β1 induces fibrosis in rat MMCs in
vitro
To determine the molecular mechanism of
TGF-β1-induced fibrosis in MMCs, it was firstly examined whether
TGF-β1 induced fibrosis in MMCs in vitro. The dissociated
MMCs were treated with different concentrations of TGF-β1 and the
effect on the induction of the fibrosis marker, CTGF was
investigated. Results of qPCR revealed that a 48-h treatment with
TGF-β1 induced the expression of CTGF in a dose-dependent manner
(P=0.031, P=0.027, and P=0.042 for 1, 2 and 4 ng/ml TGF-β1,
respectively vs. the control; Fig.
2A). Similarly, western blotting demonstrated the induction of
CTGF protein by TGF-β1 (P=0.035, P=0.0077, and P=0.00058 for 1, 2
and 4 ng/ml TGF-β1, respectively vs. the control; Fig. 2B). Together, these data indicate
that TGF-β1 may induce fibrosis in rat MMCs in vitro, and
that primary cultured MMCs may serve as an in vitro cellular
model for the study of meningeal fibrosis.
TGF-β1 induces activation of p38 MAPK
signaling
Previous studies have demonstrated that the p38
signaling pathway is closely associated with fibrosis, particularly
TGF-β1-induced fibrosis, in many cell types (18,19).
However, the association between the p38 signaling pathway and
TGF-β1-induced fibrosis of MMCs is unclear. To elucidate this
association, it was first investigated whether activation of the
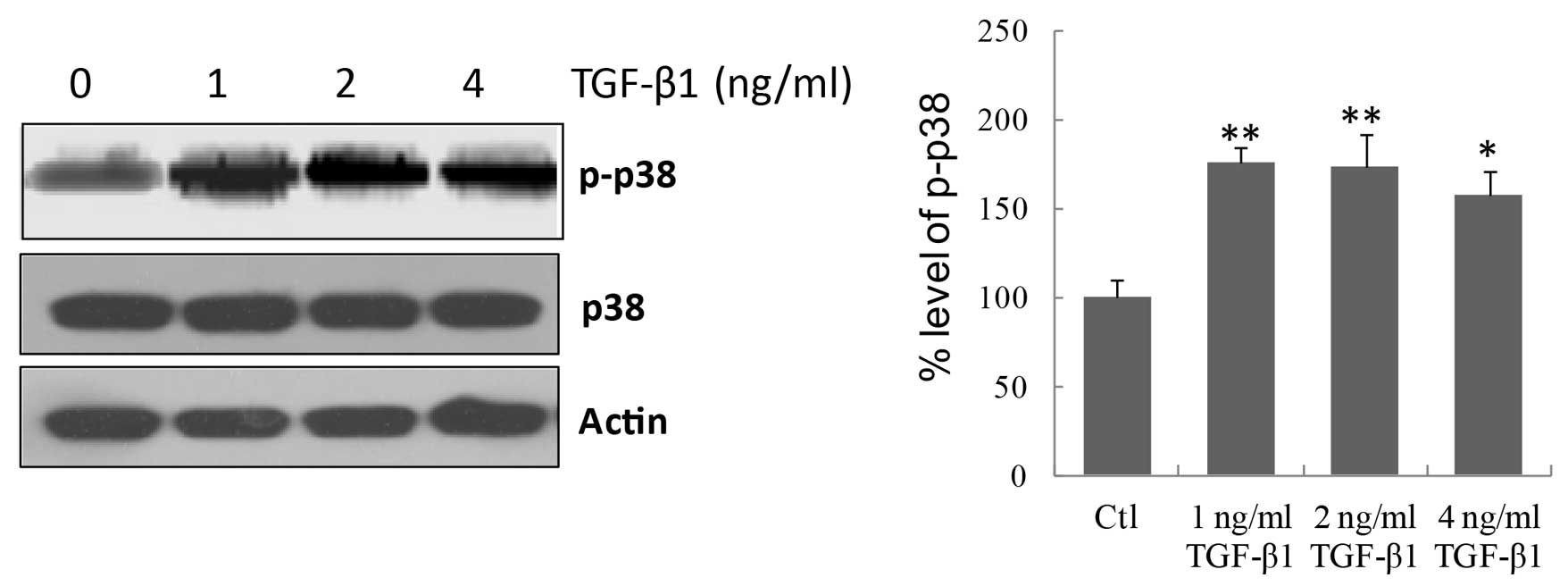
p38 signaling pathway was involved in TGF-β1-induced fibrosis. As
displayed in Fig. 3, TGF-β1 at all
concentrations significantly increased (P=0.0037, P=0.0085, and
P=0.019 for 1, 2 and 4 ng/ml TGF-β1, respectively vs. the control)
the phosphorylation of p38 in MMCs, indicating that TGF-β1
activates the p38 signaling pathway in MMCs.
TGF-β1 induces fibrosis in rat MMCs via
p38 MAPK signaling
Although TGF-β1 may induce the activation of p38
signaling in MMCs as described above, whether p38 activation is
associated with TGF-β1-induced fibrosis remains unknown. Therefore,
the effect of the p38 inhibitor, SB203580 on the TGF-β1-induced
expression of CTGF was examined. As displayed in Fig. 4A and B, SB203580 significantly
inhibited the activation of p38 (P=0.0014, P=0.00039, and
P=0.000086 for 1, 5 and 10 µM SB203580, respectively vs. the
control). In addition, SB203580 at various concentrations inhibited
the induction of CTGF (P= 0.029, P= 0.014, and P= 0.0012 for 1, 5
and 10 µM SB203580, respectively vs. the control; Fig. 4C and D). As CTGF is an indicator of
cell fibrosis, these data suggest that TGF-β1 induces fibrosis in
rat MMCs via p38 MAPK signaling.
Discussion
Communicating hydrocephalus is usually secondary to
subarachnoid hemorrhage, meningitis and traumatic brain injury,
amongst other neurological disorders, as no effective therapeutic
strategy for these conditions is available. Currently, it is
proposed that the imbalance of cerebrospinal fluid secretion and
absorption caused by fibrous adhesion of leptomeninges is the
pathological basis of chronic hydrocephalus (7); however, the specific mechanisms
remain unclear. Studies have indicated that tissue fibrosis is
crucial in the imbalance between ECM protein synthesis and
degradation, leading to the excessive deposition of ECM (20,21).
This complex pathophysiological process involves various cytokines
(22), among which TGF-β1 and its
downstream effector, CTGF are possibly the most critical
fibrosis-inducing factors. They have been demonstrated to be
important in the induction of fibrosis in various organs and
tissues, including the kidney, heart, liver, lung and skin
(23,24). Previously, the role of TGF-β1 in
diseases of the central nervous system have begun to be
investigated. It is reported that intrathecal injection of
recombinant TGF-β1 in transgenic rats resulted in impaired
cerebrospinal fluid flow and extensive meningeal adhesion,
degeneration and thickening (22,25),
indicating an important role of TGF-β1 in the occurrence of
hydrocephalus and meningeal fibrosis. However, the underlying
mechanisms of TGF-β1-induced meningeal fibrosis are not fully
understood.
The present study demonstrated that TGF-β1 induces
the expression of CTGF in MMCs in a dose-dependent manner,
suggesting that cultured MMCs could be used as a reliable model
with which to study meningeal fibrosis in vitro. To validate
this cell model, the signaling pathway that is responsible for
TGF-β1-induced fibrosis in MMCs was analyzed. As p38 signaling is
known to be a key pathway mediating TGF-β1-induced fibrosis in
cells of other types (12), the
present study determined whether the p38 signaling pathway is
involved in TGF-β1-induced fibrosis in MMCs. It was demonstrated
that TGF-β1 activates the p38 signaling pathway in MMCs. In
addition, the p38-specific inhibitor, SB203580, significantly
suppressed the induction of the fibrosis marker CTGF. This suggests
that the p38 MAPK pathway is an important signaling pathway through
which TGF-β1 induces the expression of CTGF. Thus, it may provide a
novel strategy for therapeutic intervention, by blocking the p38
signaling pathway for the treatment of meningeal fibrosis induced
by TGF-β1, rather than direct blocking of TGF-β1 function.
Studies have demonstrated that the mechanisms by
which TGF-β1 induces the expression of CTGF are cell-specific. For
example, Xie et al (26)
observed that in airway smooth muscle cells, the extracellular
signal-regulated kinase and c-Jun N-terminal kinase signaling
pathways, but not the p38 signaling pathway, are involved in
TGF-β1-induced CTGF expression. By contrast, Chang et al
(27) demonstrated that p38 is
involved in the TGF-β1-induced CTGF expression in buccal mucosa
fibroblasts. In liver progenitor cells, p38 was demonstrated to be
necessary for TGF-β1-induced CTGF expression and fibrosis (12). However, to the best of our
knowledge, the mechanisms by which TGF-β1 act on MMCs have not yet
been reported. In conclusion, the current study presents an
important axis of the TGF-β1/p38 MAPK/CTGF signaling pathway in
meningeal fibrosis, and provides a possible novel strategy for the
clinical treatment of communicating hydrocephalus following
subarachnoid hemorrhage.
References
|
1
|
Decimo I, Fumagalli G, Berton V, Krampera
M and Bifari F: Meninges: From protective membrane to stem cell
niche. Am J Stem Cells. 1:92–105. 2012.PubMed/NCBI
|
|
2
|
Etus V, Kurtkaya O, Koc K, Ciftci E, Sav A
and Ceylan S: Multisegmental spinal leptomeningeal fibrosis in
Riedel thyroiditis. Case illustration. J Neurosurg. 98(Suppl 3):
S2992003.
|
|
3
|
Robertson PL, Muraszko KM, Blaivas M and
Brunberg JA: Leptomeningeal fibrosis and the delayed diagnosis of a
central nervous system neoplasm (primitive neuroectodermal tumor).
Pediatr Neurol. 16:74–78. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou G, Su X, Ma J, Wang L and Li D:
Pioglitazone inhibits high glucose-induced synthesis of
extracellular matrix by NF-κB and AP-1 pathways in rat peritoneal
mesothelial cells. Mol Med Rep. 7:1336–1342. 2013.PubMed/NCBI
|
|
5
|
Xiao L, Sun L, Liu FY, Peng YM and Duan
SB: Connective tissue growth factor knockdown attenuated matrix
protein production and vascular endothelial growth factor
expression induced by transforming growth factor-beta1 in cultured
human peritoneal mesothelial cells. Ther Apher Dial. 14:27–34.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peng Y, Liu H, Liu F, Liu Y, Li J and Chen
X: Troglitazone inhibits synthesis of transforming growth
factor-beta1 and reduces matrix production in human peritoneal
mesothelial cells. Nephrology (Carlton). 11:516–523. 2006.
View Article : Google Scholar
|
|
7
|
Hung KY, Huang JW, Tsai TJ and Hsieh BS:
Peritoneal fibrosing syndrome: Pathogenetic mechanism and current
therapeutic strategies. J Chin Med Assoc. 68:401–405. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang Y, Zhang N, Lan F, Van Crombruggen K,
Fang L, Hu G, Hong S and Bachert C: Transforming growth factor-beta
1 pathways in inflammatory airway diseases. Allergy. 69:699–707.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prud'homme GJ: Pathobiology of
transforming growth factor beta in cancer, fibrosis and immunologic
disease, and therapeutic considerations. Lab Invest. 87:1077–1091.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wei JL, Peng YM and Liu F: Connective
tissue growth factor and fibronectin secretion in renal tubular
epithelial cells induced by TGF-beta1: Suppressive effects of
troglitazone. Cell Biol Int. 31:30–34. 2007. View Article : Google Scholar
|
|
11
|
Li T, Zhang P, Yuan B, Zhao D, Chen Y and
Zhang X: Thrombin induced TGF-β1 pathway: A cause of communicating
hydrocephalus post subarachnoid hemorrhage. Int J Mol Med.
31:660–666. 2013.PubMed/NCBI
|
|
12
|
Zarubin T and Han J: Activation and
signaling of the p38MAP kinase pathway. Cell Res. 15:11–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei LH, Han SG and Li T: Improvement on
culture in vitro of rat menigeal mesothelial cell. Chinese J
Neuroanat. 25:445–448. 2009.
|
|
14
|
Yang HJ, Wang L, Xia YY, Chang PN and Feng
ZW: NF-kappaB mediates MPP+-induced apoptotic cell death in
neuroblastoma cells SH-EP1 through JNK and c-Jun/AP-1. Neurochem
Int. 56:128–134. 2010. View Article : Google Scholar
|
|
15
|
Yang HJ, Xia YY, Wang L, Liu R, Goh KJ, Ju
PJ and Feng ZW: A novel role for neural cell adhesion molecule in
modulating insulin signaling and adipocyte differentiation of mouse
mesenchymal stem cells. J Cell Sci. 124:2552–2260. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Katz S, Balogh P, Nagy N and Kiss AL:
Epithelial-to-mesenchymal transition induced by Freund's adjuvant
treatment in rat mesothelial cells: A morphological and
immunocytochemical study. Pathol Oncol Res. 18:641–649. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rosellini A, Michelini M, Tanda G, Mandys
V and Revoltella RP: Expansion of human mesothelial progenitor
cells in a longterm three-dimensional organotypic culture of
Processus vaginalis peritonei. Folia Biol (Praha). 53:50–57.
2007.
|
|
18
|
Liu Q, Wang CY, Liu Z, Ma XS, He YH, Chen
SS and Bai XY: Hydroxysafflor yellow A suppresses liver fibrosis
induced by carbon tetrachloride with high-fat diet by regulating
PPAR-γ/p38 MAPK signaling. Pharm Biol. 52:1085–1093. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang L, Li Y, Chen M, Su X, Yi D, Lu P
and Zhu D: 15-LO/15-HETE mediated vascular adventitia fibrosis via
p38 MAPK-dependent TGF-β. J Cell Physiol. 229:245–257. 2014.
View Article : Google Scholar
|
|
20
|
Xu X, Xiao L, Xiao P, Yang S, Chen G, Liu
F, Kanwar YS and Sun L: A glimpse of matrix metalloproteinases in
diabetic nephropathy. Curr Med Chem. 21:3244–3260. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lan TH, Huang XQ and Tan HM: Vascular
fibrosis in atherosclerosis. Cardiovasc Pathol. 22:401–407. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lagèrze WA: New prospects in the treatment
of diabetic retinopathy: Current situation and pharmacological
developments. MMW Fortschr Med. 145:37–38. 2003.In German.
|
|
23
|
Tampe D and Zeisberg M: Potential
approaches to reverse or repair renal fibrosis. Nat Rev Nephrol.
10:226–237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leask A: Potential therapeutic targets for
cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF,
partners in fibroblast activation. Circ Res. 106:1675–1680. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crews L, Wyss-Coray T and Masliah E:
Insights into the pathogenesis of hydrocephalus from transgenic and
experimental animal models. Brain Pathol. 14:312–316. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie S, Sukkar MB, Issa R, Khorasani NM and
Chung KF: Mechanisms of induction of airway smooth muscle
hyperplasia by transforming growth factor-beta. Am J Physiol Lung
Cell Mol Physiol. 293:L245–L253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang JZ, Yang WH, Deng YT, Chen HM and
Kuo MY: EGCG blocks TGFβ1-induced CCN2 by suppressing JNK and p38
in buccal fibroblasts. Clin Oral Invest. 17:455–461. 2013.
View Article : Google Scholar
|