Introduction
Janus kinase (JAK)-signal transducer and activator
of transcription (STAT)-mediated signal transduction pathway is
important for regulating DNA transcription and the activities of
the cell cycle. This pathway has three main signaling components:
Receptors, JAK, and STAT (1).
Extracellular signal molecules, including interferon, interleukin
and growth factors, can bind with their receptors and cause
activation of the kinase function of JAK through
auto-phosphorylation. Consequently, STAT binds to the
phosphorylated receptor, where it is phosphorylated by JAK. The
phosphorylated STAT protein then binds to another phosphorylated
STAT protein to form STATs dimer complex. The complex can be
translocated into the nucleus and there it binds to DNA, promotes
transcription of genes and causes the expression of proteins that
affect basic cell activities, including cell growth,
differentiation and death (2).
Type II cyclic guanosine monophosphat e
(cGMP)-dependent protein kinase (PKG II) is a serine/threonine
kinase. It was identified >30 years ago and has been typically
implicated only in several physiological functions, including
intestinal secretion, bone growth, and learning and memory
(3). However, increasing evidence
has demonstrated that this kinase is involved in regulating
proliferation and apoptosis of cells, and is potentially associated
with tumorigenesis. For example, research data demonstrated that
PKG II inhibited proliferation and/or induced apoptosis of human
prostate cells, neruoglioma cells and breast cancer cells (4–6).
Additionally, the inhibitory effect of PKG II on gastric cancer
cells for several years has been investigated. An important finding
was that PKG II blocks the activation of epidermal growth factor
receptor (EGFR), the initiating event of the epidermal growth
factor (EGF)-induced signal transduction process (7,8). As
the activation of EGFR can initiate the signal transduction of
several pathways, including phospholipase Cγ-mediated,
phosphatidylinositol 3-kinase (PI3K)/Akt serine/threonine kinase 1
(Akt)-mediated and JAK-STAT-mediated pathways (9–11),
it is expected that the blockage of the EGFR activation by PKG II
will inhibit the corresponding signal transductions and exert a
wide-range of effects on the biological activities of cancer cells.
The present study was performed to investigate the inhibitory
effect of PKG II on EGF/EGFR-initiated signal transduction of the
JAK-STAT-mediated pathway.
Materials and methods
Cell line and reagents
AGS human gastric cancer cell line was provided by
the Institute of Cell Biology (Shanghai, China). Adenoviral vectors
encoding the cDNA of β-galactosidase (β-gal) and PKG II (Ad-LacZ
and Ad-PKG II, respectively), as well as the SRE-luc plasmid,
RSV-β-gal plasmid and CMV vector plasmid were gifts from Dr Gerry
Boss and Dr Renate Pilz (University of California, San Diego, CA,
USA). Dulbecco's modified Eagle's media (DMEM) and fetal bovine
serum (FBS) were obtained from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Polyclonal rabbit antibody against PKG II
was purchased from Abgent, Inc. (San Diego, CA, USA; cat no.
AP8001a; dilution, 1:200). Monoclonal mouse anti-β-actin antibody
was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA;
cat no. sc-47778; dilution, 1:1,000). Polyclonal rabbit
anti-phosphorylated (p)-EGFR (Tyr1068) was obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA; cat no. 2236;
dilution, 1:1,000). Polyclonal rabbit anti-p-JAK1 (cat no. BS4108;
dilution, 1:500), anti-p-JAK2 (cat no. BS4109; dilution, 1:500),
anti-p-STAT1 (cat no. BS4178; dilution, 1:500) and anti-p-STAT3
(cat. no. BS4180; dilution, 1:500) antibodies were purchased from
Bioworld Technology, Inc. (St. Louis Park, MN, USA). Monoclonal
mouse anti-cyclin D1 (cat. no. BMO771; dilution, 1:400) and
poly-clonal rabbit anti-cyclin E (cat no. BAO774; dilution, 1:400)
antibodies were from Wuhan Boster Biological Technology, Ltd.
(Wuhan, China). Horseradish peroxidase-conjugated goat anti-mouse
(cat. no. 115-035-003) and goat anti-rabbit (cat. no. 111-035-003)
polyclonal secondary antibodies (dilution, 1:10,000) were from
Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA).
The cellular permeable cGMP analog, 8-pCPT-cGMP, was from EMD
Millipore (Billerica, MA, USA). EGF was obtained from Sigma-Aldrich
(St. Louis, MO, USA). Electrochemilumines-cence (ECL) reagents were
from EMD Millipore. All other reagents used were of analytical
grade.
Cell culture and preparation of cell
extracts
AGS cells were cultured in DMEM supplemented with
10% FBS and maintained at 37°C in a humidified incubator, with 95%
air and 5% CO2. On the day prior to infection, cells
were plated into 6-well plates. When the cells were 70–80%
confluent, they were infected with Ad-LacZ or Ad-PKG II with a MOI
of 100% or mock infected. At 24 h after the infection, the medium
was replaced with serum-free medium, and the culture was continued
for 12 h. The infected cells were incubated with 100 or 250
µM 8-pCPT-cGMP for 1 h, and were then incubated with 100
ng/ml EGF. To observe the phosphorylation of EGFR, the EGF
incubation time was 5 min; to observe the change of protein
expression, the EGF incubation time was 12 h. Differently treated
cells were harvested at various times by aspiration of the media
and direct addition of heated 2X sodium dodecyl sulfate (SDS)
sample buffer. The cell lysate was scraped and transferred to
Eppendorf tube, heated for 5 min at 100°C and stored at −20°C.
Western blotting
A Bicinchoninic Acid Protein Assay was used to
quantify the protein concentration of the cellular extract. Then,
10 µg protein was loaded onto each lane and proteins were
separated by SDS-polyacrylamide gel electrophoresis (8–12%) gel
according to the molecular size, and transferred onto a
polyvinylidene fluoride membrane. Blots were blocked with 5% (w/v)
nonfat milk in Tris-buffered saline-Tween 20 for 1 h at room
temperature, and then incubated at 4°C overnight with the primary
antibody, followed by incubation with the secondary antibody at
room temperature for 1 h. The signal was visualized by using the
ECL detection reagents. To perform densitometry analysis, digital
images of the positive bands were obtained with a Chemidoc XRS and
analyzed using the image analysis program Quantity One v4.4.0.36
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The results were
presented as the ratio of target protein/loading control.
Transient transfection and luciferase
reporter assays
Plasmids were purified with a Qiagen Plasmid
Purification Kit (Qiagen GmbH, Hilden, Germany). Transient
transfections were performed using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.), initially using 70–80% confluent
cells/well in 24-well plate with 0.6 µg of plasmid DNA
mixture, following the manufacturer's instructions. For each well,
AGS cells were transfected with 0.1 µg SRE-luc plasmid DNA,
0.1 µg RSV-β-Gal plasmid DNA and 0.4 µg CMV-vector
plasmid DNA. After 8 h of incubation, the serum-free medium was
replaced with fresh medium containing 10% serum and cells were
infected with adenoviral vectors as described above. After 36 h of
incubation, cells were washed with phosphate-buffered saline (PBS)
and harvested, then luciferase assays were performed. The
luciferase and β-gal activities of the lysates were measured using
a chemiluminescence detector (Berthold Technologies GmbH, Zug,
Switzerland). Transcription factor activity was reported in
relative light units (RLU, luciferase/β-gal). All experiments were
performed independently at least three times, with two
parallels.
Cell cycle analysis
Cells were plated into 6-well plates and infected
with adenoviral vectors as described above. The cells at the
logarithmic growth phase were trypsinized, washed with PBS twice,
and then fixed with 70% ethanol at 4°C overnight. The fixed cells
were washed with PBS twice, re-suspended in 400 µl PBS
(containing 50 µg/ml ribonuclease A and 50 µg/ml
propidium iodide), and incubated in an ice bath away from light for
30 min. The cell cycle distribution was detected using the
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA). All experiments were performed independently at least three
times.
Statistical analysis
The data are expressed as the means ± standard
deviation. Statistical significance was detected using one way
analysis of variance followed by Student-Neuman-Keuls method with
SPSS statistical software (version 17.0; SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
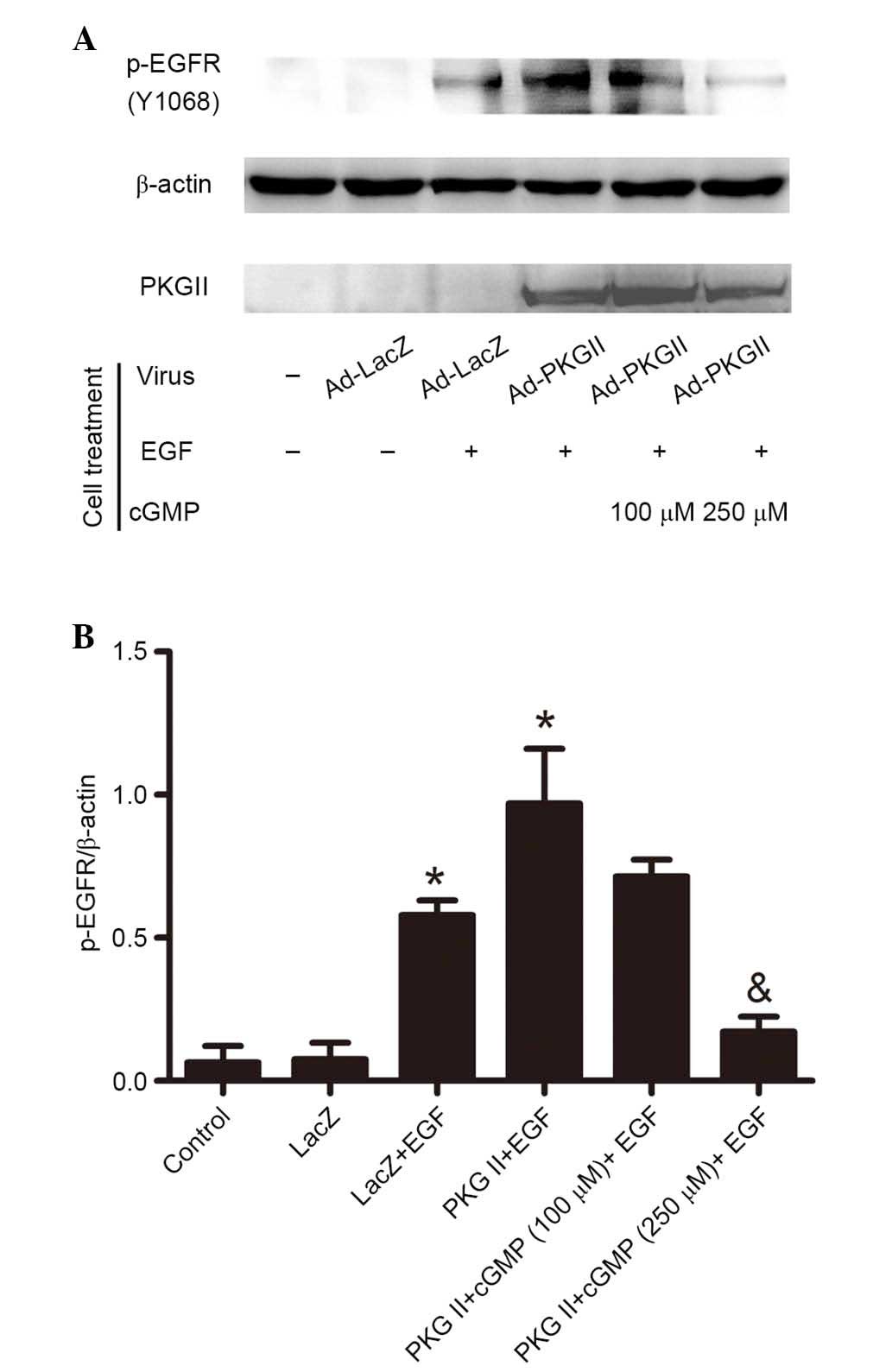
PKG II inhibits EGF-induced Tyr1068
phosphorylation of EGFR
Tyr1068 is one of the important auto-phosphorylation
sites of EGFR. Phosphorylation of this site is associated with
JAK/STAT-mediated signaling (12).
In the current study, western blotting with antibody against p-EGFR
(Tyr1068) was performed to investigate the inhibitory effect of PKG
II on Tyr1068 phosphorylation of EGFR in differently treated AGS
cells. The results demonstrated that EGF treatment (100 ng/ml, 5
min) caused a significant increase in EGFR Tyr1068 phosphorylation
compared with the control and Ad-LacZ group (P=0.001, the PKG
II+EGF group vs. the control group; P=0.001, the LacZ+EGF group vs.
the LacZ group). In cells infected with Ad-PKG II and stimulated
with 8-pCPT-cGMP (100 or 250 µM, 1 h), the EGF-induced
phosphorylation was significantly decreased compared with the PKG
II + EGF group (P=0.003, the PKG II+cGMP+EGF group vs. the PKG
II+EGF group; Fig. 1). This
indicated that PKG II inhibited the activation of EGFR caused by
EGF.
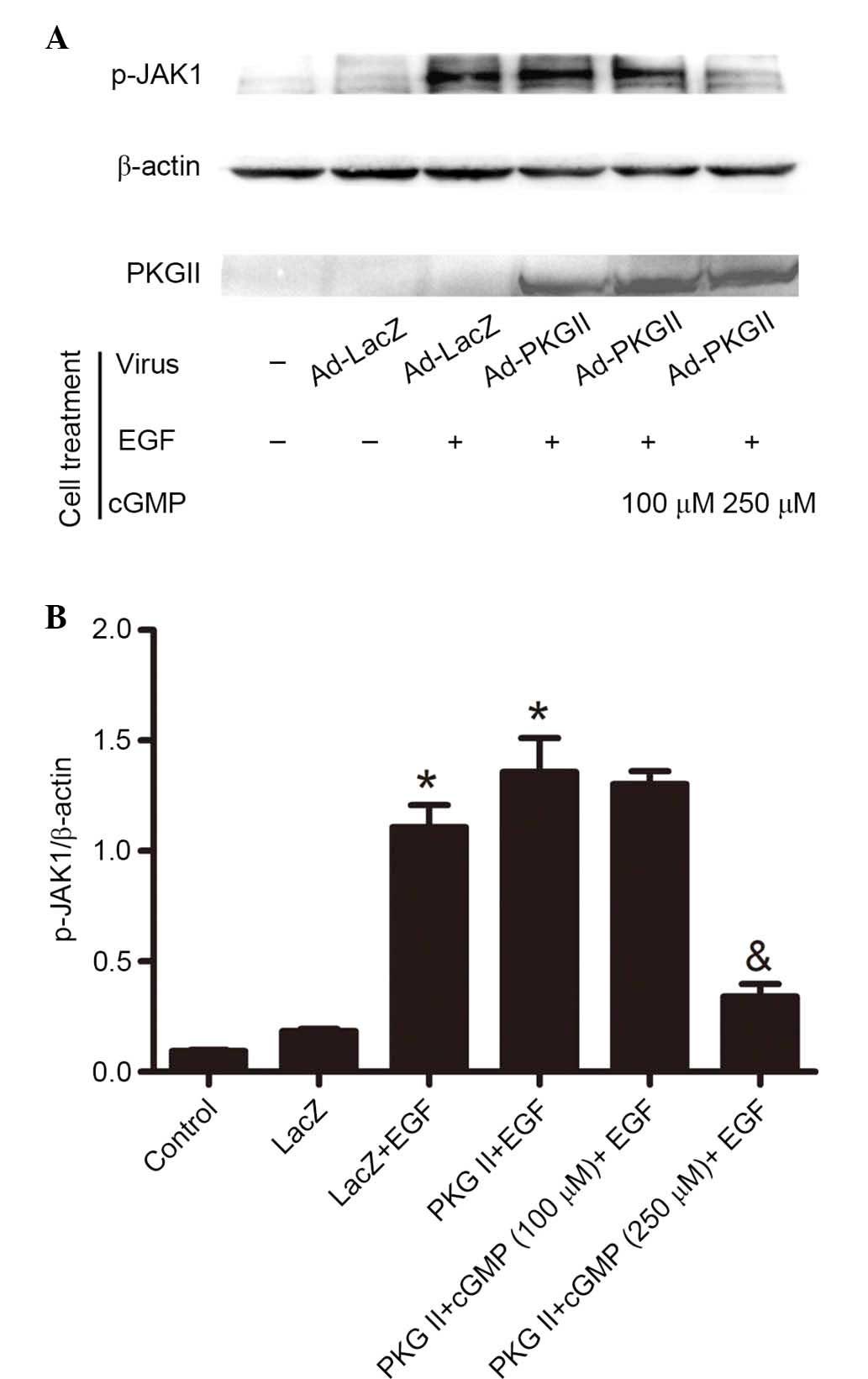
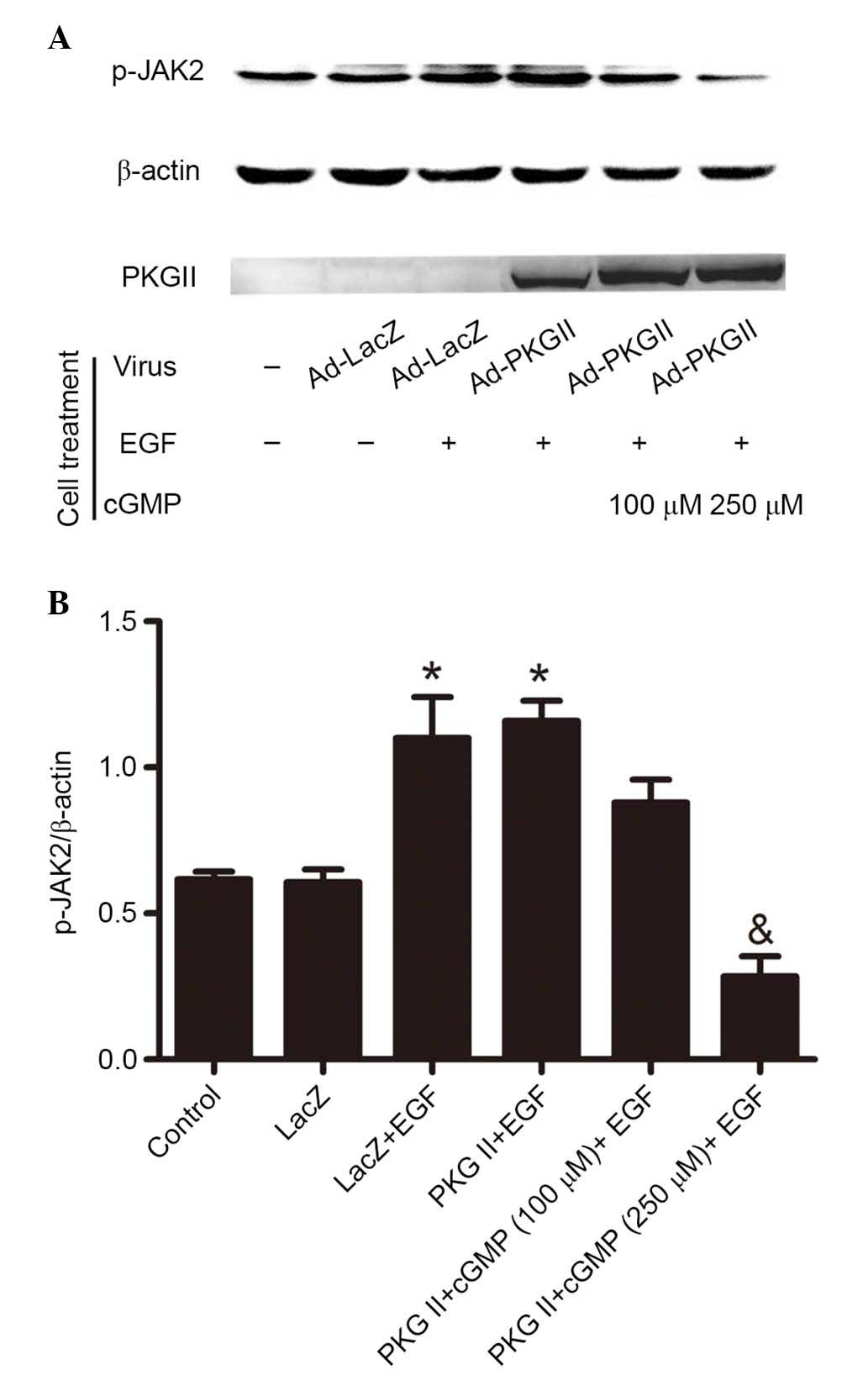
PKG II inhibits EGF-induced
phosphorylation of JAK1 and JAK2
Western blotting with antibody against p-JAK1
(Tyr1022) and p-JAK2 (Tyr1007/Tyr1008) was applied to detect the
phosphorylation/activation of these kinases. The results
demonstrated that EGF treatment (100 ng/ml, 5 min) increased the
Tyr1022 phosphorylation of JAK1 (Fig.
2) and Tyr1007/1008 phosphorylation of JAK2 (Fig. 3). In cells infected with Ad-PKG II,
stimulated with 8-pCPT-cGMP (100 or 250 µM, 1 h) and then
treated with EGF (100 ng/ml, 5 min), the phosphorylation level of
JAK1 and JAK2 was significantly lower compared with cells infected
with Ad-LacZ or Ad-PKG II and treated with EGF only (P=0.007, the
PKG II+cGMP+EGF group vs. the PKG II+EGF group, Fig. 2; P=0.025, the PKG II+cGMP+EGF group
vs. the PKG II+EGF group, Fig. 3).
These results demonstrated that PKG II inhibited the EGF-induced
activation of JAKs.
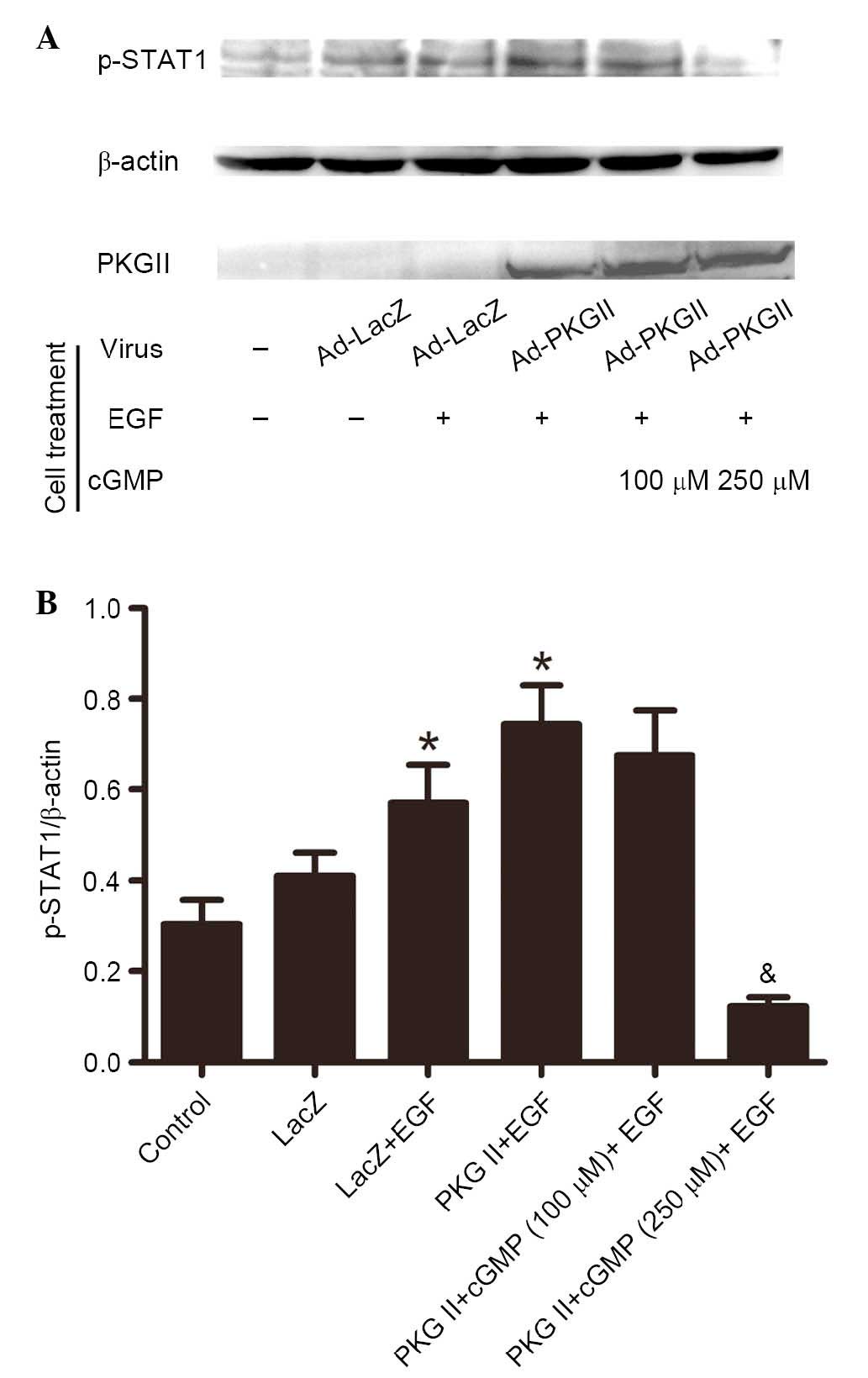
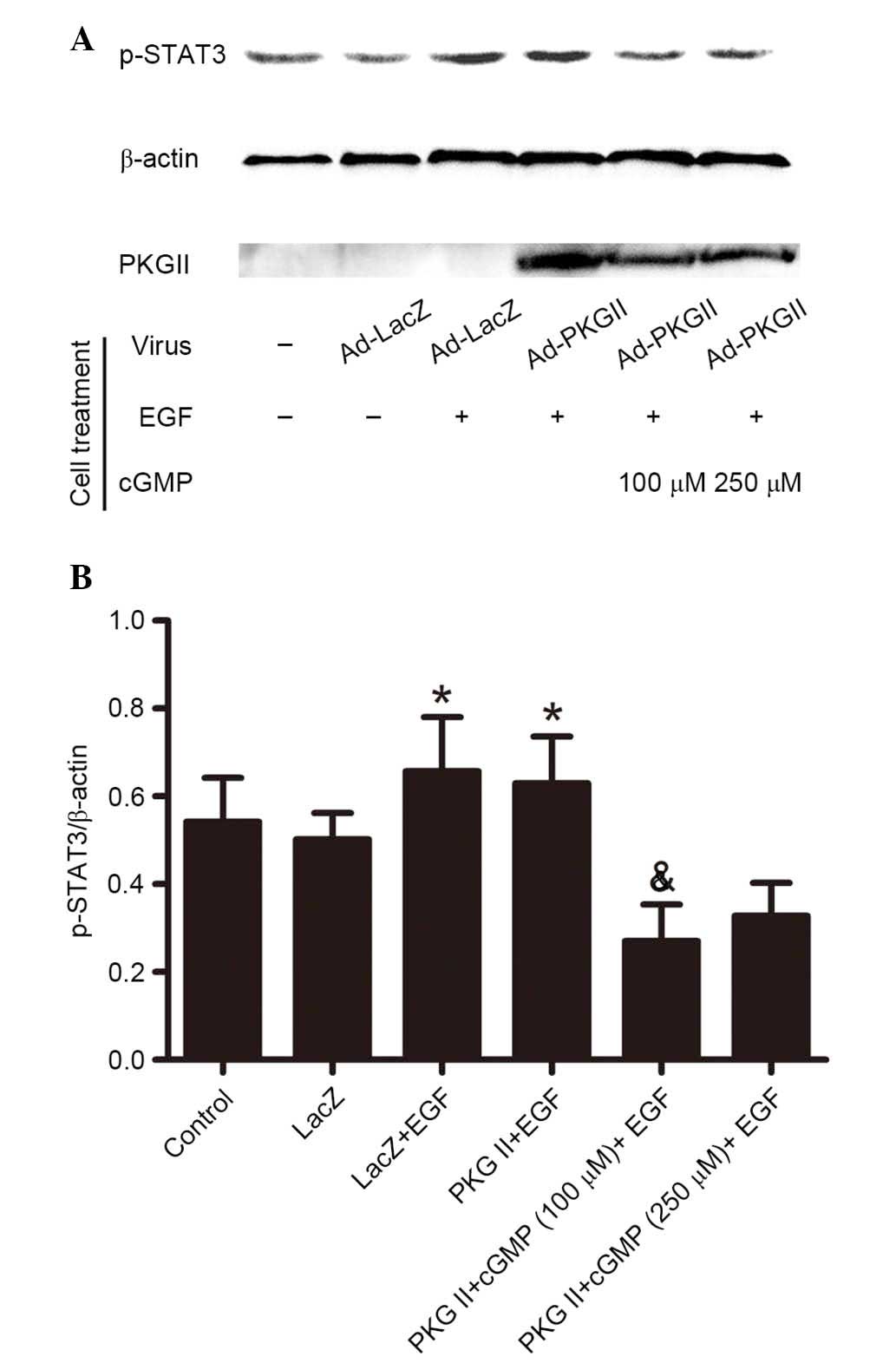
PKG II inhibits EGF-induced
phosphorylation of STAT1 and STAT3
Activated JAK can induce phosphorylation of STAT,
which then binds with another phosphorylated STAT to form a dimer.
This is an important step of JAK/STAT-mediated signaling (13). In current study, western blotting
with antibodies against p-STAT1 (Tyr701) and p-STAT3 (Tyr705) was
performed to detect the phosphorylation of STAT1 and STAT3 in
cells. The results demonstrated that in cells infected with Ad-LacZ
and treated with EGF (100 ng/ml, 5 min), Tyr701 phosphorylation of
STAT1 (Fig. 4) and STAT3 (Fig. 5) was significantly increased
(P=0.035, the PKG II+EGF group vs. the control group; P=0.029, the
LacZ+EGF group vs. the LacZ group, Fig. 4; P=0.038, the PKG II+EGF group vs.
the control group; P=0.021, the LacZ+EGF group vs. the LacZ group,
Fig. 5) compared with control and
untreated Ad-LacZ cells. In the cells infected with Ad-PKG II,
stimulated with 8-pCPT-cGMP (100 or 250 µM, 1 h) and then
treated with EGF (100 ng/ml, 5 min), the phosphorylation level of
STAT1 and STAT3 was significantly reduced compared with cells
infected with Ad-LacZ and treated with EGF only (P=0.009, the PKG
II+cGMP+EGF group vs. the PKG II+EGF group, Fig. 4; P=0.011, the PKG II+cGMP+EGF group
vs. the PKG II+EGF group, Fig. 5).
These results indicated that PKG II inhibited the EGF-induced
activation of STAT1 and STAT3.
PKG II inhibits the transcriptional
activity caused by EGF
Dimerized STATs can move into the nucleus and
associate with transcription factor to initiate transcriptional
activity (13). To investigate the
inhibitory effect of PKG II on EGF/EGFR-induced transcript
activity, reporter gene assays were performed to detect the
inhibition of PKG II on EGF/EGFR initiated transcription. The
results demonstrated that EGF treatment (100 ng/ml, 12 h)
significantly increased SRE-dependent transcriptional activity
compared with the controls (P=0.005, PKG II+EGF group vs. the
control group; P=0.008, the LacZ+EGF group vs. the LacZ group).
Additionally, increased of PKG II activity (infected with Ad-PKG II
for 24 h and treated with 100 or 250 µM cGMP for 1 h)
inhibited the stimulating effect of EGF/EGFR on transcriptional
activity compared with EGF-treated Ad-PKG II cells (P=0.033, the
PKG II+cGMP+EGF group vs. the PKG II+EGF group, Fig. 6).
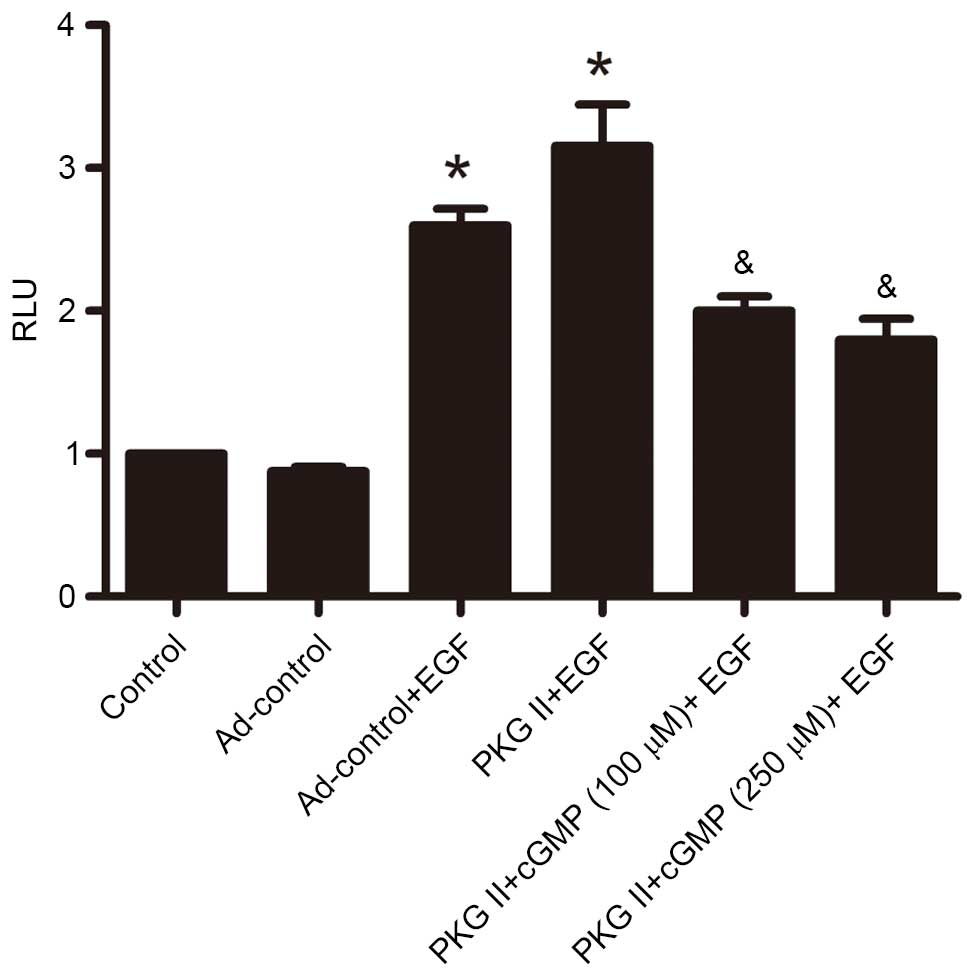 | Figure 6PKG II inhibits SRE-dependent
transcription initiated by EGF. AGS cells were transfected with
plasmid DNA mixture for 8 h and infected with Ad-LacZ or Ad-PKG II
for 24 h, serum-starved overnight, and treated as follows: In
Ad-LacZ + EGF and Ad-PKG II + EGF groups, cells were incubated with
EGF (100 ng/ml) for 12 h; in Ad-PKG II + cGMP groups, cells were
incubated with 100 or 250 µM 8-pCPT-cGMP respectively for 1
h, and then incubated with EGF for 12 h. The cell lysate was
prepared and reporter gene assay was applied to detect the
SRE-dependent transcription activity. The results were presented as
RLU (luciferase/LacZ). The data are presented as the mean ±
standard deviation from 3 independent experiments.
*P<0.05 vs. control group and LacZ group;
&P<0.05 vs. LacZ + EGF group and PKG II + EGF
group. Ad, adenovirus; EGF, epidermal growth factor; PKG II, type
II cGMP-denpendent protein kinase; LacZ, β-galactosidase; cGMP,
cyclic guanosine monophosphate. |
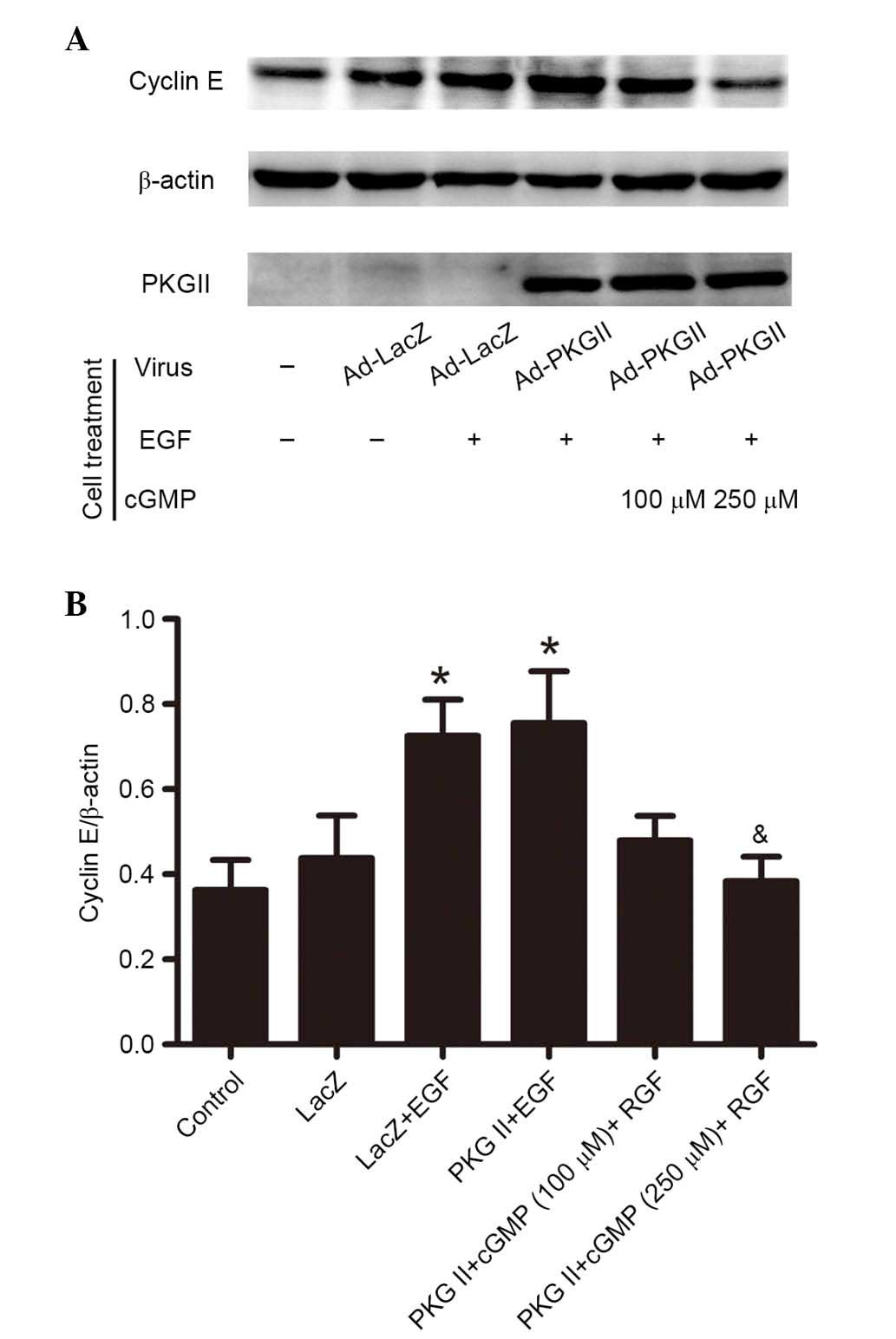
PKG II inhibits EGF-induced expression of
cyclin D1 and cyclin E
Activated STAT3 can upregulate the expression levels
of various proteins involved in cell cycle progression, including
Fos, c-Myc, and cyclin D (14). To
investigate the inhibition of PKG II on EGF/EGFR-JAK/STAT
signaling-induced expression of cell cycle-associated proteins,
western blotting was performed to detect the expression of cyclin
D1 (Fig. 7) and cyclin E (Fig. 8) in AGS cells. The results
demonstrated that EGF treatment (100 ng/ml, 12 h) caused a
significant increase in the expression levels of cyclin D1 and
cyclin E compared with untreated controls (P=0.006, the PKG II+EGF
group vs. the control group; P=0.015, the LacZ+EGF group vs. the
LacZ group, Fig. 7; P=0.023, the
PKG II+EGF group vs. the control group; P=0.000, the LacZ+EGF group
vs. the LacZ group, Fig. 8). The
increased PKG II activity caused by infecting the cells with Ad-PKG
II and stimulating with 8-pCPT-cGMP (100 or 250 µM, 1 h)
significantly inhibited the EGF-induced expression of cyclin D1 and
cyclin E compared with Ad-PKG II cells treated with EGF only
(Figs. 7 and 8).
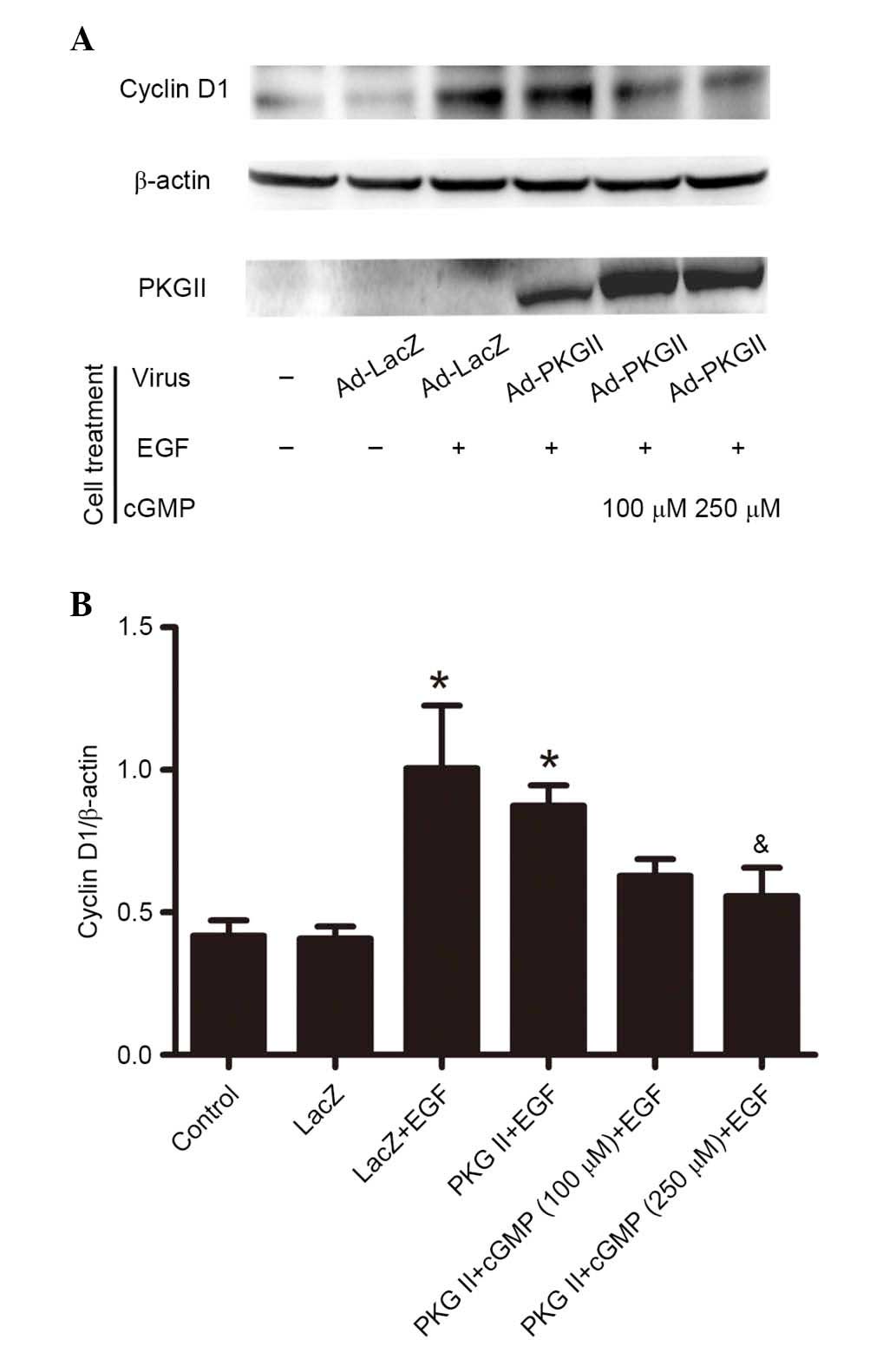 | Figure 7PKG II inhibits EGF-induced expression
of cyclin D1. AGS cells were infected with either Ad-LacZ or Ad-PKG
II, serum starved overnight, and treated differently: In Ad-LacZ
group, no drug treatment; in Ad-LacZ + EGF group, the cells were
incubated with EGF (100 ng/ml) for 12 h; in Ad-PKG II + cGMP + EGF
group, cells were incubated with 100 or 250 µM 8-pCPT-cGMP
for 1 h and followed by incubating with EGF (100 ng/ml) for 12 h.
(A) The cell lysate was subjected to western blotting to detect the
expression of cyclin D1. β-actin was detected as a loading control
and PKG II was detected to demonstrate the efficiency of the
infection with Ad-PKG II. (B) Densitometry analysis was performed
to quantify the positive bands and the ratio of Cylin D1/β-actin
was determined. The data are presented as the mean ± standard
deviation from 3 independent experiments. *P<0.05 vs.
control group and LacZ group; &P<0.05 vs. LacZ +
EGF group and PKG II + EGF group. PKG II, type II cGMP-denpendent
protein kinase; Ad, adenovirus; LacZ, β-galactosidase; EGF,
epidermal growth factor; cGMP, cyclic guanosine monophosphate. |
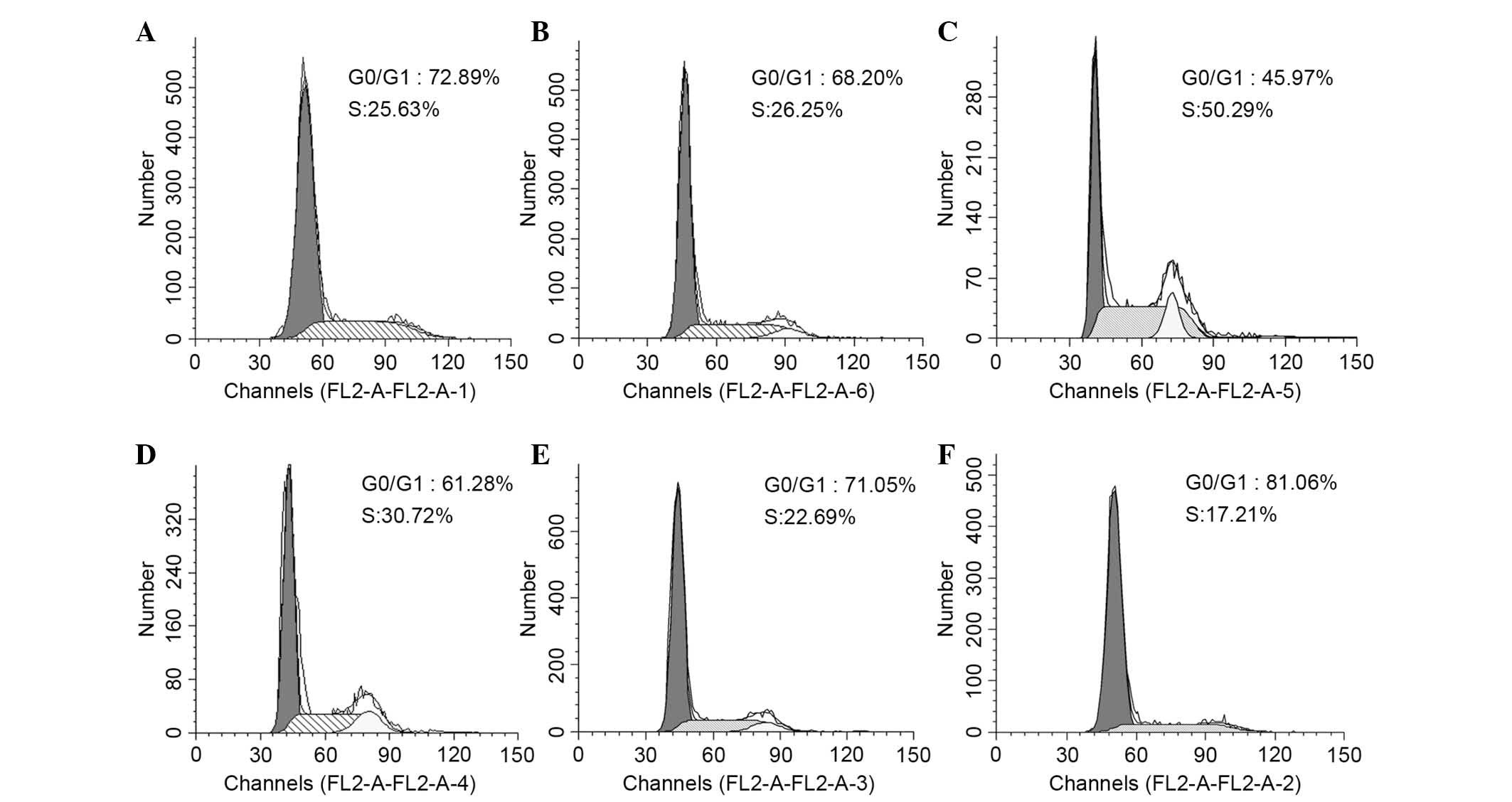
PKG II inhibits EGF-induced cell cycle
progression
STAT3 activity is critical for a wide range of
functions in numerous cell types, including cellular
differentiation, cell-cycle progression, proliferation and survival
(15). In the present study, flow
cytometry was performed to detect the changes in the cell cycle in
differently treated AGS cells. The data shown are the mean ± SD
from 3 independent experiments (P=0.028, the LacZ+EGF group vs. the
control group; P=0.016, the PKG II+cGMP (100 µM)+EGF group
vs. the PKG II+EGF group). The results demonstrated that EGF
treatment (100 ng/ml, 12 h) promoted cells to enter S-phase, with
the number of cells increased from 26.25% (Ad-LacZ group) to 50.29%
(Ad-LacZ + EGF group). The activation of PKG II (through infecting
the cells with Ad-PKG II and treating the cells with 100 or 250
µM cGMP for 1 h) inhibited the effect of EGF, inducing a
decrease from 50.29% (Ad-LacZ + EGF group) to 22.69% (Ad-PKG II +
8-pCPT-cGMP 100 µM group) and 17.21% (Ad-PKG II+8-pCPT-cGMP
250 µM group; Fig. 9).
Discussion
JAKs, which exhibit tyrosine kinase activity, are
associated with several types of cell surface receptors, including
receptors for cytokines and growth factors. The binding of a ligand
to the receptor triggers the activation of JAKs. With increased
kinase activity, JAKs phosphorylate tyrosine residues on the
receptor and create sites for interaction with proteins that
contain phosphotyrosine-binding SH2 domains (1). STATs possess SH2 domains and, thus,
are recruited to the receptors through binding with these
phosphotyrosine residues. Receptor-bound STATs are phosphorylated
by JAKs and the phospho-tyrosine then functions as binding site for
SH2 domains of other STATs, mediating their dimerization. Different
STATs form hetero or homodimers. Finally, activated STAT dimers
accumulate in the cell nucleus and activate transcription of their
target genes (2,16). JAK-STAT-mediated signaling is
associated with cell cycle activity. It may induce expression of
cell cycle-associated proteins, including cyclins, and cause
changes in the cell cycle (13).
The abnormal signal transduction activity of this pathway is
closely associated with tumorigenesis of multiple tumors, including
gastric cancer (17–19).
EGFR is an important member of the receptor tyrosine
kinase family. When a ligand, predominantly EGF, binds with EGFR,
several signal transduction pathways can be initiated (9-11).
EGFR is closely associated with tumorigenesis, and overexpression
and mutation of EGFR occurs in the majority of cancers (20). In vitro experiments have
previously confirmed that overexpression of EGFR caused
transformation of NIH-3T3, Rat-1 and NRK cells, and blocking EGFR
activation inhibited proliferation of certain tumor cells (21). Furthermore, clinical investigate
has previously demonstrated that patients with cancer that
overexpress of EGFR typically have poor prognosis. For example,
EGFR overexpression was detected in 60% of patients with non-small
cell lung cancer and the prognosis of the patients was poor, with
survival of 4–5 months (22).
Thus, EGFR is a potential cancer therapy target and the methods of
inhibiting EGFR activity are of specific significance (23).
Previous results have demonstrated that PKG II
inhibited EGF/EGFR-induced signal transduction of mitogen-activated
protein kinase (MAPK)/extracellular signal-regulated
kinase-mediated, MAPK/JNK-mediated, PLCγ1/protein kinase C-mediated
and PI3K/Akt-mediated pathways (7,8,24,25).
In the current study, as a part of a series of studies, the
inhibitory effect of PKG II on EGF/EGFR-induced signal transduction
of the JAK/STAT-mediated signaling pathway was investigated.
Tyr1068 is the phosphorylation site that is associated with
activation of JAK/STAT-mediated signal transduction (12). The results of the current study
demonstrated that the Tyr1068 phosphorylation level was
significantly increased when the cells were stimulated with EGF,
demonstrating the activation of EGFR and the initiation of
JAK/STAT-mediated signaling. Increased PKG II activity efficiently
inhibited this phosphorylation. For the important signaling
components of this signal transduction pathway, including JAK1 and
JAK2, STAT1 and STAT3, EGF-induced phosphorylation (activation) was
detected and the inhibitory effect of PKG II was determined. For
the EGF-induced gene transcription activity, expression of cell
cycle-associated proteins and cell cycle process, the inhibitory
effect of PKG II was also clearly demonstrated in the study. These
results demonstrated the inhibitory effect of PKG II on
EGF/EGFR-initiated signaling of the JAK/STAT-mediated pathway and
further confirmed that PKG II exerts direct inhibitory activity of
EGFR, and thus, exhibits wide-ranging inhibition of
EGF/EGFR-initiated signal transduction and biological activities.
The present results, and findings from previous studies, strongly
indicate that PKG II is a potential inhibitor of EGFR and will
provide novel insights into anti-cancer strategies.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81272755 and 81201959);
Natural Science Foundation of Colleges and Universities in Jiangsu
Province (grant no. 12KJB310001); the Specialized Research Fund for
Senior Personnel Program of Jiangsu University (grant no.
11JDG114); Chinese Postdoctoral Science Foundation (grant no.
2014M561599); Postdoctoral research grants program of Jiangsu
Province (grant no. 1401144C). The Innovation Grant of Jiangsu
University (grant no. CXZZ13-0701). We thank Dr Gerry Boss and Dr
Renate Pilz (University of California, San Diego, USA). for the
kind gifts of adenoviral constructs.
References
|
1
|
Aaronson DS and Horvath CM: A road map for
those who don't know JAK-STAT. Science. 296:1653–1655. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hebenst reit D, Horejs-Hoeck J and Duschl
A: JAK/STAT-dependent gene regulation by cytokines. Drug News
Perspect. 18:243–249. 2005. View Article : Google Scholar
|
|
3
|
Hofmann F: The biology of cyclic
GMP-dependent protein kinases. J Biol Chem. 280:1–4. 2005.
View Article : Google Scholar
|
|
4
|
Cook AL and Haynes JM: Protein kinase G
II-mediated proliferative effects in human cultured prostatic
stromal cells. Cell Signal. 16:253–261. 2004. View Article : Google Scholar
|
|
5
|
Swartling FJ, Ferletta M, Kastemar M,
Weiss WA and Westermark B: Cyclic GMP-dependent protein kinase II
inhibits cell proliferation, Sox9 expression and Akt
phosphorylation in human glioma cell lines. Oncogene. 28:3121–3131.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fallahian F, Karami-Tehrani F, Salami S
and Aghaei M: Cyclic GMP induced apoptosis via protein kinase G in
oestrogen receptor-positive and-negative breast cancer cell lines.
FEBS J. 278:3360–3369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Y, Chen Y, Qu R, Lan T and Sang J: Type
II cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cells. Oncol Rep. 27:553–558. 2012.
|
|
8
|
Lan T, Chen Y, Sang J, Wu Y, Wang Y, Jiang
L and Tao Y: Type II cGMP-dependent protein kinase inhibits
EGF-induced MAPK/JNK signal transduction in breast cancer cells.
Oncol Rep. 27:2039–2044. 2012.PubMed/NCBI
|
|
9
|
Xie Z, Peng J, Pennypacker SD and Chen Y:
Critical role for the catalytic activity of phospholipase C-gamma1
in epidermal growth factor-induced cell migration. Biochem Biophys
Res Commun. 399:425–428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dong P, Xu Z, Jia N, Li D and Feng Y:
Elevated expression of p53 gain-of-function mutation R175H in
endometrial cancer cells can increase the invasive phenotypes by
activation of the EGFR/PI3K/AKT pathway. Mol Cancer. 8:1032009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quesnelle KM, Boehm AL and Grandis JR:
STAT-mediated EGFR signaling in cancer. J Cell Biochem.
102:311–319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morandell S, Stasyk T, Skvortsov S, Ascher
S and Huber LA: Quantitative proteomics and phosphoproteomics
reveal novel insights into complexity and dynamics of the EGFR
signaling network. Proteomics. 8:4383–4401. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Makki R, Meister M, Pennetier D, Ubeda JM,
Braun A, Daburon V, Krzemień J, Bourbon HM, Zhou R, Vincent A and
Crozatier M: A short receptor downregulates JAK/STAT signalling to
control the Drosophila cellular immune response. PLoS Biol.
8:e10004412010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barré B, Avril S and Coqueret O: Opposite
regulation of myc and p21waf1 transcription by STAT3 proteins. J
Biol Chem. 278:2990–2996. 2003. View Article : Google Scholar
|
|
15
|
Nichane M, Ren X and Bellefroid EJ:
Self-regulation of Stat3 activity coordinates cell-cycle
progression and neural crest specification. EMBO J. 29:55–67. 2010.
View Article : Google Scholar :
|
|
16
|
Wang YH and Huang ML: Organogenesis and
tumorigenesis: Insight from the JAK/STAT pathway in the Drosophila
eye. Dev Dyn. 239:2522–2533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation, and
cancer: How intimate is the relationship? Ann N Y Acad Sci.
1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiong H, Du W, Wang JL, Wang YC, Tang JT,
Hong J and Fang JY: Constitutive activation of STAT3 is predictive
of poor prognosis in human gastric cancer. J Mol Med (Berl).
90:1037–1046. 2012. View Article : Google Scholar
|
|
19
|
Deng JY, Sun D, Liu XY, Pan Y and Liang H:
STAT-3 correlates with lymph node metastasis and cell survival in
gastric cancer. World J Gastroenterol. 16:5380–5387. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Normanno N, Bianco C, De Luca A, Maiello
MR and Salomon DS: Target-based agents against ErbB receptors and
their ligands: A novel approach to cancer treatment. Endocr Relat
Cancer. 10:1–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu T and Li C: Convergence between
Wnt-β-catenin and EGFR signaling in cancer. Mol Cancer. 9:2362010.
View Article : Google Scholar
|
|
22
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Quatrale AE, Porcelli L, Silvestris N,
Colucci G, Angelo A and Azzariti A: EGFR tyrosine kinases
inhibitors in cancer treatment: In vitro and in vivo evidence.
Front Biosci (Landmark Ed). 16:1962–1972. 2011. View Article : Google Scholar
|
|
24
|
Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu
M, Tao Y, Wang Y, Qian H and Gu L: PKG II inhibits EGF/EGFR-induced
migration of gastric cancer cells. PLoS One. 8:e616742013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu M, Chen Y, Jiang L, Li Y, Lan T, Wang Y
and Qian H: Type II cGMP-dependent protein kinase inhibits
epidermal growth factor-induced phosphatidylinositol-3-kinase/Akt
signal transduction in gastric cancer cells. Oncol Lett.
6:1723–1728. 2013.PubMed/NCBI
|